
Abstract
Acute myeloid leukemia (AML) is an aggressive hematopoietic malignancy that is cured in as few as 15%–40% of cases. Tremendous improvements in AML prognostication arose from a comprehensive analysis of leukemia cell genomes. Among normal karyotype AML cases, mutations in the FLT3 gene are the ones most commonly detected as having a deleterious prognostic impact. FLT3 is a transmembrane tyrosine kinase receptor, and alterations of the FLT3 gene such as internal tandem duplications (FLT3-ITD) deregulate FLT3 downstream signaling pathways in favor of increased cell proliferation and survival. FLT3 tyrosine kinase inhibitors (TKI) emerged as a new therapeutic option in FLT3-ITD AML, and clinical trials are ongoing with a variety of TKI either alone, combined with chemotherapy, or even as maintenance after allogenic stem cell transplantation. However, a wide range of molecular resistance mechanisms are activated upon TKI therapy, thus limiting their clinical impact. Massive research efforts are now ongoing to develop more efficient FLT3 TKI and/or new therapies targeting these resistance mechanisms to improve the prognosis of FLT3-ITD AML patients in the future.
Introduction
Acute myeloid leukemia (AML) is an aggressive hematopoietic cancer arising from bone marrow immature myeloid progenitor cells transformed by recurrent genetic alterations that cooperate to induce cell proliferation and survival. These driver genetic events may include gene fusion (such as PML-RARA) or alterations of genes involved in epigenetic (eg, DNMT3A), metabolism (eg, IDH1/2), transcription (eg, NPM1), and signaling (eg, FLT3, fms-like tyrosine kinase 3) regulation.Citation1,Citation2 Among these genes, FLT3 is the one most frequently altered in AML: up to 30% of cases harbor an internal tandem duplication (FLT3-ITD), or a tyrosine kinase domain (TKD) amino acid substitution (FLT3-TKD) is seen in 5%–10%.Citation3,Citation4 Currently, only a minority of patients with AML are cured by highly intensive therapies that encompass high-dose cytotoxic chemotherapy and hematopoietic stem cell transplantation.Citation5 The detection of a FLT3-ITD mutation is a hallmark of adverse prognosis in AML as less than 15% of these patients experience long-term survival despite intensive therapies.Citation6 In contrast, the detection of a FLT3-TKD mutation does not have the same negative impact on survival.Citation7 Hence, finding new therapeutic options for the FLT3-ITD AML patient represents a major goal for physicians and scientists since years. In this review, we will focus on a novel therapeutic modality, the FLT3 tyrosine kinase inhibitors (TKI), and discuss the mechanisms involved in the frequent clinical resistance to these molecules.
FLT3 mutations in AML
FLT3 structure and function
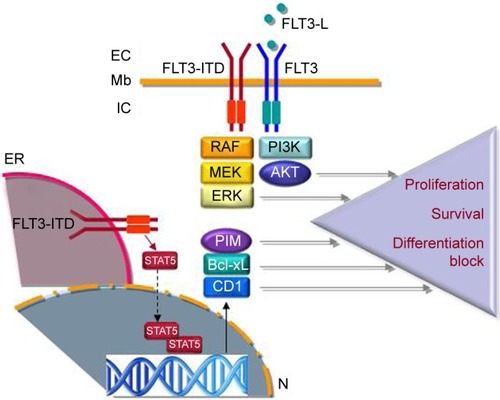
FLT3 belongs to a family of 58 protein tyrosine kinase receptors (TKR) characterized by an extracellular domain for ligand binding, a transmembrane helix, and a C-terminal intracytoplasmic part supporting the tyrosine kinase.Citation8 FLT3 is a Class III TKR characterized by the presence of five immunoglobulin-like motifs within their extracellular part that are exclusively expressed in hematopoietic cells and induce the activation of intracellular signaling pathways such as PI3K/AKT or ERK/MAPK upon ligand binding ().Citation9 Mice knockout experiments revealed the critical role of FLT3 in normal hematopoiesis, as FLT3−/− hematopoietic progenitors fail to compensate for hematopoietic deficiency in lethally irradiated mice.Citation10,Citation11 Moreover, Boyer et alCitation12 demonstrated that in mice, FLT3 is expressed on very immature hematopoietic progenitor cells lacking self-renewing potential but retaining capacity of differentiating into all hematopoietic lineages including megakaryocytes and erythrocytes, highlighting the critical role of FLT3 in the early stages of hematopoiesis.
Figure 1 Schematic view of FLT3 and FLT3-ITD signaling.
Abbreviations: Mb, membrane; EC, extracellular; FLT3-L, FLT3 ligand; IC, intracellular; TKD, tyrosine kinase domain; ER, endoplasmic reticulum; CD1, cyclin D1; AML, acute myeloid leukemia; Pim, proviral integration site.
FLT3 in AML: clinical perspectives
Early reports demonstrated an overexpression of FLT3 mRNA in AML and acute B-cell (but not T-cell) leukemiaCitation13 as well as an overexpression of FLT3 ligand (FLT3-L).Citation14 However, a breakthrough in our understanding of AML pathophysiology came from the discovery of mutations within the FLT3 gene located on chromosome 13q12. Nakao et alCitation15 identified FLT3 FLT3-ITD, detected in up to 30% of diagnosis AML cases, and then Yamamoto et alCitation16 found point mutations within FLT3-TKD, leading to activating amino acid substitutions, which are detected in 5%–10% of AML samples.
From genome-wide sequencing studies, we learned that FLT3 is the most frequently mutated gene in AML, generally detected in samples with normal karyotype, and these mutations frequently co-occur with alterations of other genes such as DNMT3A or NPM1.Citation3,Citation4 The presence of a FLT3-ITD mutation (but not of FLT3-TKD mutations) adversely impacts on prognosis, with a less than 20% long-term overall survival.Citation3,Citation6 In their recent, very large-scale genomic study on AML, Papaemmanuil et alCitation3 observed a FLT3-ITD alteration in as much as 22% of a cohort of 1,540 AML patients. Interestingly, these mutations frequently co-occurred with NPM1 and DNMT3A mutations (39%) and chromatin or RNA splicing gene mutations (15%), and were also found to be associated with t(15;17) and t(6;9) translocations (35% and 80%, respectively). In this large study, FLT3-TKD mutations were found in less than 5% of the cohort, mostly co-occurring with MLL-PTD (1%) and NPM1 (3%) abnormalities. This study unambiguously confirmed the adverse impact of FLT3-ITD mutations, particularly when co-occurring with both DNMT3A and NPM1 mutations, and observed an adverse impact of the gene–gene interaction between MLL-PTD and FLT3-TKD mutations.Citation3
While FLT3-ITD patients experience the same complete remission rate after induction therapy compared with other AML patients, they frequently relapse even after receiving allogeneic stem cell transplantation. The prognosis of relapsing patients is particularly poor as virtually none of the currently used therapies – including TKI that will be discussed later – induces long-term remissions.Citation17 New therapeutic approaches based on a better understanding of FLT3-ITD AML biology are, therefore, warranted to improve patient outcome.
FLT3 in AML: biological perspectives
Recent evidence suggests that AML arises from a clonal hematopoietic compartment referred to as preleukemic stem cells, which is expanded due to early genetic events such as DNMT3A mutations and thereafter transformed due to the occurrence of secondary driver mutations in other genes such as NPM1 or FLT3.Citation18 From the comparison between AML genomes, we also learned that FLT3 mutations occur as secondary events during AML clonal evolution.Citation2 While these data suggest that FLT3 mutations are secondary, late events during leukemogenesis, Chu et alCitation19 demonstrated that FLT3-ITD drives a myeloproliferative disorder in mice due to proliferation and subsequent depletion of a quiescent long-term hematopoietic stem cell population, which was reverted by the FLT3 inhibitor sorafenib. A model to reconcile these findings may involve a preleukemic hematopoietic population amplified by initiating genetic lesions including DNMT3A, ASXL1, or TET2 mutations, which will be transformed into AML due to mutations – such as FLT3-ITD – acquired later in the differentiation process.Citation20 In the absence of a survival advantage during the early stages of hematopoiesis, the occurrence of a FLT3-ITD mutation may in contrast deplete the hematopoietic stem cell compartment, preventing the development of a FLT3-ITD-driven leukemic clone.Citation19
FLT3-ITD mutation produces receptors with a constitutive tyrosine kinase activity due to homodimerization or heterodimerization with the normal FLT3 receptors.Citation21 While FLT3 receptors induce signal transduction from the cell membrane, FLT3-ITD receptors are mostly located in the endoplasmic reticulum (ER) due to glycosylation defects leading to a distinct signaling pattern including the phosphorylation and activation of the STAT5 transcription factor.Citation22 Interestingly, Chu et alCitation19 have shown that FLT3 receptors are not detected at the cell surface of hematopoietic stem cell, but they found FLT3 mRNA and proteins and evidence for FLT3-driven signal transduction in this cellular compartment, suggesting that intracellular ER-dependent FLT3-ITD signaling may be critical in disease propagation.
The constitutive activation of STAT5 by FLT3-ITD promotes AML cell proliferation and survival through the transactivation of target genes including BCL2L1, PIM1, CDKN1A, and CCND1.Citation23 Surprisingly, among STAT5 targets is the suppressor of cytokine signaling 1 gene encoding a negative regulator of cytokine-induced signaling, which promoted FLT3-ITD-induced myeloproliferative disease through escape from interferon growth inhibitory effects during cellular transformation.Citation24 Recently, Chatterjee et alCitation25 demonstrated that STAT5 nuclear translocation was due to a FAK/Rac1-Tiam1/PAK1 axis in FLT3-ITD and also in KIT D816V-positive cells and that targeting FAK, Tiam1, or PAK1 significantly reduced leukemia propagation in part through STAT5 inhibition.
FLT3-ITD also regulates myeloid cell differentiation, which is made evident by the observation of differentiation syndromes in AML patients treated by FLT3 inhibitors.Citation26 In fact, FLT3-ITD blocks the activity of the C/EBPα transcription factor through direct phosphorylation of serine 21 and indirectly through CDK1 activation, inhibiting myeloid differentiation.Citation27,Citation28 In addition, other FLT3-ITD target genes are involved in differentiation including RGS2 and GADD45.Citation29,Citation30
Interestingly, metabolic pathways – ie, glycolysis and mitochondrial tricarboxylic acid cycle – recently emerged as new targets for cancer therapy.Citation31 In FLT3-ITD AML cells, Alvarez-Calderon et alCitation32 demonstrated that targeting mitochondrial metabolism using oligomycin A, an inhibitor of the mitochondrial ATP synthase, enhanced the antileukemic activity of the FLT3 TKI quizartinib against primary AML samples in vitro and in vivo in humanized patient-derived xenograft mouse models, suggesting that FLT3-ITD could induce an addiction of AML cells to mitochondrial metabolism.
Finally, recent data implicated a deregulation of oxidative metabolism in FLT3-ITD-induced leukemogenesis. FLT3-ITD receptors increase the expression of reactive oxygen species (ROS) due to increased nicotinamide adenine dinucleotide phosphate (NADPH) oxidase isoform 4 expression through STAT5 and also p22(phox), a small membrane-bound subunit of the NADPH oxidase complex.Citation33,Citation34 Besides DNA damage and genomic instability, ROS production may affect the function of signaling effectors as illustrated by Godfrey et al,Citation35 showing that ROS overproduction due to FLT3-ITD inactivates the protein tyrosine phosphatase DEP-1 – a negative regulator of FLT3-ITD signaling – through oxidation of its catalytic cysteine.
FLT3 tyrosine kinase inhibitors
The characterization of frequent FLT3 mutations in AML patients and their correlation to adverse clinical outcome in the case of FLT3-ITD prompted the search for FLT3 TKI. These TKI are heterocyclic compounds blocking FLT3 activity through competition with ATP at the TKD of FLT3.Citation36 While generally demonstrating in vitro activity against multiple tyrosine kinases, their efficacy in AML patients is markedly enhanced in FLT3-ITD cases and correlates with inhibition of FLT3 phosphorylation and downstream signaling relays.Citation36 Comprehensive reviews on the use of FLT3 TKI in clinical trials have been recently published.Citation17,Citation36
Schematically, three categories of studies currently encompass FLT3 inhibitors in AML. First, studies using FLT3 TKI as monotherapy were reported mostly in a refractory/relapse context of FLT3-ITD AML. Some of them are already reported and show that, while of significant clinical activity, FLT3 TKI, such as quizartinib or sorafenib, fail to induce long-lasting remissions in this context, while some patients were successfully bridged to allogenic transplant after achieving TKI-induced remission.Citation37,Citation38 Second, several clinical trials were recently designed to combine a TKI to Vidaza, a hypomethylating agent with activity in AML, due to a possible synergistic activity of these compounds in FLT3-ITD AML.Citation17 Third, important results recently came from clinical trials combining a TKI with conventional chemotherapy in AML. In a Phase II randomized trial (SORAML), 276 AML patients were treated by conventional chemotherapy plus sorafenib (400 mg twice daily, n=134) or plus placebo (n=133), resulting in a 40% versus 22% 3-years event-free survival in favor of the sorafenib group. In this study, a trend to improved survival was detected in patients treated by sorafenib when a FLT3-ITD was present, although the results were not statistically significant possibly due to the limited size of the cohort (n=46).Citation39 In a recent abstract, Stone et alCitation40 reported the results of a Phase III study (RATIFY) that combined midostaurine or placebo to a standard chemotherapy in 717 FLT3-ITD AML patients. They showed that the addition of midostaurine extended the median overall survival from 25 to 75 months, which represents the first demonstration of a significant activity of a TKI in FLT3-ITD+ AML in randomized trials. These results prompted the designation of midostaurine as a breakthrough therapy in FLT3-mutated AML by the US Food and Drug Administration in 2016. Finally, as posttransplant relapses have been successfully managed by the FLT3 TKI sorafenib,Citation41,Citation42 the idea emerged to administer a FLT3 TKI in patients achieving a complete remission after allogenic transplant. In fact, several recent studies report promising results using this maintenance approach, albeit follow-up intervals remain short.Citation43–Citation45 In a recent retrospective study, sorafenib posttransplant maintenance was associated with an improved 2-year progression-free survival and a reduced risk of relapse.Citation46 However, these preliminary results warrant confirmation in prospective clinical trials, and several questions need to be addressed in this perspective such as the timing of TKI initiation and the duration of therapy.Citation17
Resistance to therapy in FLT3-ITD AML
The presence of a FLT3-ITD mutation has a strong negative prognosis impact in AML due to an increased risk of relapse after chemotherapy. The current treatment guidelines in FLT3-ITD AML have been recently reviewed by Konig and LevisCitation17 and support the use of allogenic stem cell transplantation whenever possible in first remission to decrease the risk of relapse. For refractory/relapse patients, no guidelines are available and their survival is generally very short regardless of the therapeutic intervention used.Citation17
Resistance to cytotoxic chemotherapy
Genomic pairwise comparisons between diagnosis and relapse AML samples revealed the expansion of a founding clone or subclone, which may acquire new oncogenic mutations at relapse.Citation47 In FLT3-ITD AML, relapse-specific mutations were found at low frequency in several genes including FLT3 (FLT3-TKD mutations), WT1, ASXL1, MLL3, and FAT4, which may provide some insights on the mechanisms of resistance to chemotherapy.Citation48 Interestingly, MLL3 and FAT4 were tumor suppressors in the FLT3-ITD context, as knockdown of their expression by RNA interference stimulated AML propagation including in mouse models, suggesting that inactivating mutations in these genes may have favored AML relapse after chemotherapy.Citation48
Interestingly, FLT3-ITD AML samples collected at the time of relapse appear to be more sensitive to FLT3 TKI in vitro compared to diagnosis samples, suggesting that chemotherapy is selective for an AML clone addicted to FLT3-ITD signaling.Citation49 This clonal selection might be directly favored by chemotherapy, which markedly enhance the production of FLT3-L by the bone marrow microenvironment and activate FLT3-ITD-dependent signaling pathways in AML cells.Citation50
Oncogene addiction to FLT3-ITD
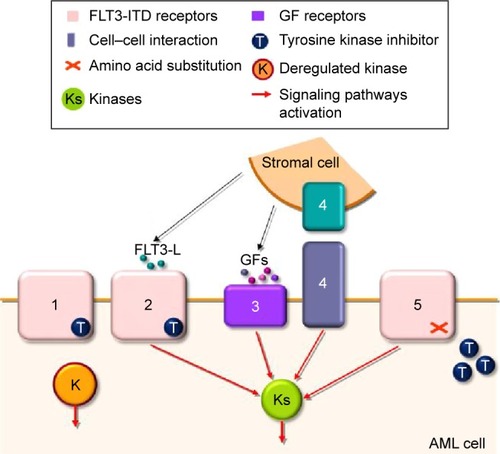
A significant number of reports have shown that FLT3 TKI frequently induce clinical responses in FLT3-ITD AML, including some complete responses, when used as a monotherapy.Citation17 While some transient responses in blast count are reported in the absence of FLT3-ITD mutation, probability due to the activity of TKI against other kinases, complete responses are seen only in FLT3-ITD patients, attesting for an oncogenic addiction to FLT3-ITD in their AML cells.Citation36 However, responses are generally lost within month, even when the TKI is given continuously.Citation38 The addiction of AML cells to FLT3-ITD suggests that this deregulated TKR may represent a very attractive target for therapy, and in fact, important efforts were undertaken to understand the molecular basis of resistance to TKI in FLT3-ITD AML to develop new efficient therapeutic strategies in the future. The current state of the art of these resistance mechanisms is depicted in .
Figure 2 Overview of the resistance mechanisms to FLT3 TKI in AML.
Abbreviations: TKI, tyrosine kinase inhibitors; AML, acute myeloid leukemia; FLT3-L, FLT3 ligand; Pim, proviral integration site.
FLT3 allele burden
FLT3-ITD allele burden is defined by the ratio between the mutated and the wild-type allele. A high (>0.5) FLT3-ITD burden is associated with an adverse prognosis after conventional therapy, even if allogenic stem cell transplant may benefit this category of AML patients.Citation51,Citation52 In contrast, the antileukemic activity of six different FLT3 TKI appeared superior in vitro against samples with a high FLT3-ITD allele burden.Citation49 Interestingly in this report, FLT3 inhibition did not correlate to TKI-induced cytotoxicity in all cases, suggesting that some samples may not have been addicted to FLT3-ITD-driven signaling.Citation49 The follow-up of FLT3 allele burden in the course of conventional or TKI therapy will be facilitated by new sensitive sequencing techniques, and the impact of minimal residual disease monitoring will probably be widely evaluated in FLT3-ITD AML in the future.Citation53,Citation54
Overexpression of oncogenic kinases
The activation of bypass signaling pathways in response to target inhibition of an oncogenic signaling pathway is a well-recognized mechanism of resistance to therapy in cancer, as illustrated, for example, by the activation of CRAF in BRAF V600-mutated cells treated with vemurafenib or by the activation of the MET tyrosine kinase in EGFR-mutated cells treated by gefitinib.Citation55 In FLT3-ITD AML, the cytosolic tyrosine kinase SYK is overactivated and directly activates FLT3-ITD by direct binding and phosphorylation. Moreover, SYK appears critical for myeloid disease propagation in mice, with potential implications in resistance to TKI.Citation56 AXL is a receptor tyrosine kinase overexpressed in many cancer types including in AML. Upon incubation with TKI, FLT3-ITD cells – and particularly cells that were resistant to TKI – demonstrated an increased expression and phosphorylation of AXL, and AXL inhibition by small molecules and by RNA interference facilitated the activity of FLT3 TKI in vitro.Citation57
The impact expression of the PIM serine/threonine kinase family members have been extensively studied in cancer and AML.Citation58,Citation59 PIM kinases are FLT3-ITD targets dependent on – at least for PIM1 – STAT5 activity that controls several oncogenic relays including transcription, protein translation, cell cycle, apoptosis, and cell migration.Citation58 Early reports showed that PIM kinases overexpression may contribute to FLT3-ITD-induced cell transformation and resistance to FLT3 TKI in vitro.Citation60–Citation62 Several groups reported a significant antileukemic activity of PIM inhibitors in preclinical models of AML,Citation63–Citation65 and currently a clinical trial is ongoing with the LGH447 pan-PIM kinase inhibitor in AML regardless of their FLT3 mutational status (NCT02078609). We recently observed that PIM kinases overexpression decreases the efficacy of FLT3 TKI due to a direct action of PIM kinases on FLT3-ITD receptors. In AML cells resistant to TKI, PIM kinase inhibition by RNA interference or by the pan-PIM kinase inhibitor LGB321 (similar to LGH447) restored the cytotoxic activity of FLT3 TKI against these cells, providing the molecular basis to understand the synergy between FLT3 and PIM kinase inhibition in FLT3-ITD+ AML.Citation66 In fact, the SGI-1776 compound is a dual PIM/FLT3 inhibitor that reached a clinical trial development stage. Unfortunately, unanticipated cardiac toxicity – possibly due to an off-target activity against the hERG potassium channel – resulted in withdrawal of SGI-1776 from clinical use.Citation67 Future development of dual PIM/FLT3 inhibitors might be of great interest after TKI failure in FLT3-ITD AML patients.
FLT3-L
While the expression of FLT3 receptors is restricted to hematopoietic cells, FLT3-L is expressed by a wide variety of cells, particularly by bone marrow stroma and T lymphocytes after chemotherapy.Citation50 In fact, increased levels of FLT3-L significantly impaired the ability of FLT3 TKI to inhibit FLT3-ITD activity and induce cytotoxicity in FLT3-ITD AML.Citation50 As FLT3-L levels gradually increased after repeated cycles of chemotherapy, these results suggested to combine chemotherapy with a TKI frontline, an approach that seems fruitful in the clinics as attested by the recent results of the RATIFY clinical trial as discussed earlier.Citation40 In addition, evidence suggests that, in contrast to cytotoxic chemotherapy, treatment with hypomethylating agents such as 5-azacytidine may prevent the release of FLT3-L. On the basis of these observations, several clinical trials are currently ongoing combining Vidaza to different TKI molecules in FLT3-ITD AML.Citation17 An interesting approach to bypass FLT3-L-induced resistance to therapy is the development of anti-FLT3 antibodies. These antibodies bind FLT3 regardless the concentration of FLT3-L and induce cytotoxicity mainly through antibody-dependent cell-mediated cytotoxicity.Citation68 FLYSYN is such a therapeutic antibody that will be used in clinical trials to target minimal residual disease in AML in complete remission (NCT02789254).
Microenvironment
The bone marrow microenvironment has long been recognized as a feeder for hematopoietic cancer cell growth and has been involved in resistance to FLT3 TKI in FLT3-ITD AML.Citation69 Soluble molecule secretion as well as cellular interactions between AML and stromal cells was reported as cytoprotective factors toward AML cells including against TKI, particularly through a sustained activity of signaling pathways such as ERK.Citation70 A recent report also suggests an important role for integrin αvβ3 in activating β-catenin signaling in FLT3-ITD AML cells, promoting resistance to FLT3 TKI.Citation71 Clinical use of TKI revealed a differential activity of these molecules between the blood and the bone marrow compartment. First-generation FLT3 TKI frequently produced a peripheral blast cell clearance, while the bone marrow blast cells were generally persistent.Citation17 Second-generation TKI more frequently induced complete bone marrow blast cell clearance, but occurring later compared to the blood compartment (several weeks compared to few days). In fact, bone marrow blast cell clearance seems to be due to progressive myeloid differentiation rather than apoptosis with the implication of C/EBPα downstream of FLT3-ITD inhibition.Citation26 These data suggest that leukemia-permissive interactions with the bone marrow microenvironment are important mediators of FLT3 TKI resistance for which therapeutic strategies must be refined by combining TKI to agents that target these interactions, such as the CXCR4 inhibitor plerixafor.
FLT3 mutation
FLT3 TKIs are small molecules that bind to the TKD through establishing hydrogen bond interactions. The TKI/kinase complex is thereby stabilized, which prevents ATP from reaching its binding site.Citation72 Mutations occurring within the binding site of TKI may reduce their affinity for these molecules, hence reducing their antileukemic potential. This therapeutic resistance mechanism by modification of the target is commonly found in bacteria with acquired resistance to antibiotics and has been widely studied in the face of resistance to TKI in chronic myelogenous leukemia (CML).Citation55 While occurring in virtually all cases of TKI resistance in CML, with a particular mention of the T315I “gatekeeper” mutation preventing the binding of most TKI to ABL TKD, the detection of mutations within FLT3-TKD only recently emerged as an important mechanism of resistance to FLT3 TKI.Citation36
These FLT3-TKD mutations may be detected at diagnosis in the absence of FLT3-ITD mutation in 5%–10% of AML cases, and the nature as well as the clinical impact of these mutations was recently reviewed by Leung et al.Citation36 The same mutations may occur on FLT3-ITD alleles and induce amino acid substitutions within the kinase domain generally at position D835.Citation38 The F691L amino acid substitution, which is found less frequently than D835 mutations, is a “gatekeeper” mutation similar to the T315I mutation found in CML and also induces a global resistance to FLT3 TKI.Citation72 Other TKD mutations were reported at low frequency in FLT3-ITD relapse samples, and overall, Alvarado et alCitation73 reported the occurrence of TKD mutations in 22% of relapse samples of FLT3-ITD AML patients treated by quizartinib, sorafenib, lestaurtinib, or KW-2449 in clinical trials. Smith et alCitation74 reported FLT3-TKD mutations in all samples from patients relapsing from quizartinib therapy and demonstrated that these mutations were not detectable in diagnosis samples, suggesting a strong selective pressure against FLT3 during the course of TKI therapy that supported the notion of oncogene addiction to FLT3-ITD of AML cells, pinpointing FLT3-ITD as an important therapeutic target in AML.
However, the mechanisms by which these additional mutations confer resistance to TKI are not fully understood. TKD mutations may stabilize the FLT3 kinase domain in an open ATP-binding configuration that enhance FLT3 activity and reduce TKI binding.Citation36 In fact, Type 2 TKI such as quizartinib or sorafenib bind the TKD in an inactive conformation state, while Type 1 TKI (midostaurine, crenolanib) may target both active and inactive conformations of tyrosine kinases.Citation75 Interestingly, Smith et alCitation74 reported that crenolanib, a Type 1 TKI with a strong selectivity for FLT3, was powerful to induce cytotoxicity in FLT3-ITD cells harboring TKD resistance mutations.
How to target these mutations arising after TKI therapy is a central question that should be addressed to improve long-term disease control.Citation36 First, new, more potent TKI may be discovered, as discussed earlier in the case of crenolanib. In fact, several other TKI, such as AUZ454 or ponatinib, are currently being developed to challenge these FLT3-TKD mutations, with ponatinib being routinely used in BCR-ABL T315I-mutated leukemia.Citation76 Second, multikinase inhibitor or dual therapy with a FLT3 TKI and another inhibitor may be considered to circumvent signaling pathways activated in TKD-mutated AML cells. Following this concept, several approaches have been developed including combination of FLT3 inhibitors with mTOR, NFkB, MEK, or PI3K inhibitors and also new compounds with dual activity against FLT3 and PIM, JAK, or aurora kinases.Citation36 Virtually any pathway involved in TKI resistance may be considered as a potential target for dual targeting, and a rigorous preclinical evaluation of these new therapeutic options is required to select the best candidates for further clinical development in the future.
Other mechanisms
Other resistance mechanisms may involve changes in the regulation of target genes and/or in activation of signaling pathways downstream of FLT3-ITD.Citation38 Interestingly, microarray analysis on primary samples from FLT3-ITD AML patients who became refractory to sorafenib showed an upregulation of the TESC gene, which encodes a calcium-interacting protein with impact on intracellular pH regulation and resistance to sorafenib.Citation77 Agnostic approaches such as those developed in this study may represent powerful approaches to identify new strategies to target TKI resistance mechanisms in FLT3-ITD AML cells. Interestingly, Lam et alCitation78 recently identified (by using a high-content chemical screen) homoharringtonine, a plant alkaloid with cytotoxic activity in AML achieved through protein translation inhibition, as having a synergistic activity with sorafenib in vitro, in mouse models, and in a clinical trial conducted in patients with refractory/relapsed FLT3-ITD AML, some of whom were resistant to FLT3 TKI.
Perspectives
AML is currently cured in only a minority of patients, and this is even more so when an FLT3-ITD mutation is detected in leukemic blasts. Tremendous efforts led to the development of highly active and clinically pertinent FLT3 TKI, as accumulated evidences pinpointed that AML cells may be addicted to FLT3-ITD-dependent signaling. However, relapses are commonly observed after TKI therapy due to different resistance mechanisms that encompass FLT3 TKD amino acid substitutions, bypass activation of oncogenic kinases, and leukemia-permissive interactions with the bone marrow microenvironment. In contrast to CML in which resistance mechanisms appear to be uniform, the pattern of resistance mechanisms in FLT3-ITD AML is far from being fully understood and represent an active field of research. However, recent results from a Phase III clinical trial in which FLT3-ITD AML patients were treated with chemotherapy and midostaurine or placebo strongly suggested a survival benefit to FLT3 inhibition. Future strategies will include more potent and/or specific FLT3 inhibitors and integration into a global strategy in which TKI may be given in combination with chemotherapy and/or as a consolidation strategy, particularly after allogenic stem cell transplantation or even as a maintenance therapy, possibly combined to hypomethylating agents or other molecules for which a synergy may be found with FLT3 TKI.
Disclosure
The authors report no conflicts of interest in this work.
References
- MuratiABrecquevilleMDevillierRMozziconacciMJGelsi-BoyerVBirnbaumDMyeloid malignancies: mutations, models and managementBMC Cancer20121230422823977
- WelchJSLeyTJLinkDCThe origin and evolution of mutations in acute myeloid leukemiaCell2012150226427822817890
- PapaemmanuilEGerstungMBullingerLGenomic classification and prognosis in acute myeloid leukemiaN Engl J Med2016374232209222127276561
- PatelJPGönenMFigueroaMEPrognostic relevance of integrated genetic profiling in acute myeloid leukemiaN Engl J Med2012366121079108922417203
- BuchnerTSchlenkRFSchaichMAcute Myeloid Leukemia (AML): different treatment strategies versus a common standard arm – combined prospective analysis by the German AML IntergroupJ Clin Oncol201230293604361022965967
- ThiedeCSteudelCMohrBAnalysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosisBlood200299124326433512036858
- JankeHPastoreFSchumacherDActivating FLT3 mutants show distinct gain-of-function phenotypes in vitro and a characteristic signaling pathway profile associated with prognosis in acute myeloid leukemiaPLoS One201493e8956024608088
- Blume-JensenPHunterTOncogenic kinase signallingNature2001411683535536511357143
- LevisMFLT3/ITD AML and the law of unintended consequencesBlood2011117266987699021586749
- MackarehtschianKHardinJDMooreKABoastSGoffSPLemischkaIRTargeted disruption of the flk2/flt3 gene leads to deficiencies in primitive hematopoietic progenitorsImmunity1995311471617621074
- MatthewsWJordanCTWiegandGWPardollDLemischkaIRA receptor tyrosine kinase specific to hematopoietic stem and progenitor cell-enriched populationsCell1991657114311521648448
- BoyerSWSchroederAVSmith-BerdanSForsbergECAll hematopoietic cells develop from hematopoietic stem cells through Flk2/Flt3-positive progenitor cellsCell Stem Cell201191647321726834
- BirgFCourcoulMRosnetOExpression of the FMS/KIT-like gene FLT3 in human acute leukemias of the myeloid and lymphoid lineagesBlood19928010258425931384791
- ZhengRLevisMPilotoOFLT3 ligand causes autocrine signaling in acute myeloid leukemia cellsBlood2004103126727412969963
- NakaoMYokotaSIwaiTInternal tandem duplication of the flt3 gene found in acute myeloid leukemiaLeukemia19961012191119188946930
- YamamotoYKiyoiHNakanoYActivating mutation of D835 within the activation loop of FLT3 in human hematologic malignanciesBlood20019782434243911290608
- KonigHLevisMTargeting FLT3 to treat leukemiaExpert Opin Ther Targets2015191375425231999
- ShlushLIZandiS1MitchellAIdentification of pre-leukaemic haematopoietic stem cells in acute leukaemiaNature2014506748832833324522528
- ChuSHHeiserDLiLFLT3-ITD knockin impairs hematopoietic stem cell quiescence/homeostasis, leading to myeloproliferative neoplasmCell Stem Cell201211334635822958930
- HirschPZhangYTangRGenetic hierarchy and temporal variegation in the clonal history of acute myeloid leukaemiaNat Commun201671247527534895
- KiyoiHTowatariMYokotaSInternal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the productLeukemia1998129133313379737679
- Schmidt-ArrasDBöhmerSAKochSAnchoring of FLT3 in the endoplasmic reticulum alters signaling qualityBlood2009113153568357619204327
- ChoudharyCBrandtsCSchwableJActivation mechanisms of STAT5 by oncogenic Flt3-ITDBlood2007110137037417356133
- ReddyPNSarginBChoudharyCSOCS1 cooperates with FLT3-ITD in the development of myeloproliferative disease by promoting the escape from external cytokine controlBlood201212081691170222517899
- ChatterjeeAGhoshJRamdasBRegulation of Stat5 by FAK and PAK1 in oncogenic FLT3- and KIT-driven leukemogenesisCell Rep2014941333134825456130
- SexauerAPerlAYangXTerminal myeloid differentiation in vivo is induced by FLT3 inhibition in FLT3/ITD AMLBlood2012120204205421423012328
- RadomskaHSAlberich-JordàMWillBGonzalezDDelwelRTenenDGTargeting CDK1 promotes FLT3-activated acute myeloid leukemia differentiation through C/EBPalphaJ Clin Invest201212282955296622797303
- ZhengRFriedmanADLevisMLiLWeirEGSmallDInternal tandem duplication mutation of FLT3 blocks myeloid differentiation through suppression of C/EBPalpha expressionBlood200410351883189014592841
- PeruginiMKokCHBrownALRepression of Gadd45alpha by activated FLT3 and GM-CSF receptor mutants contributes to growth, survival and blocked differentiationLeukemia200923472973819151789
- SchwableJChoudharyCThiedeCRGS2 is an important target gene of Flt3-ITD mutations in AML and functions in myeloid differentiation and leukemic transformationBlood200510552107211415536149
- PusapatiRVDaemenAWilsonCmTORC1-dependent metabolic reprogramming underlies escape from glycolysis addiction in cancer cellsCancer Cell201629454856227052953
- Alvarez-CalderonFGregoryMAPham-DanisCTyrosine kinase inhibition in leukemia induces an altered metabolic state sensitive to mitochondrial perturbationsClin Cancer Res20152161360137225547679
- StanickaJRussellEGWoolleyJFCotterTGNADPH oxidase-generated hydrogen peroxide induces DNA damage in mutant FLT3-expressing leukemia cellsJ Biol Chem2015290159348936125697362
- JayaveluAKMüllerJPBauerRNOX4-driven ROS formation mediates PTP inactivation and cell transformation in FLT3ITD-positive AML cellsLeukemia201630247348326308771
- GodfreyRAroraDBauerRCell transformation by FLT3 ITD in acute myeloid leukemia involves oxidative inactivation of the tumor suppressor protein-tyrosine phosphatase DEP-1/PTPRJBlood2012119194499451122438257
- LeungAYManCHKwongYLFLT3 inhibition: a moving and evolving target in acute myeloid leukaemiaLeukemia201327226026822797419
- CortesJEKantarjianHForanJMPhase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication statusJ Clin Oncol201331293681368724002496
- ManCHFungTKHoCSorafenib treatment of FLT3-ITD+ acute myeloid leukemia: favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutationBlood2012119225133514322368270
- RolligCServeHHüttmannAAddition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trialLancet Oncol201516161691169926549589
- StoneRMMandrekarSSanfordBLThe multi-kinase inhibitor midostaurin (M) prolongs survival compared with placebo (P) in combination with daunorubicin (D)/cytarabine (c) induction (ind), high-dose c consolidation (consol), and as maintenance (maint) therapy in newly diagnosed acute myeloid leukemia (AML) patients (pts) age 18–60 with FLT3 mutations (muts): an international prospective randomized (rand) P-controlled double-blind trial (CALGB 10603/RATIFY [Alliance])Paper presented at: 57th Annual ASH Meeting2015Orlando, FL
- De FreitasTMarktelSPiemonteseSHigh rate of hematological responses to sorafenib in FLT3-ITD acute myeloid leukemia relapsed after allogeneic hematopoietic stem cell transplantationEur J Haematol201696662963626260140
- MetzelderSWangYWollmerECompassionate use of sorafenib in FLT3-ITD-positive acute myeloid leukemia: sustained regression before and after allogeneic stem cell transplantationBlood2009113266567657119389879
- TarlockKChangBCooperTSorafenib treatment following hematopoietic stem cell transplant in pediatric FLT3/ITD acute myeloid leukemiaPediatr Blood Cancer20156261048105425662999
- AntarAKharfan-DabajaMAMahfouzRBazarbachiASorafenib maintenance appears safe and improves clinical outcomes in FLT3-ITD acute myeloid leukemia after allogeneic hematopoietic cell transplantationClin Lymphoma Myeloma Leuk201515529830225550214
- ChenYBLiSLaneAAPhase I trial of maintenance sorafenib after allogeneic hematopoietic stem cell transplantation for fms-like tyrosine kinase 3 internal tandem duplication acute myeloid leukemiaBiol Blood Marrow Transplant201420122042204825239228
- BrunnerAMLiSFathiATHaematopoietic cell transplantation with and without sorafenib maintenance for patients with FLT3-ITD acute myeloid leukaemia in first complete remissionBr J Haematol Epub7192016
- DingLLeyTJLarsonDEClonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencingNature2012481738250651022237025
- GargMNagataYKanojiaDProfiling of somatic mutations in acute myeloid leukemia with FLT3-ITD at diagnosis and relapseBlood2015126222491250126438511
- PratzKWSatoTMurphyKMStineARajkhowaTLevisMFLT3-mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AMLBlood201011571425143220007803
- SatoTYangXKnapperSFLT3 ligand impedes the efficacy of FLT3 inhibitors in vitro and in vivoBlood2011117123286329321263155
- PratcoronaMBrunetSNomdedéuJFavorable outcome of patients with acute myeloid leukemia harboring a low-allelic burden FLT3-ITD mutation and concomitant NPM1 mutation: relevance to post-remission therapyBlood2013121142734273823377436
- SchlenkRFKayserSBullingerLDifferential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantationBlood2014124233441344925270908
- BibaultJEFigeacMHélevautNNext-generation sequencing of FLT3 internal tandem duplications for minimal residual disease monitoring in acute myeloid leukemiaOncotarget2015626228122282126078355
- TholFKölkingBDammFNext-generation sequencing for minimal residual disease monitoring in acute myeloid leukemia patients with FLT3-ITD or NPM1 mutationsGenes Chromosomes Cancer201251768969522454318
- PagliariniRShaoWSellersWROncogene addiction: pathways of therapeutic response, resistance, and road maps toward a cureEMBO Rep201516328029625680965
- PuissantAFenouilleNAlexeGSYK is a critical regulator of FLT3 in acute myeloid leukemiaCancer Cell201425222624224525236
- ParkIKMundy-BosseBWhitmanSPReceptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemiaLeukemia201529122382238926172401
- BraultLGasserCBracherFHuberKKnappSSchwallerJPIM serine/threonine kinases in the pathogenesis and therapy of hematologic malignancies and solid cancersHaematologica201095610041001520145274
- TamburiniJGreenASBardetVProtein synthesis is resistant to rapamycin and constitutes a promising therapeutic target in acute myeloid leukemiaBlood200911481618162719458359
- KimKTBairdKAhnJYPim-1 is up-regulated by constitutively activated FLT3 and plays a role in FLT3-mediated cell survivalBlood200510541759176715498859
- AdamMPogacicVBenditMTargeting PIM kinases impairs survival of hematopoietic cells transformed by kinase inhibitor-sensitive and kinase inhibitor-resistant forms of Fms-like tyrosine kinase 3 and BCR/ABLCancer Res20066673828383516585210
- AgrawalSKoschmiederSBäumerNPim2 complements Flt3 wild-type receptor in hematopoietic progenitor cell transformationLeukemia2008221788617943165
- ChenLSRedkarSTavernaPCortesJEGandhiVMechanisms of cytotoxicity to Pim kinase inhibitor, SGI-1776, in acute myeloid leukemiaBlood2011118369370221628411
- KeetonEKMcEachernKDilmanKSAZD1208, a potent and selective pan-Pim kinase inhibitor, demonstrates efficacy in preclinical models of acute myeloid leukemiaBlood2014123690591324363397
- GarciaPDLangowskiJLWangYPan-PIM kinase inhibition provides a novel therapy for treating hematologic cancersClin Cancer Res20142071834184524474669
- GreenASMacielTTHospitalMAPim kinases modulate resistance to FLT3 tyrosine kinase inhibitors in FLT3-ITD acute myeloid leukemiaSci Adv201518e150022126601252
- FoulksJMCarpenterKJLuoBA small-molecule inhibitor of PIM kinases as a potential treatment for urothelial carcinomasNeoplasia201416540341224953177
- HofmannMGroße-HovestLNüblingTGeneration, selection and preclinical characterization of an Fc-optimized FLT3 antibody for the treatment of myeloid leukemiaLeukemia20122661228123722289926
- ParmarAMarzSRushtonSStromal niche cells protect early leukemic FLT3-ITD+ progenitor cells against first-generation FLT3 tyrosine kinase inhibitorsCancer Res201171134696470621546568
- WeisbergELiuQNelsonEUsing combination therapy to override stromal-mediated chemoresistance in mutant FLT3-positive AML: synergism between FLT3 inhibitors, dasatinib/multi-targeted inhibitors and JAK inhibitorsLeukemia201226102233224422469781
- YiHZengDShenZIntegrin alphavbeta3 enhances beta-catenin signaling in acute myeloid leukemia harboring Fms-like tyrosine kinase-3 internal tandem duplication mutations: implications for microenvironment influence on sorafenib sensitivityOncotarget2016
- TreiberDKShahNPIns and outs of kinase DFG motifsChem Biol201320674574623790484
- AlvaradoYKantarjianHMLuthraRTreatment with FLT3 inhibitor in patients with FLT3-mutated acute myeloid leukemia is associated with development of secondary FLT3-tyrosine kinase domain mutationsCancer2014120142142214924737502
- SmithCCLasaterEALinKCCrenolanib is a selective type I pan-FLT3 inhibitorProc Natl Acad Sci U S A2014111145319532424623852
- FathiATEmergence of crenolanib for FLT3-mutant AMLBlood2013122223547354824263951
- SmithCCLasaterEAZhuXActivity of ponatinib against clinically-relevant AC220-resistant kinase domain mutants of FLT3-ITDBlood2013121163165317123430109
- ManCHLamSSSunMKA novel tescalcin-sodium/hydrogen exchange axis underlying sorafenib resistance in FLT3-ITD+ AMLBlood2014123162530253924608976
- LamSSYHoESKHeB-LIn vitro drug screening identified homoharringtonine as an effective adjunct for treatment of FLT3-ITD acute myeloid leukemiaSci Transl Med20168359359ra129