
Abstract
Osteogenesis imperfecta (OI) is a group of genetic skeletal disorders, with a prevalence of 1 in 15,000–20,000 births. OI type V has been described in approximately 150 cases and all patients carry the variant (c.-14C> T) in the IFITM5 gene. However, two other variants, p.S40L and p.N48S have been reported in this gene, leading to clinical phenotypes different from OI type V. Here we described a patient with multiple bone fractures, scoliosis, skull alteration (plagiocephaly), bone deformation, bone rickets, and intramedullary epithelioid osteosarcoma that bears the recently reported heterozygous variant c.143A>G (p.N48S) in the IFITM5 gene. This case supports the pathogenicity of this new variant in the IFITM5 gene and adds information regarding its clinical phenotype.
Introduction
Osteogenesis imperfecta (OI), also known as brittle bone disease, is a group of disorders characterized by bone fragility, susceptibility to bone fractures, bone deformity, and growth deficiency, with variable genetic and clinical presentations, usually related to collagen type 1 biosynthesis (OI types I–IV, most frequent genes: COL1A1 and COL1A2) but other forms related to mineralization defect (OI type VI, gene SERPINF1), 3-hydroxylation defects (OI types VII–IX, genes CRTAP, LEPRE1, and PPIB, respectively) and chaperone defects (OI types X and XI, genes SERPINH1 and FKBP10, respectively) have been described.Citation1 Other general manifestations of OI include blue sclerae, dentinogenesis imperfecta, joint laxity, and hearing loss.Citation2 The prevalence of these disorders is 1 in 15,000–20,000 births,Citation1 with classical, collagen alteration-related OI (types I–IV) comprising about 90% of cases.Citation2
Another OI not mentioned above is OI type V (OMIM: 610967), the only type, aside from types I–IV, to have a dominant inheritance pattern.Citation1 The causative gene for OI type V was published in 2012 by two different groups and was mapped to the interferon-induced transmembrane protein 5 (IFITM5) gene, in which patients present the point mutation (c.-14C> T) in its 5′ untranslated region (UTR) that adds five amino acids (Met-Ala-Leu-Glu-Pro) to the N-terminal region of the protein, altering its function.Citation3,Citation4 Clinical manifestations of OI type V comprise hyperplastic callus formation, radial head dislocation, radioulnar interosseous membrane ossification, limitation in forearm rotation, irregularly arranged lamellae, and a mesh-like pattern of lamellation.Citation5
The other, less common variant, described in the IFITM5 gene leading to OI, is the missense c.119C> T (p.S40L) variant. The patients that bear this mutation are classified as atypical OI type VI, given the bone histology that resembles the one found in patients with OI type VI.Citation6 Recently, a third variant, c.143A>G (p.N48S), was described in the IFITM5 gene, leading to a phenotype different from the one seen in type V, with fish-scale pattern in lamellar bone, osteoporosis, and calvarial doughnut lesions.Citation7 The disorders caused by variants in the IFITM5 gene are rare, with the most common (c.-14C> T) variant described in approximately 150 cases up to 2019,Citation8 which made the description of the patients bearing variants in this gene of particular interest for physicians.Citation9 Here we described a Colombian male patient diagnosed with osteogenesis imperfecta with the heterozygous variant c.143A>G (p.N48S) in the IFITM5 gene.
Case Presentation
A 12-year-old male patient was adopted at three months old, his adoptive mother referred that he was born at term without complications and that his family history was unremarkable. According to the adoptive mother and the clinical history, at 3 months of age, the proband already had bone rickets and multiple bones and skull fractures that led to a diagnosis of osteogenesis imperfecta (OI); however, the OI type was not determined.
At 9 years of age, a biopsy taken from a lesion in the right humerus, and the computerized axial tomography (CT) revealed a clinical finding suggestive of conventional intramedullary osteosarcoma, epithelioid subtype. Thorax CT did not show the presence of osteosarcoma in other structures and the bone gammagraphy confirmed the presence of cancer only in the right humerus; this test also showed intense captation by the column, maxillary, and sacrum, although this was attributed to the OI. Given the extent of the osteosarcoma, the right arm was amputated and ten cycles of chemotherapy were given without complications derived from the immunosuppression. Anatomo-pathological evaluation of the amputated arm showed a large and homogeneous tumor lesion (lesion length, width, and height: 14, 7.5, and 7 centimeters, respectively) originating from the shaft of the humerus, extending to the metaphysis, and expansively penetrating the surrounding soft tissue without compromising vascular structures. Histological examination also showed a malignant mesenchymal neoplastic lesion made up of epithelioid cells with increased nucleus–cytoplasm ratio, anisocytosis, anisokaryosis, and evident nucleoli. These cells were positive for vimentin and CD99 markers.
At 10 years old, a clinical whole-exome sequencing test was performed, revealing the heterozygous variant c.143A>G (p.N48S) in the IFITM5 gene and classified as a variant of uncertain significance (VUS) according to the guidelines of the College of Medical Genetics and Genomics ACMG.Citation10 This IFITM5 variant is predicted to be damaging by SIFT (0.012) and PROVEAN, probably damaging (0.896) by Polyphen2, disease-causing by MutationTaster2, and have a CADD score of 22.8.
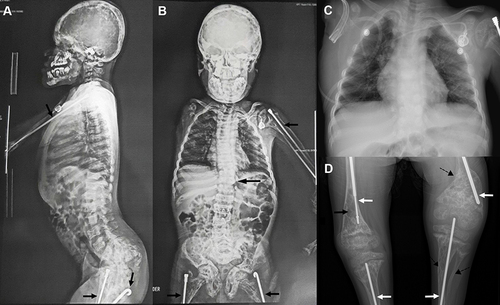
Physical examination at age 12 showed gait disorder (the patient did not walk), cranial deformities, flattened head (plagiocephaly), and an umbilical hernia. Other alterations included a flat nasal bridge, gingival melanosis, thick lips, and a soft abdomen without palpable hepatosplenomegaly. No blue sclerae or hearing loss were noticed. Weight was 25.4 kg (1st percentile), height was 106 cm (<1st percentile), and head circumference was 52 cm (11th percentile). Radiological findings from CT of the extremities and spine orthography were suggestive of decreased bone mineralization (), multiple past fractures, and defects in bone remodeling in all the bones (); beaded ribs and thoracolumbar scoliosis (Cobb angle: 11) were also noticed (). Neither physical examination nor radiological findings showed signs of osteosarcoma recurrence. To date, the patient has had 12 long-bone fractures leading to deformities, requiring correction by osteotomy and insertion of intramedullary rods, and bowing of the left femur, tibia, and fibula (). Fractures are commonly located in the diaphysis of the left and right (before arm amputation) humerus, radius, ulna, femur, tibia, and fibula. Neither vertebral compression fractures nor hyperplastic callus formation has been noticed so far. Paraclinical tests showed normal levels of blood calcium (9,6 mg/dL), ionized calcium (1.34 millimol/L), phosphate in the urine (0.44 g/24 h), parathyroid hormone (51.7 pg/mL), vitamin D (52.3 ng/mL), blood magnesium (1.83 mg/dL), and lactate dehydrogenase (193 IU/L); decreased levels of calcium in the urine (29.7 mg/24 hours), and elevated levels of alkaline phosphatase (901 IU/L). The proband has been treated with intramedullary nails, osteotomies for bone deformation, vitamin D, intravenous zoledronic acid, and growth hormone since the diagnosis of OI at 3 months of age. Currently, he is in pediatric gastroenterology, oncohematology, nephrology, endocrinology, and palliative care follow-up.
Figure 1 Skeletal radiographs of the patient. (A and B) Mild scoliosis and intramedullary rods in the long bones (black arrows). (C) Thorax CT-scan, and (D) Intramedullary rods (white arrows), diaphyseal bowing (dashed arrows), and osteopenia (black arrow).
Discussion
Genetic skeletal disorders are a highly heterogeneous group of diseases in which a large number of genes are involved, with more new genes and new variants, in those or already described genes, been reported every year.Citation11 Currently, 437 disease-causing genes have being identified in these disorders, with many of them involved in processes different than bone and cartilage formation or skeletal development,Citation11 complicating the elucidation of the pathophysiological mechanism driving the disease, and therefore, making complex the anticipation of future clinical manifestations in the patients.
Among genetic skeletal disorders, OI type V is a highly phenotypically heterogeneous entity, even in patients from the same family, but with a homogeneous genetic etiology; almost all patients present with the point mutation (c.-14C> T) in the IFITM5 gene, but clinical manifestations can vary between facial gestalt, knee contracture, susceptibility to bones fractures (some patients do not present this typical OI manifestation), scoliosis, radioulnar interosseous membrane ossification, bowing of the long bones, and so on.Citation8,Citation12 Our patient had multiple bone fractures since 3 months old, especially in long bones, therefore a finding of elevated alkaline phosphatase was expected; he also had bone rickets, mild scoliosis, bowing of the lower long bones, plagiocephaly, no capacity to ambulate, and the most interesting clinical manifestation noticed, intramedullary osteosarcoma (epithelioid subtype) in his right humerus, leading to arm amputation given its aggressive growth. Osteosarcoma diagnosis in patients with OI type V has to be established carefully, as one of the differential diagnoses of this malignancy is hyperplastic callus formation (HPC), a relatively common feature of patients with this bone disease.Citation8 However, in this case, the pathology results of the biopsy taken and the amputated limb revealed intramedullary osteosarcoma, and patients reported to have HPC mimicking osteosarcoma can be misdiagnosed with periosteal or cortical osteosarcoma, given the benign mass that forms on the bone surface, giving an appearance, in radiography and CT, similar to this type of osteosarcoma.Citation13,Citation14 Furthermore, this patient, given the variant found in the IFITM5 gene and the absence of classical features of the disease, cannot be classified as OI type V but as IFITM5-related OI until a proper evaluation and classification of patients with this new variant is performed.
On the other hand, the protein coded by the IFITM5 gene, bone-restricted Ifitm-like (BRIL), is expressed only in bone tissue, and particularly in osteoblasts where it appears to have a role in the bone matrix mineralization process in mice.Citation15 However, ifitm5 knock-out mice do not show major abnormalities in bone synthesis. Furthermore, patients bearing mutations in this gene, such as those with OI type V or OI due to the variant c.119C> T (p.S40L) can have a wide heterogeneity and severity of the disease, with under or over mineralization of the bones and also extra-skeletal alterations (eg, blue sclera and dentinogenesis imperfecta) in the latter,Citation9,Citation16 which implies that different mutations in this gene lead to different clinical phenotypes but also that the same mutation produces a wide phenotypic spectrum. In this manner, no conclusion can be drawn regarding the role of the variant c.143A>G (p.N48S) in the extra-skeletal alterations found in our patient, as only two patients have been described previously, and no OI-related extra-skeletal manifestations were found in them.
As mentioned above, genetically, OI type V has a predominant variant causing the disease in the majority of patients, the (c.-14C> T) variant in the IFITM5 gene. However, two other variants have been published to be related to OI in this same gene: the c.119C> T (p.S40L)Citation6 and, the recently described c.143A>G (p.N48S).Citation7 The latter was reported in a mother and her daughter with a novel form of OI named osteoporosis with calvarial doughnut lesions (OP-CDL) that present with skeletal fragility, multiple fractures (especially long bones), scoliosis, bone deformities, and sclerotic skull lesions; a phenotype different from those arising from the classical (c.-14C> T) variant.Citation7 This variant, c.143A>G, was also found in our patient, and as reported it was associated with a deleterious effect by the predicting algorithms such as SIFT, PROVEAN, and the CADD score. Although no sclerotic skull lesions were found, the plagiocephaly, as a cranial alteration, may be associated with this novel variant. Furthermore, osteosarcoma was not reported in the two patients described, however, this malignancy could be one clinical feature of the variant described, but most probably, was a sporadic event rather than an OI-related manifestation.Citation17 The association of OI with cancer has been investigated for several years, however, a clear association has not been established. The protein coded by the COL1A1 gene seems to be involved in some cancer such as colon,Citation18 gastric,Citation19 and ovarian cancersCitation20 but not as a causative factor. On the other hand, neither of the genes involved in OI have been postulated as predisposition genes to cancer in the pediatric populationCitation21 and the cumulative risk of cancer development due to the frequent exposition of these patients to X-ray and CT scans is minimal,Citation22 which supports that the presence of osteosarcoma in our patient with OI probably is a casual relationship.
More cases describing the clinical history of patients with this variant are necessary to establish the full spectrum of its clinical manifestations and a possible genotype–phenotype correlation. Furthermore, a significant limitation in this case report is that no functional evaluation was performed. Thus, it is not possible to conclude if this variant causes a gain-of-function, a loss-of-function, or if the new protein has abnormal interactions with other proteins and if this reflects the distinct clinical manifestations compared to other IFITM5-related OI.
Conclusion
Here we report a patient with an IFITM5-related OI that presented multiple bone fractures since the age of 3 months, scoliosis, plagiocephaly, bone deformation, bone rickets, and conventional, epithelioid osteosarcoma. The heterozygous variant c.143A>G (p.N48S) in the IFITM5 gene was found. As far as we know, this is the first report of conventional osteosarcoma in a patient with OI caused by variants in the IFITM5 gene, although whether it is related to the genetic condition or a sporadic event remains unclear. This report contributes to expanding the phenotype of this newly IFITM5-related OI and also supports its pathogenicity, given that currently it is classified as a VUS variant. More case reports or case series are needed to establish a genotype-phenotype for this new alteration in the IFITM5 gene.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Informed Consent
This study was approved by the Ethics Committee of Fundación Valle del Lili, Colombia (human study protocol #1504) and performed in accordance with the Declaration of Helsinki. Written informed consent was obtained from the parents of the patient. Information revealing the subject’s identity was not included in the manuscript. The patient was identified by number and not by his real name.
Consent for Publication
Written informed consent for publication of clinical details and images/photographs was obtained from the patient.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest.
Acknowledgments
The authors would like to thank the patient and his parents for agreeing to the publication of this report. We also thank the people who have contributed to the development and execution of this study.
References
- Forlino A, Cabral WA, Barnes AM, Marini JC. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol. 2011;7:540–557. doi:10.1038/nrendo.2011.81
- Van Dijk FS, Cobben JM, Kariminejad A, et al. Osteogenesis imperfecta: a review with clinical examples. Mol Syndromol. 2011;2:1–20. doi:10.1159/000332228
- Semler O, Garbes L, Keupp K, et al. A mutation in the 5′-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type v with hyperplastic callus. Am J Hum Genet. 2012;91:349–357. doi:10.1016/j.ajhg.2012.06.011
- Cho TJ, Lee KE, Lee SK, et al. A single recurrent mutation in the 5′-UTR of IFITM5 causes osteogenesis imperfecta type v. Am J Hum Genet. 2012;91:343–348. doi:10.1016/j.ajhg.2012.06.005
- Glorieux FH, Rauch F, Plotkin H, et al. Type V osteogenesis imperfecta: a new form of brittle bone disease. J Bone Miner Res. 2000;15:1650–1658. doi:10.1359/jbmr.2000.15.9.1650
- Reich A, Bae AS, Barnes AM, et al. Type V OI primary osteoblasts display increased mineralization despite decreased COL1A1 expression. J Clin Endocrinol Metab. 2015;100:E325–E332. doi:10.1210/jc.2014-3082
- Mäkitie RE, Pekkinen M, Morisada N, et al. A novel IFITM5 variant associated with phenotype of osteoporosis with calvarial doughnut lesions: a case report. Calcif Tissue Int. 2021;109:626–632. doi:10.1007/s00223-021-00878-5
- Cao YJ, Wei Z, Zhang H, Zhang ZL. Expanding the clinical spectrum of osteogenesis imperfecta type V: 13 additional patients and review. Front Endocrinol. 2019;10:1–9. doi:10.3389/fendo.2019.00375
- Zhytnik L, Maasalu K, Duy BH, et al. IFITM5 pathogenic variant causes osteogenesis imperfecta V with various phenotype severity in Ukrainian and Vietnamese patients. Hum Genomics. 2019;13:25.
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi:10.1038/gim.2015.30
- Mortier GR, Cohn DH, Cormier-Daire V, et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet Part A. 2019;179:2393–2419. doi:10.1002/ajmg.a.61366
- Glorieux FH, Moffatt P. Phenotypic variability of osteogenesis imperfecta type V caused by an IFITM5 mutation. J Bone Miner Res. 2013;28:1519–1522. doi:10.1002/jbmr.1982
- Vonderlind HC, Jessel M, Knobel A, Juergensen I, Struewer J. Late onset hyperplastic callus formation in osteogenesis imperfecta type V simulating osteosarcoma—A case report. Int J Surg Case Rep. 2020;69:83–86. doi:10.1016/j.ijscr.2020.03.024
- Deng Y, Huo Y, Li J. Case report: hyperplastic callus of the femur mimicking osteosarcoma in osteogenesis imperfecta type V. Front Endocrinol. 2021;12:1–6. doi:10.3389/fendo.2021.622674
- Moffatt P, Gaumond MH, Salois P, et al. Bril: a novel bone-specific modulator of mineralization. J Bone Miner Res. 2008;23:1497–1508. doi:10.1359/jbmr.080412
- Hanagata N. IFITM5 mutations and osteogenesis imperfecta. J Bone Miner Metab. 2016;34:123–131. doi:10.1007/s00774-015-0667-1
- Takahashi S, Okada K, Nagasawa H, Shimada Y, Sakamoto H, Itoi E. Osteosarcoma occurring in osteogenesis imperfecta. Virchows Arch. 2004;444:454–458. doi:10.1007/s00428-004-0985-5
- Wang X, Song Z, Hu B, Chen Z, Chen F, Cao C. MicroRNA-642a-5p inhibits colon cancer cell migration and invasion by targeting collagen type I α1. Oncol Rep. 2021;45:933–944. doi:10.3892/or.2020.7905
- Guo Y, Lu G, Mao H, et al. Mir-133b suppresses invasion and migration of gastric cancer cells via the col1a1/tgf-β axis. Onco Targets Ther. 2020;13:7985–7995. doi:10.2147/OTT.S249667
- Li M, Wang J, Wang C, et al. Microenvironment remodeled by tumor and stromal cells elevates fibroblast-derived COL1A1 and facilitates ovarian cancer metastasis. Exp Cell Res. 2020;394:112153. doi:10.1016/j.yexcr.2020.112153
- Zhang J, Walsh MF, Wu G, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373:2336–2346. doi:10.1056/NEJMoa1508054
- Thorby-Lister A, Högler W, Hodgson K, et al. Cumulative radiation exposure from medical imaging and associated lifetime cancer risk in children with osteogenesis imperfecta. Bone. 2018;114:252–256. doi:10.1016/j.bone.2018.06.021