
Abstract
Choroideremia is a complex and rare disease that is frequently misdiagnosed due to its similar appearance to classic retinitis pigmentosa. Recent advances in genetic testing have identified specific genetic mutations in many retinal dystrophies, and the identification of the mutation of the CHM gene on the X chromosome 25 years ago has paved the way for gene replacement therapy with the first human trials now underway. This article reviews the epidemiological and pathological features of choroideremia and new prospects in imaging to monitor disease progression, as well as potential treatment approaches for choroideremia.
Introduction
Choroideremia is an X-linked, recessive disease of the retina resulting in progressive degeneration of the retina, the retinal pigment epithelium (RPE), and the choroid. The prevalence of choroideremia is estimated to be approximately 1:50,000 in people of European descent.Citation1
The disease is characterized by the clinical appearance of atrophy of the choroid resulting in a characteristic pale color of the fundus, originating from the translucence of the white sclera (). In males, the condition gradually advances and starts with night blindness and eventually results in progressive loss of peripheral vision and total blindness in the late stages, although some extreme peripheral field detection may remain.Citation2 However, disease progression is relatively slow and the fovea is generally only affected in the end stages of the disease. Therefore, patients may retain good central visual function as late as 50–70 years old.Citation3,Citation4
Figure 1 Fundus color photographs of the right and left eye of a patient with advanced choroideremia.
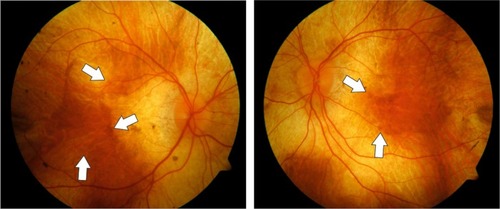
Although X-linked diseases preferentially occur in men (as they have only one X chromosome) choroideremia may also present in female carriers. Carrier females may show characteristic pigmentary changes of the fundus with patchy chorioretinal degeneration. This is due to the process of lyonization in which one copy of the X chromosome is silencedCitation5 and therefore cells carrying the mutation are intermixed with cells expressing the normal X chromosome resulting in a mosaic pattern of the disease. Therefore, most female carriers are asymptomatic and maintain normal visual acuity. The characteristic fundus appearance and a family history consistent with X-linked disease are sufficient for the diagnosis of choroideremia. Other retinal dystrophies with similar fundus appearance can usually be ruled out by family history or laboratory testing. These include autosomal recessive gyrate atrophy which can usually be ruled out by family history and testing for hyperornithinemia, Oliver–McFarlane syndrome, a PNPLA6-related disorder and dominantly negative mutations in RPE65 or bifocal chorioretinal atrophy, and an autosomal dominant disease with a gene mutation on chromosome 6.Citation6–Citation8
There are several reports on the histological changes in choroideremia – this is critically important for designing treatments, because it is essential to know which retinal cells are affected by the CHM gene deficiency and therefore need to be targeted by gene therapy vectors. In postmortem histological sections, independent degeneration of choriocapillaris, RPE, and neurosensory retina have been described. Furthermore, areas of relatively well-preserved retinal architecture were found adjacent to areas of severe degeneration, corresponding to the clinical fundus appearance.Citation9,Citation10 Photoreceptor degeneration was less pronounced above preserved RPE, but occasionally photoreceptor loss was seen in areas with preserved RPE.Citation11 Within the affected areas, the retina was thinned due to thinning of outer segments of the photoreceptors and loss of nuclei within the outer nuclear layer. Subsequently, loss of nuclei within the inner nuclear layer was found as well.Citation11 In addition, there was profound rarefication of most vasculature and melanocytes within the choroid. There is some evidence that inflammation occurs within the retina in patients with choroideremia: a recent report has provided evidence of a mild T-lymphocytic infiltration within the choroid.Citation11 Furthermore, pigment filled macrophages have been identified in the subretinal space associated with rosette formation of the retina.Citation12 Whereas inflammation is not likely to play an active part in the pathogenesis of choroideremia, there seems to be a reactive inflammatory response in the areas of active disease as photoreceptor debris usually is phagocytized and removed by macrophages.Citation13
Retinal imaging in choroideremia and implications for gene replacement therapy
In the last decade, advances in retinal imaging have led to an improved understanding of morphological changes occurring during the disease course of choroideremia. Optical coherence tomography (OCT) can be used to diagnose macular changes associated with choroideremia such as choroidal neovascularization,Citation14 cystoid macular edema,Citation15 epiretinal membrane formation,Citation16 outer retinal tubulations,Citation17 macular hole formation,Citation18 and macular hole complicated by retinal detachment.Citation19 Fundus autofluorescence shows a characteristic speckled pattern of low- and high-density fundus autofluorescence ().Citation20 Increased lipofuscin accumulation originating from digestion of degenerating rods by RPE cells is thought to cause increased autofluorescence in choroideremia, whereas low density fluorescence reflects loss of the RPE layer whilst the choroid is still intact. This is corroborated by functional tests, showing an association of multifocal electroretinogram (mfERG) deterioration with major fundus autofluorescence changes.Citation20,Citation21 Despite high resolution OCT and genetic characterization of choroideremia, the sequence of morphological changes leading to vision loss remains unknown. There are several theories on which retinal layers are primarily affected by choroideremia. Whereas some reports show that the RPE is the first layer to degenerate, followed by loss of photoreceptors,Citation22 others claim that the photoreceptor layer,Citation23 in particular the rods,Citation24 is primarily affected, followed by degeneration of the RPE and the choriocapillaris. The latter hypothesis is not however supported by the observations from patients with dominant RPE65 mutations who have a very similar choroidal atrophy appearance to CHM. Since the RPE65 gene is known to be expressed only in the RPE and being dominantly negative, we assume it causes RPE cell death; this phenotype provides an alternative pathway for the choroidal atrophy being RPE-driven in both diseases. Conversely, if CHM were a disease primarily of rod photoreceptor loss, then we might expect it to appear similar to the other forms of rod-cone dystrophy (retinitis pigmentosa).
Figure 2 Multimodal imaging of a patient with choroideremia.
Abbreviations: AF, autofluorescence; OCT, optical coherence tomography; IR, infrared.
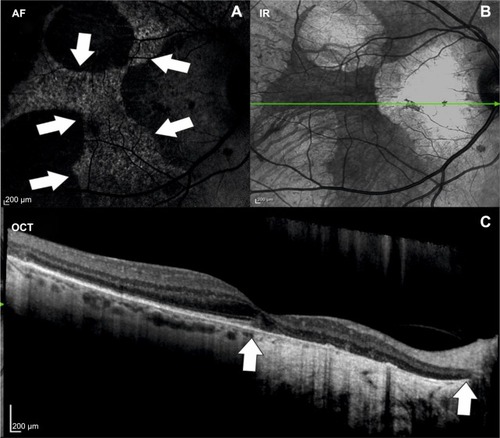
Cone loss at the fovea and smaller atrophic-appearing cones at the edge of degeneration have also been found using adaptive optics scanning laser ophthalmoscopy.Citation25 Functional tests such as mfERG and microperimetry are already employed to monitor therapeutic responses after gene replacement therapy. Future developments in imaging of retinal dystrophies should aim at identifying risk factors and reliable markers for disease progression. Here, several imaging modalities such as OCT angiography or fluorescence lifetime imaging ophthalmoscopy may provide disease specific information. In a recent report using en face OCT angiography, it has been shown that choriocapillaris nonperfusion was more pronounced than the retinal nonperfusion in patients with choroideremia. The observed choriocapillaris nonperfusion is thought to follow the RPE loss and may therefore possibly represent a marker for disease progression and/or response to treatment.Citation26 Fluorescence lifetime imaging ophthalmoscopy is a novel imaging modality able to quantify fluorescence lifetimes in the retina.Citation27,Citation28 In contrast to fundus autofluorescence, fluorescence lifetimes are influenced by metabolic changes within the retina. Therefore, this imaging modality may be valuable to identify areas at risk for disease progression in retinal dystrophies and may be a valuable tool to assess therapeutic responses. The exact sequel of structural changes occurring within the retina has received increasing interest in the last years in light of emerging therapies for retinal diseases.
Genetics of choroideremia
Choroideremia is caused by mutations in the CHM gene, located on the long arm of the X chromosome, encoding the 95 kDa protein Rab escort protein 1 (REP1). The REP1 gene is ubiquitously expressed and acts as an escort protein of native regulators of intracellular trafficking. The deletions in the CHM gene causing choroideremia were first described in 1990.Citation29,Citation30 Mutations in the CHM gene have been identified in European,Citation31–Citation35 Canadian,Citation36 American,Citation37 Japanese,Citation38 and Chinese families.Citation39 These mutations cause truncation or absence of REP1, which plays an essential role in the intracellular vesicular transport. REP1 is involved in lipid modification of Rab proteins by binding newly synthesized, unprenylated Rabs and presenting them to a catalytic Rab geranylgeranyltransferase or farnesyltransferase subunit for the geranylgeranyl transfer reaction.Citation40 Furthermore, REP1 facilitates transport of prenylated Rabs to their specific destination membrane by binding to the hydrophobic prenylation motifs at their C termini.Citation41 Rab proteins are present in all cells and so far more than 50 Rab proteins have been identified.Citation42 Within the eye, many processes such as transport of proteins within the photoreceptors or regulation of phagocytosis and degradation of disc membranes shed from the outer segments of photoreceptors by RPE are reliant on Rab protein function. Generally, loss of REP1 is compensated by Rab escort protein 2, a protein that closely resembles REP1 and is encoded by the X-linked retrogene CHML (choroideremia-like), which is located on chromosome 1.Citation43 Most newly synthesized Rabs bind to one of the two REP proteins and therefore mutations in CHM generally do not lead to systemic disease. A possible explanation for the eye restricted phenotype is that some Rabs, such as Rab 27a which has a key role within the retina,Citation44 are preferentially prenylated by REP1 or that REP2 prenylated Rab27a complexes may have a lower affinity to geranylgeranyltransferase.Citation45 Therefore, the phenotype of choroideremia may be mainly caused by a tissue specific inability of REP2 to compensate for the function of REP1.
Review of recent developments in therapies for choroideremia
The retina is unusually accessible in the evaluation of trials of gene therapy. Not only can disease progression be imaged in nearly histological detail using high resolution OCT, but also there are several functional tests available to monitor therapeutic responses. Furthermore, modern vitreoretinal techniques allow for controlled and local delivery of gene replacement products to the affected tissue. Extensive preliminary work in mouse and dog models of retinal degeneration have shown that the use of adeno-associated vectors to deliver gene replacement therapy in the retina is safe and may result in stable and substantial restoration of the gene product and functional improvement.Citation46 Of the many different adenoassociated virus (AAV) subtypes, AAV2 has a reasonable affinity for primate photoreceptors and RPE,Citation47 but has the advantage over other serotypes in being well characterized in a number of animal models. Therefore, AAV2 has been used as the vector for gene replacement therapy in several gene therapy trials for retinal dystrophies.Citation48–Citation51 Because the amount of genetic material is limited by the packaging capacity of the vector, the size of gene to be replaced is of critical relevance. The limit for AAV vectors is estimated to be around 5 kb for single stranded DNA.Citation52 With a total size of approximately 1.9 kb, the coding sequence of REP1 fits well within the packaging limit of AAV2 with spare space for other regulatory elements, such as the promoter and poly-A signal sequences.
Preclinical studies have shown the feasibility of REP1 replacement therapy using AAV2 in Chmnull/+ mice. In Chmnull/+ mice, selective ablation of Chm in photoreceptor cells or the RPE results in independent degeneration of both of these layers resembling the clinical phenotype of choroideremia.Citation53 Using AAV2-REP1 delivered subretinally, a dose-dependent improvement of retinal function was observed in Chmnull/+ mice. These findings served as proof of concept for future clinical gene replacement therapies in humans.Citation54
Retinal gene therapy in humans involves delivery of a viral vector to the photoreceptors and the RPE. In order to reach this tissue, a pars plana vitrectomy is necessary, followed by a small retinotomy and subretinal injection of fluid containing the vector. This procedure is highly complex because it must be done without damaging the fovea in a degenerate retina. The injection, to some extent, resembles subretinal injection of recombinant tissue plasminogen activator for the treatment and displacement of subretinal hemorrhage, although in these cases the retina is already detached and the visual acuity is severely compromised. In CHM, the potential risks of subretinal gene therapy should not be underestimated as the delivery of the vector entails detachment of the fovea which may lead to reduction of contrast sensitivity or even visual acuity. Furthermore, it is unknown whether or not the gene product or the vector might cause an immune reaction with potentially detrimental effects to the retina in some individuals.
Recently, preliminary results of a Phase I–II clinical trial assessing the effects of the AAV2-REP1 vector in six patients with choroideremia have been published.Citation55 In this report, gene therapy with AAV2-REP1 was shown to be safe with a greater than 2- and 4-line improvement of visual acuity in two patients. Furthermore, it was shown that the five patients who received the full dose of AAV vector had improvements in mean retinal sensitivity. In this study, only patients with advanced disease with macular involvement were included. Four patients had near normal best corrected visual acuity at baseline and detachment of the fovea, which was required for gene replacement delivery, was shown to be well tolerated in these patients. Future prospects of gene replacement therapy in patients with choroideremia should target patients at an early disease stage when the retinal architecture and function are still intact. This has several implications for the clinician. Because there is now a potential therapy for patients with choroideremia and other retinal dystrophies, counseling of patients in clinics should include information on current results of gene replacement trials. Furthermore, genetic testing and molecular diagnosis of the DNA or messenger RNA causing retinal dystrophy should be used to support the clinical diagnosis. Even in the case of clinically confirmed choroideremia, genetic confirmation should be obtained in order to determine whether a patient is suitable for future gene replacement therapy trials. In addition, it is important to establish a database of patients with confirmed genetic mutations to inform patients of forthcoming gene replacement therapies and offer them the possibility of participating in future trials.
Disclosure
REM is a founder and director of NightstaRx Ltd, a choroi-deremia gene therapy company established by the University of Oxford and funded by the Wellcome Trust. MSZ reports no conflicts of interest in this work.
References
- MacDonaldIMSeredaCMcTaggartKMahDChoroideremia gene testingExpert Rev Mol Diagn2004447848415225095
- MacDonaldIMMahDYHoYKLewisRASeabraMCA practical diagnostic test for choroideremiaOphthalmology1998105163716409754170
- KarnaJChoroideremia. A clinical and genetic study of 84 Finnish patients and 126 female carriersActa Ophthalmol Suppl19861761683014804
- CoussaRGKimJTraboulsiEIChoroideremia: effect of age on visual acuity in patients and female carriersOphthalmic Genet201233667322060191
- LyonMFGene action in the X-chromosome of the mouse (Mus musculus L.)Nature196119037237313764598
- GodleyBFTiffinPAEvansKKelsellREHuntDMBirdACClinical features of progressive bifocal chorioretinal atrophy: a retinal dystrophy linked to chromosome 6qOphthalmology19961038938988643244
- BowneSJHumphriesMMSullivanLSA dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvementEur J Hum Genet2011191074108121654732
- KmochSMajewskiJRamamurthyVMutations in PNPLA6 are linked to photoreceptor degeneration and various forms of childhood blindnessNat Commun20156561425574898
- McCullochJCThe pathologic findings in two cases of choroideremiaTrans Am Acad Ophthalmol Otolaryngol19505456557215424921
- RafuseEVMcCullochCChoroideremia. A pathological reportCan J Ophthalmol196833473525727760
- MacDonaldIMRussellLChanCCChoroideremia: new findings from ocular pathology and review of recent literatureSurv Ophthalmol20095440140719422966
- RodriguesMMBallintineEJWiggertBNLeeLFletcherRTChaderGJChoroideremia: a clinical, electron microscopic, and biochemical reportOphthalmology1984918738836089068
- JolySFranckeMUlbrichtECooperative phagocytes: resident microglia and bone marrow immigrants remove dead photoreceptors in retinal lesionsAm J Pathol20091742310232319435787
- SawaMTamakiYKlancnikJMJrYannuzziLAIntraretinal foveal neovascularization in choroideremiaRetina20062658558816770269
- GeneadMAFishmanGACystic macular oedema on spectral-domain optical coherence tomography in choroideremia patients without cystic changes on fundus examinationEye (Lond)201125849020966974
- CameronJDFineBSShapiroIHistopathologic observations in choroideremia with emphasis on vascular changes of the uveal tractOphthalmology1987941871963574884
- GoldbergNRGreenbergJPLaudKTsangSFreundKBOuter retinal tubulation in degenerative retinal disordersRetina2013331871187623676993
- ZinkernagelMSGroppeMMacLarenREMacular hole surgery in patients with end-stage choroideremiaOphthalmology20131201592159623562166
- ShinodaHKotoTFujikiKMurakamiATsubotaKOzawaYClinical findings in a choroideremia patient who underwent vitrectomy for retinal detachment associated with macular holeJpn J Ophthalmol20115516917121400066
- PreisingMNWegscheiderEFriedburgCPoloschekCMWabbelsBKLorenzBFundus autofluorescence in carriers of choroideremia and correlation with electrophysiologic and psychophysical dataOphthalmology200911612011209.e1–e219376587
- EdwardsTLGroppeMJollyJKDownesSMMacLarenRECorrelation of retinal structure and function in choroideremia carriersOphthalmology20151221274127625682176
- FlanneryJGBirdACFarberDBWeleberRGBokDA histopatho-logic study of a choroideremia carrierInvest Ophthalmol Vis Sci1990312292362303326
- JacobsonSGCideciyanAVSumarokaARemodeling of the human retina in choroideremia: rab escort protein 1 (REP-1) mutationsInvest Ophthalmol Vis Sci2006474113412016936131
- SyedNSmithJEJohnSKSeabraMCAguirreGDMilamAHEvaluation of retinal photoreceptors and pigment epithelium in a female carrier of choroideremiaOphthalmology200110871172011297488
- SyedRSundquistSMRatnamKHigh-resolution images of retinal structure in patients with choroideremiaInvest Ophthalmol Vis Sci20135495096123299470
- JiaYBaileySTHwangTSQuantitative optical coherence tomography angiography of vascular abnormalities in the living human eyeProc Natl Acad Sci U S A2015112E2395E240225897021
- DysliCDysliMEnzmannVWolfSZinkernagelMSFluorescence lifetime imaging of the ocular fundus in miceInvest Ophthalmol Vis Sci2014557206721525249601
- DysliCQuellecGAbeggMQuantitative analysis of fluorescence lifetime measurements of the macula using the fluorescence lifetime imaging ophthalmoscope in healthy subjectsInvest Ophthalmol Vis Sci2014552106211324569585
- CremersFPSankilaEMBrunsmannFDeletions in patients with classical choroideremia vary in size from 45 kb to several megabasesAm J Hum Genet1990476226282220804
- van de PolTJCremersFPBrohetRMWieringaBRopersHHDerivation of clones from the choroideremia locus by preparative field inversion gel electrophoresisNucleic Acids Res1990187257311969148
- BeaufrereLTufferySHamelCThe protein truncation test (PTT) as a method of detection for choroideremia mutationsExp Eye Res1997658498549441709
- PascalODonnellyPFouanonCHerbertOLe RouxMGMoisanJPA new (old) deletion in the choroideremia geneHum Mol Genet1993214898242078
- SankilaEMTolvanenRvan den HurkJACremersFPde la ChapelleAAberrant splicing of the CHM gene is a significant cause of choroideremiaNat Genet199211091131302003
- SchwartzMRosenbergTvan den HurkJAvan de PolDJCremersFPIdentification of mutations in Danish choroideremia familiesHum Mutat1993243478477262
- van BokhovenHSchwartzMAndreassonSMutation spectrum in the CHM gene of Danish and Swedish choroideremia patientsHum Mol Genet19943104710517981671
- NesslingerNMitchellGStrasbergPMacDonaldIMMutation analysis in Canadian families with choroideremiaOphthalmic Genet19961747528832720
- ForsythePMaguireAFujitaRMoenCSwaroopABennettJA carboxy-terminal truncation of 99 amino acids resulting from a novel mutation (Arg555 – > stop) in the CHM gene leads to choroideremiaExp Eye Res1997644874909196401
- FujikiKHottaYHayakawaMREP-1 gene mutations in Japanese patients with choroideremiaGraefes Arch Clin Exp Ophthalmol199923773574010447648
- ZhouQLiuLXuFGenetic and phenotypic characteristics of three Mainland Chinese families with choroideremiaMol Vis20121830931622355242
- Pereira-LealJBHumeANSeabraMCPrenylation of Rab GTPases: molecular mechanisms and involvement in genetic diseaseFEBS Lett200149819720011412856
- PylypenkoORakAReentsRStructure of rab escort protein-1 in complex with rab geranylgeranyltransferaseMol Cell20031148349412620235
- Pereira-LealJBSeabraMCThe mammalian Rab family of small GTPases: definition of family and subfamily sequence motifs suggests a mechanism for functional specificity in the Ras superfamilyJ Mol Biol20003011077108710966806
- CremersFPArmstrongSASeabraMCBrownMSGoldsteinJLREP-2, a Rab escort protein encoded by the choroideremia-like geneJ Biol Chem1994269211121178294464
- TolmachovaTAndersRAbrinkMIndependent degeneration of photoreceptors and retinal pigment epithelium in conditional knockout mouse models of choroideremiaJ Clin Invest200611638639416410831
- LarijaniBHumeANTarafderAKSeabraMCMultiple factors contribute to inefficient prenylation of Rab27a in Rab prenylation diseasesJ Biol Chem2003278467984680412941939
- AclandGMAguirreGDBennettJLong-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindnessMol Ther2005121072108216226919
- BennettJMaguireAMCideciyanAVStable transgene expression in rod photoreceptors after recombinant adeno-associated virus-mediated gene transfer to monkey retinaProc Natl Acad Sci U S A1999969920992510449795
- MaguireAMHighKAAuricchioAAge-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trialLancet20093741597160519854499
- JacobsonSGCideciyanAVRatnakaramRGene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 yearsArch Ophthalmol201213092421911650
- HauswirthWWAlemanTSKaushalSTreatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trialHum Gene Ther20081997999018774912
- BainbridgeJWMehatMSSundaramVLong-term effect of gene therapy on leber’s congenital amaurosisN Engl J Med2015372201887189725938638
- WuZYangHColosiPEffect of genome size on AAV vector packagingMol Ther201018808619904234
- TolmachovaTWavre-ShaptonSTBarnardARMacLarenREFutterCESeabraMCRetinal pigment epithelium defects accelerate photoreceptor degeneration in cell type-specific knockout mouse models of choroideremiaInvest Ophthalmol Vis Sci2010514913492020445111
- TolmachovaTTolmachovOEBarnardARFunctional expression of rab escort protein 1 following AAV2-mediated gene delivery in the retina of choroideremia mice and human cells ex vivoJ Mol Med (Berl)20139182583723756766
- MacLarenREGroppeMBarnardARRetinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trialLancet20143831129113724439297