
Abstract
Overexpression of EGFR is commonly seen in gastric cancer (GC). However, patients with GC show resistance to anti-EGFR treatments. RAS mutations are rare in GC and cannot explain de novo resistance to EGFR treatments. Therefore, it is particularly important to explore the mechanisms of resistance to anti-EGFR treatments. The RANKL activates the EGFR pathway in osteoclasts, and the RANK is expressed in gastric carcinoma. Whether the RANKL/RANK pathway has an effect on the EGFR pathway in GC remains unknown. Expressions of EGFR and RANK in GC tissues were detected using immunohistochemical staining. Nineteen patients (28%) showed high-level RANKL expression, and 33 patients (48%) showed high-level RANK expression. There was a positive correlation between expression of EGFR and RANK (P<0.001). In an in vitro study, RANKL induced activation of the EGFR pathway and further abrogated cetuximab sensitivity in GC cells. Knockdown of RANK or use of the RANKL inhibitor enhanced cetuximab effect by decreasing RANKL-induced EGFR activation. Furthermore, we showed that c-SRC mediated the EGFR activation induced by the RANKL/RANK pathway and that c-SRC inhibitor reversed the suppression of RANKL on the effect of cetuximab. In conclusion, our results suggest that in GC cells, the RANKL/RANK pathway activates the EGFR pathway and thereby causes resistance to anti-EGFR treatments.
Introduction
Gastric cancer (GC) is the fourth commonly diagnosed cancer and the second most frequent cause of cancer death worldwide.Citation1 Although comprehensive therapies have improved the response rate, patients with advanced GC continue to have a poor prognosis.Citation2 Overexpression of EGFR is associated with poor prognosis in GCs,Citation3,Citation4 yet clinical studies using anti-EGFR agents in the general population have failed to show a significant improvement.Citation5,Citation6 Therefore, it is necessary to explore the resistance mechanisms to anti-EGFR agents in these patients. Mutations in RAS oncogenes serve as the major predictive biomarker of anti-EGFR treatments in colorectal cancer. However, the mutation rate for RAS is ~5% in GC.Citation5 Therefore, searching for alternative resistance mechanisms to anti-EGFR agents is of critical importance in GC patients.
One crucial resistance mechanism to anti-EGFR agents is aberrant activation of EGFR by alternative pathways, such as c-Met and IGF-IR,Citation7,Citation8 that activate many of the same downstream signaling pathways as EGFR. RANKL is involved in osteoclast differentiation and induces EGFR activation.Citation9 RANKL is a member of the tumor necrosis factor family of cytokines, and its receptor RANK was previously found to promote osteoclast precursor maturation. Expression of RANK was found in several solid tumors such as breast, prostate and hepatocellular carcinomas.Citation10–Citation12 Recently, the RANKL/RANK pathway was shown to promote cancer cell migration by stimulating AKT and ERK, which are regarded as the most important downstream mediators of EGFR signaling.Citation13 Therefore, we speculated that the RANKL/RANK pathway might prove vital for EGFR activation in GC cells.
In this study, we explored the correlation between RANKL and EGFR expression, and the role of the RANKL/RANK pathway on the activation of EGFR signaling and resistance to anti-EGFR agents in GC. Our results might suggest a novel mechanism of resistance to anti-EGFR therapies in the treatment of GC.
Materials and methods
Reagents
Cetuximab was purchased from EMD Millipore (Billerica, MA, USA). Recombinant sRANKL and rOPG was purchased from CytoLab/PeproTech Asia (USA). PP2 was obtained from Sigma-Aldrich Co. (St Louis, MO, USA). Dasatinib was obtained from Selleck Chemicals (Houston, TX, USA). S-P immunohistochemical kit and 3,3′-diaminobenzidine tetrahydrochloride kit were obtained from Maixin Bio (Fuzhou Maixin Biological Technology Ltd., Fujian, People’s Republic of China).
Cell cultures
GC cells such as SGC-7901, MGC-803, BGC-823, MKN-45, and KATO-III and colon cancer cells such as Caco-2 were obtained from The Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (People’s Republic of China). Cells were cultured in Roswell Park Memorial Institute 1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (FBS), penicillin (100 U/mL), and streptomycin (100 mg/mL) in an atmosphere of 95% air and 5% CO2 at 37°C.
Patients and tissue samples
Specimens of gastric adenocarcinoma tissue were collected from 68 patients who underwent surgical resection at the First Affiliated Hospital of China Medical University. None of them received preoperative radiotherapy, chemotherapy or immunotherapy. Age, sex, pathological tumor–node–metastasis (pTNM) stage, and Lauren grade were evaluated following medical charts and pathology records. pTNM stage was examined according to the seventh edition of AJCC Cancer Staging Manual. Lauren grade was according to the classification of World Health Organization. The First Affiliation Hospital of China Medical University Ethical Committee approved this study, and consent was not needed due to the retrospective nature of the research.
Cell viability assay
The effect of cell proliferation was measured using an MTT assay. Cells were seeded at 3,000–5,000/well in 96-well plates and incubated at 37°C for 24 hours in 10% FBS medium. After pretreatment with dasatinib for 1 hour or not, the cells were incubated with the indicated doses of cetuximab, RANKL or a combination of them for 48 hours. MTT assay was performed as previously described.Citation14
Colony-forming assay
Cells were seeded at 300 cells (SGC-7901, MGC-803), 500 cells (Caco-2) per well in 12-well plates and exposed to 10 μg/mL cetuximab, 1 μg/mL RANKL or a combination of them treatment after plating for 24 hours in 10% FBS medium. Then followed by staining with Wright–Giemsa after 14 days, the number of colonies was counted.
Western blot
Western blot was performed as previously described.Citation14 The following antibodies were used: Anti-RANK antibody was obtained from Bethyl Laboratories, Inc. (Montgomery, TX, USA). Anti-actin antibody was produced by Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Anti-EGFR, anti-phospho-EGFR (Tyr1068), anti-AKT, anti-p-AKT (Ser473), anti-ERK1/2, anti-p-ERK1/2 (Thr202/Tyr204), anti-c-Src, and anti-p-c-Src (Y416) antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). Secondary goat anti-rabbit and goat anti-mouse antibodies were purchased from Santa Cruz Biotechnology Inc.
Immunohistochemistry
Formalin-fixed, paraffin-embedded primary gastric carcinoma tissues were cut into 3 mm sections. The method of immunohistochemistry (IHC) is discussed in our previous study.Citation15 Immunohistochemical staining was performed using the following antibodies: anti-RANK antibody from RD Company and anti-EGFR antibody from Santa Cruz Biotechnology Inc. For the evaluation of immunohistochemical, sections were observed through microscopic examining (×20 and ×40) assigned by two independent pathologists. From each section, five visual fields were randomly selected, and the score for each visual field was based on the percentage of positive cells and the staining intensity. For the percentage of positive cells, <10%, 10%–25%, 26%–50%, 51%–75%, and >76% were recorded as 0, 1, 2, 3, and 4 points, respectively. A score >2 was classified as a high-level expression and 0–2 as a low-level expression.
Small interfering RNA transfections
RANK small interfering RNA (siRNA) was obtained form RiboBio Co., Ltd. (Guangzhou, People’s Republic of China). RANK siRNA was synthesized: 5′-GGCAGGUGAUGAACUUCAA-3′. The control siRNA was AATTCTC CGAACGTGT CACGT. Cells were transfected with siRNAs using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. After 48 hours of transient transfection, the cells were analyzed by Western blot to verify gene-silencing efficiency.
Statistic analysis
Data were confirmed in three independent experiments and were expressed as mean ± standard deviation. Differences between groups were compared using Student’s t-test. The correlation between EGFR and RANK expression was assessed using Spearman’s rank correlation for continuous variables. P<0.05 was considered statistically significant. Statistic analysis was carried out using SPSS 21.0 software package (IBM Corporation, Armonk, NY, USA).
Results
Expression of EGFR shows a positive correlation with RANK expression in GC patients
A total of 68 primary GC samples were obtained, and the expression of EGFR and RANK was determined by IHC. High-level EGFR expression was detected in 19 patients (28%), and 33 patients (48%) showed high-level RANK expression (). Correlations between expression of EGFR and RANK and clinical characteristics of primary GC patients are given in . Importantly, there was a significant positive correlation between the expression of EGFR and RANK (P<0.001; ). Furthermore, we characterized a panel of five human GC cell lines (SGC-7901, MGC-803, BGC-823, MKN-45, and KATO-III) by Western blot to detect the cellular expression levels of EGFR and RANK. The results showed that EGFR and RANK expression was observed in most of the cell lines ().
Figure 1 The expression of RANKL in GC tissues.
Abbreviation: GC, gastric cancer.
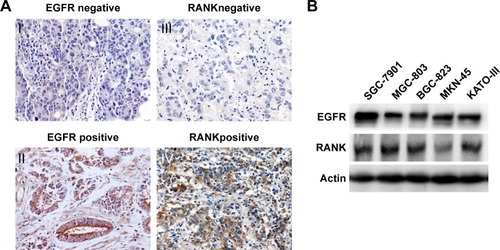
Table 1 Correlation between the expression of EGFR or RANK and the clinicopathological factors in primary GC patients
Table 2 Spearman’s correlations between EGFR and RANK expression in primary GC patients
Taken together, our results demonstrated that EGFR expression was positively correlated with RANK expression, suggesting an association between the RANKL/RANK pathway and EGFR signaling in GC.
Stimulation with RANKL activates EGFR and downstream pathways in GC cells
We continued to investigate the role of RANKL/RANK pathway in the EGFR pathway in vitro. We used SGC-7901 and MGC-803, which were frequently used in the People’s Republic of China as RAS wild-type GC cell lines. Following stimulation with RANKL, we observed phosphorylation of EGFR, AKT, and ERK, indicating that they were transiently activated at different time points in both cell lines ().
Figure 2 RANKL stimulated EGFR phosphorylation and downstream signaling in GC cells.
Abbreviation: GC, gastric cancer.
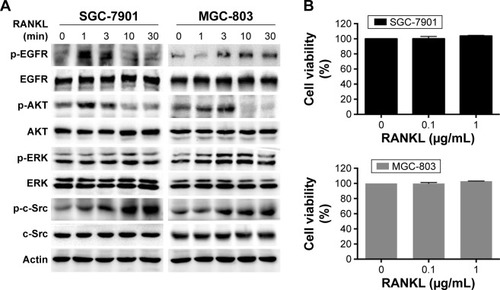
To observe the effect of RANKL on cell proliferation, we treated the two cell lines with different concentrations of RANKL for 48 hours. An MTT assay revealed that RANKL exhibited no distinct effect on cell proliferation at the concentration of 0.1 μg/mL or 1 μg/mL ().
RANKL decreases sensitivity to cetuximab in GC cells
Because RANKL activated the EGFR pathway, we speculated that the RANKL/RANK pathway could affect sensitivity to cetuximab.
First, we examined the proliferation of cells treated with cetuximab. Both cells exhibited mild growth inhibition after addition of cetuximab (21.7%±4.7% in SGC-7901 and 16.8%±2.3% in MGC-803) and indicated that they were relatively insensitive to cetuximab ().
Figure 3 The effect of RANKL on sensitivity to cetuximab in GC cells.
Abbreviation: GC, gastric cancer.
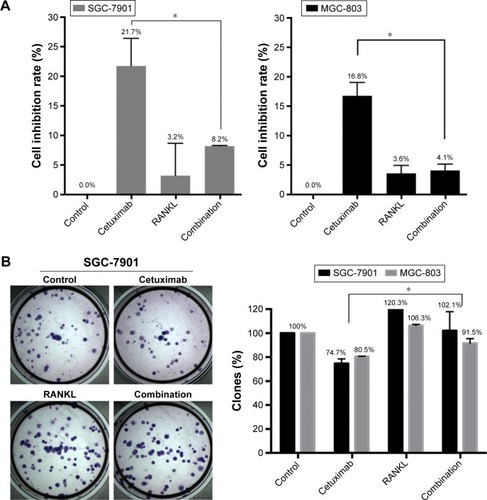
Next, to test whether RANKL modified the responsiveness of GC cell lines to cetuximab, the cells were preincubated with RANKL before addition of cetuximab. Compared with the treatment of cetuximab alone, the combination of RANKL and cetuximab significantly decreased inhibition of cell proliferation by cetuximab in SGC-7901 cells (8.2%±0.1% in the combined-treated arm vs 21.7%±4.7% in the cetuximab-treatment arm, P=0.009) and MGC-803 cells (4.1%±1.1% in the combined-treated arm vs 16.8%±2.3% in the cetuximab-treatment arm, P=0.0007; ). Furthermore, colony-forming assays showed that the combination of cetuximab and RANKL increased colony formation ability compared with cetuximab alone ().
We then explored the change in signaling when combined treated with cetuximab and RANKL in SGC-7901 cells. Cetuximab treatment alone mildly suppressed phosphorylation of EGFR, AKT and ERK, whereas combination of cetuximab and RANKL reversed this suppression ().
Figure 4 The effect of RANKL/RANK pathway on cetuximab sensitivity in SGC-7901 cells.
Abbreviations: NS, non-silencing; siRNA, small interfering RNA.
These results showed that RANKL depressed sensitivity to cetuximab in GC cells through activation of EGFR and its downstream pathways.
Blockade of RANKL/RANK pathway restores RANKL-induced cetuximab resistance in GC cells
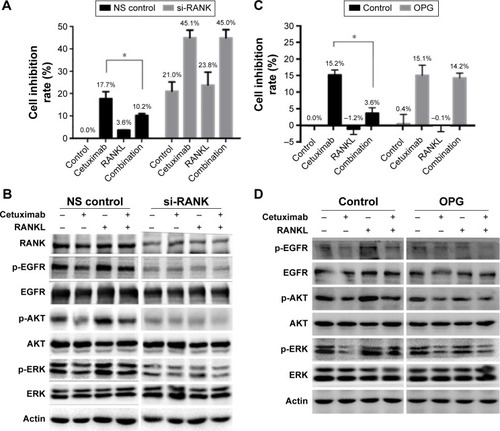
To further confirm the role of the RANKL/RANK pathway in sensitivity to cetuximab, we downregulated RANK by siRNA and then treated with cetuximab, RANKL, or a combination of them. MTT assay revealed that RANK knockdown resulted in reduction of cell proliferation by ~20%. Moreover, in the non-silencing (NS) control arm, cetuximab reduced proliferation by 17.7%±4.4% and the combination of RANKL and cetuximab reduced proliferation by only 10.2%±1.1%. In the si-RANK arm, treatment with cetuximab inhibited proliferation by 45.1%±4.8% compared with the non-treated control, while treatment combined with cetuximab and RANKL inhibited proliferation by 45.0%±5.1%. In the NS control arm, the inhibition rate of proliferation after cetuximab treatment was significantly higher than in the combination treatment, while there was no obvious change in the si-RANK arm (). Therefore, knockdown of RANK restored the RANKL-induced resistance to cetuximab in SGC-7901 cells. Furthermore, we used OPG that negatively regulates RANKL binding to RANK. As expected, in the control arm, the inhibition rate of proliferation in cetuximab-treated cells was significantly higher than that of the combined-treated cells, while there was no obvious change in the OPG arm. This indicated that inhibition of RANKL reduced RANKL-induced cetuximab resistance in SGC-7901 cells (). The results showed that both knockdown of RANK and inhibition of RANKL could reverse the effect of growth inhibition induced by cetuximab in SGC-7901 cells.
Changes in EGFR signaling by si-RANK or OPG in SGC-7901 cells were also determined. Compared to the NS control, reduced phosphorylation of EGFR, AKT and ERK was clearly detected when RANK was knocked down. Furthermore, RANK knockdown suppressed the effect of RANKL on EGFR, AKT and ERK activation (). Similarly, OPG reversed RANKL-induced EGFR, AKT and ERK activation that mediated resistance to cetuximab ().
These results suggested that inhibition of the RANKL/RANK pathway by RANK knockdown or RANKL inhibitor restored RANKL-induced cetuximab resistance through depression of EGFR, AKT and ERK phosphorylation.
C-Src activation induced by RANKL is involved in resistance to cetuximab in GC cells
We speculated that c-Src, an essential intermediate in the crosstalk between EGFR pathways and other tyrosine kinase receptors, might be involved in the interaction between EGFR and RANKL/RANK pathway. Activation of c-Src was previously observed when SGC-7901 and MGC-803 cells were stimulated with RANKL (). Then we used two selective c-Src kinase inhibitors, dasatinib and PP2, to examine the effect of c-Src on EGFR signaling in GC cells. The activation of c-Src, EGFR, AKT and ERK was dramatically attenuated in SGC-7901 and MGC-803 cells after treating with c-Src inhibitors ().
Figure 5 The effect of dasatinib on RANKL-induced cetuximab resistance in SGC-7901 cells.
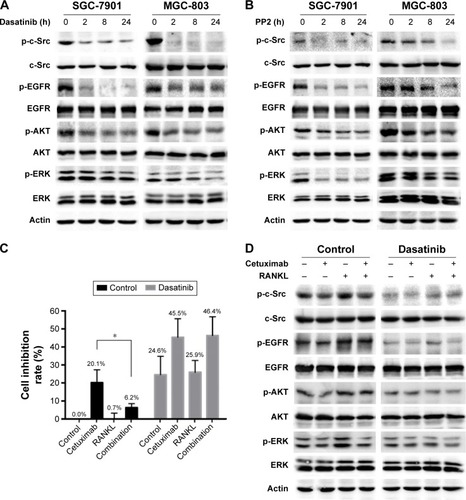
Next, we assessed the effect of c-Src on RANKL-regulated cetuximab sensitivity. Dasatinib alone decreased the proliferation of SGC-7901 cells by 24.6%±10.2%. In the control arm, cetuximab treatment inhibited proliferation by 20.1%±7.1%, while the treatment combined with cetuximab and RANKL inhibited proliferation by 6.3%±2.2%. In the dasatinib arm, cetuximab treatment inhibited proliferation by 45.5%±10.2%, while the combination treatment inhibited proliferation by 46.4%±10.4%. In the control arm, the inhibitory effect of cetuximab on proliferation was much higher than that of the combination treatment, while no significant difference was found in the dasatinib arm. These results suggested that preincubated with dasatinib reversed RANKL-induced cetuximab resistance ().
Then, we examined the signaling change of EGFR signaling. Dasatinib not only resulted in the inhibition of EGFR, AKT and ERK phosphorylation but also suppressed RANKL-stimulated EGFR activation (). These results suggested that c-Src might be one of the key mediators in RANKL-induced EGFR activation that results in RANKL-induced cetuximab resistance.
RANKL/RANK pathway also induces resistance to cetuximab in colon cancer cells
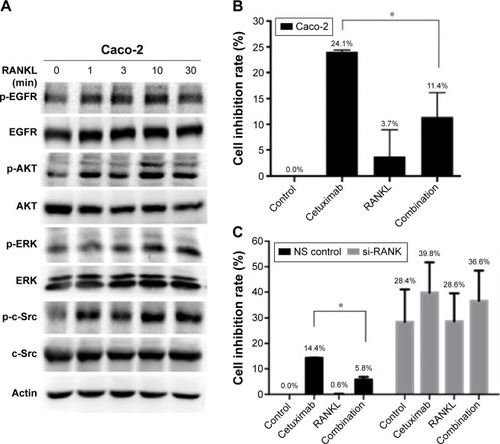
We further explored whether RANKL/RANK pathway was involved in resistance to cetuximab in colon cancer cells. We chose Caco-2 cells with wild type of RAS and RAF. Similarly, phosphorylation of EGFR, AKT, ERK, and c-Src was upregulated by RANKL (). Preincubation with RANKL also decreased sensitivity to cetuximab in Caco-2 cells (inhibition rate of 11.4%±4.7% in the combined-treated arm vs 24.1%±0.3% in the cetuximab-treatment arm, P=0.01; ). Furthermore, downregulation of RANK with siRNA restored inhibition of cell proliferation by cetuximab ().
Figure 6 The effect of RANKL/RANK pathway on sensitivity to cetuximab in Caco-2 cells.
Abbreviations: NS, non-silencing; siRNA, small interfering RNA.
Discussion
EGFR served as one of the most relevant factors in the cell proliferation, survival, apoptosis, migration, and tumorigenesis of various cancers. Overexpression of EGFR has a significant predictive value for poor survival in GC patients.Citation3 Subsequently, EGFR inhibition using cetuximab showed promise in combination with chemotherapy in several Phase II clinical studies for the treatment of metastatic GC.Citation16–Citation19 Unfortunately, no improvement in the overall survival was shown in facing the general population.Citation5,Citation6 Therefore, understanding the mechanisms of resistance to anti-EGFR treatments is of considerable significance and will help us to improve the response rate of anti-EGFR therapies. Among the various mechanisms of cetuximab resistance, aberrant activation of EGFR and downstream signaling pathways has been identified as a crucial resistance mechanism to anti-EGFR agents. In this study, we first demonstrated that EGFR expression was positively correlated with RANK expression in GC patients, and RANKL activated EGFR signaling in GC cells that contributed to cetuximab resistance.
The RANKL/RANK pathway is associated with metastasis-related factors in several malignant tumorsCitation20–Citation22 and hormone-induced tumorigenesis of breast cancer.Citation23,Citation24 The RANKL/RANK pathway has also been shown to regulate EGFR activation by cross talk during osteoclast differentiation.Citation9 However, limited studies have focused on the relationship between the RANKL/RANK pathway and EGFR signaling in primary cancer cells. Our study originally explored the relationship between the RANKL/RANK pathway and EGFR in GC patients. Our IHC results showed that RANK and EGFR were highly expressed in GC tissues. We then showed that EGFR and RANK was expressed in several different GC cell lines (). In addition to this finding, we found a positive relationship between the expression of RANK and EGFR in GC tissues (). Furthermore, RANKL stimulation induced activation of EGFR and downstream signaling (), which indicated the cross talk between the RANKL/RANK pathway and EGFR signaling.
Additionally, we observed that EGFR and downstream activation induced by RANKL decreased sensitivity to cetuximab in GC cells ( and ). Treatment with RANK siRNA or the RANKL inhibitor OPG attenuated phosphorylation of EGFR, AKT, and ERK and additionally reversed RANKL-induced resistance to cetuximab (). Our results showed that the RANKL/RANK pathway phosphorylated EGFR-signaling cascade molecules, including EGFR itself, and further decreased sensitivity to cetuximab in GC cells. Furthermore, we investigated the role of the RANKL/RANK pathway after cetuximab treatment of colon cancer using the RAS wild-type cell line Caco-2. We observed that sensitivity to cetuximab was similarly regulated by the RANKL/RANK pathway in this cell line (). To our knowledge, this is the first evidence of a functional contribution of the RANKL/RANK pathway to cetuximab resistance that therefore makes it a promising target for developing novel therapeutic strategies to improve sensitivity to cetuximab.
To investigate the link between the RANKL/RANK pathway and EGFR signaling, we considered the role of c-Src. It is well established that c-Src and EGFR phosphorylate each other in a positive feedback loop.Citation25,Citation26 Our previous study showed that c-Src activation mediated cetuximab resistance in colon cancer cells,Citation14 which have not been confirmed in GC yet. Moreover, our study showed that c-Src was activated by RANKL in the migration of breast cancer cells.Citation13 Furthermore, c-Src expression showed a significantly positive linear relationship with RANK in breast cancer patients,Citation27 indicating the relationship between c-Src and the RANKL/RANK pathway. In this study, we observed that c-Src was stimulated by RANKL () and that c-Src inhibition led to repression of EGFR phosphorylation and its downstream factors in GC cells (). Consequently, inhibition of c-Src activity reversed RANKL-induced resistance to cetuximab (). These results suggested that c-Src is an intermediate between RANKL and EGFR and that c-Src activation contributes to cetuximab resistance via downregulation of EGFR signaling.
Our results showed that exogenous RANKL stimulated the RANKL/RANK pathway and then activated the EGFR pathway in GC cells. However, the accumulation of RANKL in gastric carcinoma remains elusive. The RANKL/RANK pathway plays an important role in the immune system, and RANKL expression is associated with T lymphocytes in breast cancer cells.Citation28 GC tissues were found infiltrated with T lymphocytes and associated with poor prognosis.Citation29 Herein, we initially speculated that T lymphocytes infiltrated into GC tissues would have high level of RANKL expression, and these might stimulate the RANKL/RANK pathway in the surrounding GC cells. As a result, the stimulated RANKL/RANK pathway might activate EGFR signaling and furthermore regulate the sensitivity of anti-EGFR therapies in GC.
Taken together, our results provided a potential mechanism for the resistance to anti-EGFR treatments in gastrointestinal tumors and a conclusion that RANKL/RANK and c-Src could be essential therapeutic targets to enhance sensitivity to anti-EGFR treatments.
Author contributions
Xiujuan Qu and Yunpeng Liu conceived and designed the study. Xiaomeng Zhang, Na Song, Ye Zhang, Lingyun Zhang and Yan Wang performed the experiments. Yongxi Song and Zhenning Wang provided the samples and collected patient information. Xiaomeng Zhang wrote the paper. Xiujuan Qu, Yunpeng Liu and Yongxi Song reviewed and edited the manuscript. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Acknowledgments
This study was supported by Chinese National Foundation of National Sciences grants (No 81172198, No 81172369, No 81270036, No 81302128). Science and Technology Plan Project of Liaoning Province (No 2011404013-1 No 2014021089 No 2014225013). Liaoning BaiQianWan Talents Program (No 2014921032).
Disclosure
The authors report no conflicts of interest in this work.
References
- TorreLABrayFSiegelRLFerlayJLortet-TieulentJJemalAGlobal cancer statistics, 2012CA Cancer J Clin20156528710825651787
- WagnerADGrotheWHaertingJKleberGGrotheyAFleigWEChemotherapy in advanced gastric cancer: a systematic review and meta-analysis based on aggregate dataJ Clin Oncol200624182903290916782930
- AtmacaAWernerDPauligkCThe prognostic impact of epidermal growth factor receptor in patients with metastatic gastric cancerBMC Cancer20121252423153332
- KimJSKimMAKimTMBiomarker analysis in stage III–IV (M0) gastric cancer patients who received curative surgery followed by adjuvant 5-fluorouracil and cisplatin chemotherapy: epidermal growth factor receptor (EGFR) associated with favourable survivalBr J Cancer2009100573273819259093
- LordickFKangYKChungHCCapecitabine and cisplatin with or without cetuximab for patients with previously untreated advanced gastric cancer (EXPAND): a randomised, open-label phase 3 trialLancet Oncol201314649049923594786
- WaddellTChauICunninghamDEpirubicin, oxaliplatin, and capecitabine with or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): a randomised, open-label phase 3 trialLancet Oncol201314648148923594787
- YanoSWangWLiQHepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutationsCancer Res200868229479948719010923
- MorgilloFKimWYKimESCiardielloFHongWKLeeHYImplication of the insulin-like growth factor-IR pathway in the resistance of non-small cell lung cancer cells to treatment with gefitinibClin Cancer Res20071392795280317473213
- YiTLeeHLChaJHEpidermal growth factor receptor regulates osteoclast differentiation and survival through cross-talking with RANK signalingJ Cell Physiol2008217240942218543257
- SantiniDPerroneGRoatoIExpression pattern of receptor activator of NFkappaB (RANK) in a series of primary solid tumors and related bone metastasesJ Cell Physiol2011226378078420857484
- ArmstrongAPMillerREJonesJCZhangJKellerETDougallWCRANKL acts directly on RANK-expressing prostate tumor cells and mediates migration and expression of tumor metastasis genesProstate20086819210418008334
- JonesDHNakashimaTSanchezOHRegulation of cancer cell migration and bone metastasis by RANKLNature2006440708469269616572175
- ZhangLTengYZhangYC-Src-mediated RANKL-induced breast cancer cell migration by activation of the ERK and Akt pathwayOncol Lett20123239540022740919
- SongNLiuSZhangJCetuximab-induced MET activation acts as a novel resistance mechanism in colon cancer cellsInt J Mol Sci20141545838585124714091
- LiHXuLLiCUbiquitin ligase Cbl-b represses IGF-I-induced epithelial mesenchymal transition via ZEB2 and microRNA-200c regulation in gastric cancer cellsMol Cancer20141313624885194
- LordickFLuberBLorenzenSCetuximab plus oxaliplatin/leucovorin/5-fluorouracil in first-line metastatic gastric cancer: a phase II study of the Arbeitsgemeinschaft Internistische Onkologie (AIO)Br J Cancer2010102350050520068568
- MoehlerMMuellerATrarbachTCetuximab with irinotecan, folinic acid and 5-fluorouracil as first-line treatment in advanced gastroesophageal cancer: a prospective multi-center biomarker-oriented phase II studyAnn Oncol20112261358136621119032
- PintoCDi FabioFBaroneCPhase II study of cetuximab in combination with cisplatin and docetaxel in patients with untreated advanced gastric or gastro-oesophageal junction adenocarcinoma (DOCETUX study)Br J Cancer200910181261126819773760
- PintoCDi FabioFSienaSPhase II study of cetuximab in combination with FOLFIRI in patients with untreated advanced gastric or gastroesophageal junction adenocarcinoma (FOLCETUX study)Ann Oncol200718351051717164226
- TsubakiMKomaiMFujimotoSActivation of NF-kappaB by the RANKL/RANK system up-regulates snail and twist expressions and induces epithelial-to-mesenchymal transition in mammary tumor cell linesJ Exp Clin Cancer Res2013326224011086
- WangJLiuYWangLSunXWangYClinical prognostic significance and pro-metastatic activity of RANK/RANKL via the AKT pathway in endometrial cancerOncotarget2016755564557526734994
- Odero-MarahVAWangRChuGReceptor activator of NF-kappaB Ligand (RANKL) expression is associated with epithelial to mesenchymal transition in human prostate cancer cellsCell Res200818885887018645583
- SchramekDLeibbrandtASiglVOsteoclast differentiation factor RANKL controls development of progestin-driven mammary cancerNature201046873209810220881962
- Gonzalez-SuarezEJacobAPJonesJRANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesisNature2010468732010310720881963
- BiscardiJSMaaMCTiceDACoxMELeuTHParsonsSJc-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor functionJ Biol Chem1999274128335834310075741
- StoverDRBeckerMLiebetanzJLydonNBSrc phosphorylation of the epidermal growth factor receptor at novel sites mediates receptor interaction with Src and P85 alphaJ Biol Chem19952702615591155977797556
- LiRZhangKPenedoTLThe RANK pathway in advanced breast cancer: does Src play a role?Appl Immunohistochem Mol Morphol2016241425026200837
- TanWZhangWStrasnerAFibroblast-recruited, tumor-infiltrating CD4+ T cells stimulate mammary cancer metastasis through RANKL-RANK signalingNature2011470733554855321326202
- OhnoSTachibanaMFujiiTUedaSKubotaHNagasueNRole of stromal collagen in immunomodulation and prognosis of advanced gastric carcinomaInt J of Cancer200297677077411857352