
Abstract
Large granular lymphocytic leukemia (LGLL) is a rare lymphoproliferative disorder of transformed natural killer or T-cells attributed to chronic exposure to the proinflammatory cytokine IL-15. Diagnosis of the majority of T-cell LGLL is established by documenting clonal large granular lymphocytes (LGLs) in peripheral blood, by morphology and immunophenotype. The proteasome inhibitor bortezomib is known to target molecular pathways downstream of the IL-15 receptor signaling and has been proposed as a therapy in these patients. We report an uncommon presentation of LGLL with chronic neutropenia lacking typical blood LGLs, which failed to respond to bortezomib but obtained a very good partial remission with a classical methotrexate regimen.
Introduction
T-cell large granular lymphocytic leukemia (T-LGLL) is a rare lymphoproliferative disease characterized by the accumulation of a monoclonal T- or natural killer (NK)-lymphocyte population in the bone marrow, blood, spleen, and liver.Citation1,Citation2
Large granular lymphocytes (LGLs) are large white blood cells (WBCs) with reniform or round nuclei and abundant cytoplasm, which typically contains azurophilic granules. In normal adults, LGLs account for up to 10%–15% of peripheral blood mononuclear cells.Citation3 This population can be further classified into the following two different lineages of lymphocytes based on their cell surface markers: CD3− NK cells and CD3+ cytotoxic T-lymphocytes.Citation4,Citation5
Current World Health Organization (WHO) classification recognizes the following three distinct disorders of LGLs: T-LGLL, chronic lymphoproliferative disorders of NK cells (CLPD-NK), and aggressive NK-cell leukemia. Despite the different cellular origin, there is considerable overlap between T-LGLL and CLPD-NK in terms of clinical presentation and response to therapy.Citation6,Citation7
T-LGLL and CLPD-NK are indolent diseases frequently associated with cytopenias and a wide spectrum of autoimmune manifestations.Citation8 Neutropenia can lead to recurrent bacterial infections, representing an indication for treatment initiation in most of the cases.Citation9 Immunosuppressive therapies are usually used in this context, including methotrexate, cyclophosphamide, and cyclosporine.Citation10
With the advent of potential new therapeutic options, a recent surge in the interest for the pathophysiology of these diseases is noted.Citation5 We here report an uncommon presentation of T-LGLL, where the diagnosis was missed because of the lack of the typical appearance of circulating and bone marrow LGLs.
Case report
A 60-year-old Caucasian woman was referred to the Department of Hematology of Ion Chiricuta Oncology Institute because of recurrent respiratory and urinary infections and with a diagnosis of chronic leukopenia. She had a documented history of neutropenia with absolute neutrophil count <1 G/L for >6 years, but no abnormal cells were noted on past cytological blood and bone marrow examinations.
On admission, clinical appearance was normal except for a palpable spleen. Abdominal ultrasound described an enlarged spleen of 172/60 mm with a 90 cm2 surface. A chest abdomen pelvis computed tomography scan described mild hepatosplenomegaly with no lymph node enlargement. Blood tests confirmed leukopenia with severe neutropenia and lymphopenia (WBC 0.88 G/L, neutrophils 0.28 G/L, and lymphocytes 0.3× G/L) and mild normocytic anemia (hemoglobin 112 g/L and mean corpuscular volume 88 fL) and thrombocytopenia (122× G/L). The reticulocyte count was slightly increased, as were lactate dehydrogenases and unconjugated bilirubin levels. Direct Coombs’ test was negative, but haptoglobin was low and cold agglutinins were detected. The presence of cryoglobulins was also revealed. Serum protein immunofixation electrophoresis was negative for monoclonality, as was screening for viral infections (CMV, EBV, HTLV, and HIV) and autoimmunity (rheumatoid factor, anti-double-stranded DNA antibodies, and antinuclear autoantibodies, the latter at the upper limit of normal). No atypical cells were noted on the peripheral blood smear.
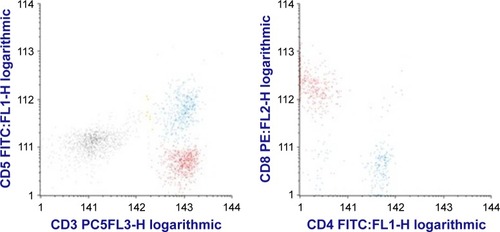
Examination of the bone marrow identified a population of atypical lymphocytes, but no azurophilic granules were described. The infiltrate was difficult to quantify because of the presence of >90% smudge cells (). The lymphocytic infiltration was identified by bone marrow flow cytometry as including a 20% population of atypical suppressor CD3+/CD8+/CD5− T-cells, coexpressing CD7, CD43, and CD2. A bone marrow biopsy confirmed the presence of an infiltration with lymphocytes of the above phenotype, described as large cells with an abundant cytoplasm. This immunophenotype is compatible with the expression of LGL surface markers, and the aberrant lack of CD5 expression sustained the clonality of the mentioned population ().
Figure 1 Bone marrow examination showing infiltrate with cells of lymphoid morphology, intact (arrows), or smudged.
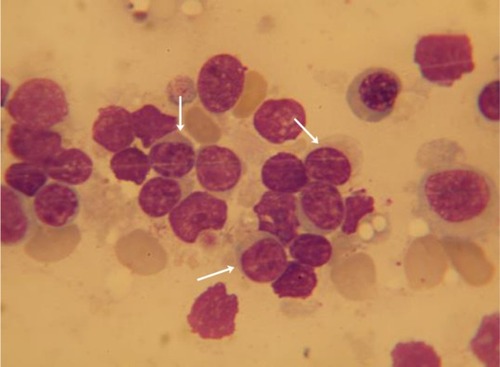
Figure 2 Dot plot histograms showing atypical suppressor CD5−CD3+CD8+ T cells (in red) and normal CD3+CD5+ T-cells (blue).
The patient was diagnosed with T-LGLL, secondary cryoglobulinemia, and associated cold agglutinin hemolytic anemia. Treatment with corticosteroids (prednisone 1 mg/kg) was initiated. After 4 weeks, there was no response and, in the presence of recurrent respiratory infection symptoms, we decided to opt for a therapy that could yield a rapid response. Thus, a second-line treatment with bortezomib associated with dexamethasone (bortezomib 1.3 mg/m2 intravenous on days 1, 4, 8, and 11 and dexamethasone 40 mg/day intravenous on days 1–4 and 8–11) was initiated. The patient underwent two cycles of therapy, with no biological or clinical response. She required hospitalization in our department four times during the two chemotherapy cycles for microbiologically negative febrile neutropenia, treated with broad-spectrum antibiotics and granulocyte-colony stimulating factor. Subsequently, treatment with low-dose methotrexate (10 mg/m2/week orally) was commenced with a partial response that was rapidly acquired and maintained. The patient continues to have a very good partial response, with a stable absolute neutrophil count of >1 G/L, and did not present any clinically manifest infections requiring antibiotics or hospitalization for over 2 years.
Discussion
Historically, a circulating LGL count of >2 G/L was considered as mandatory for the diagnosis of large granular lymphocytic leukemia (LGLL), but current diagnostic requirements lowered this threshold to >0.4 or 0.5 G/L together with a suggestive clinical presentation, which may include splenomegaly, cytopenias including neutropenia and/or anemia, and autoimmune diseases. For T-LGLL, an activated T-cell phenotype is expected, expressing CD3, CD8, CD57, and/or CD16, demonstrated by flow cytometry and/or bone marrow biopsy. T-cell clonality assays are an important contributor toward diagnosis.Citation4
Our observation reports an unusual presentation of a very rare disease. The particularity of the case is the absence of circulating LGLs, no cytoplasmic azurophilic granules in the bone marrow LGLs, and the particular treatment choice. Our patient did not display any atypical lymphocytes in the peripheral blood. On the contrary, she presented lymphopenia with normal lymphocytic morphology. She did not respond to proteasome inhibitor treatment, although bortezomib is known to target some of the pathways implicated in the pathogenesis of this disease.
An evolution similar to our patient’s is seen in Felty’s syndrome, condition with a pathogenesis closely related to LGLL as both diseases have a common genetic determinant, the HLA-DR4 haplotype.Citation11 Classically, the criteria for Felty’s syndrome required the triad rheumatoid arthritis, leukopenia, and splenomegaly.Citation12 Nowadays, leukopenia is clearly defined as neutropenia, and it became clear that patients displaying only rheumatoid arthritis and neutropenia have a similar clinical evolution regardless of the presence or absence of splenic enlargement.Citation12 A total of 11%–36% of LGLL patients have been reported to have rheumatoid arthritis, and 30%–40% of the patients diagnosed with Felty’s syndrome have LGL expansion.Citation1,Citation4,Citation13,Citation14
A new vision unifying the hematologic and rheumatologic pathologies could improve the treatment of LGLL and warrants the consideration of different interesting therapeutic options. Patients with unexplained neutropenia with or without serologically positive rheumatoid arthritis should be tested for circulating or bone marrow LGLs.
Interestingly, LGLL has also been reported to be associated with B-cell lymphoproliferations. In a single-center study reviewing 126 patients with LGL leukemia or expansion, 39.4% of patients also presented a lymphoproliferative disease including monoclonal gammopathy of unknown significance and monoclonal B-cell lymphocytosis, either concomitantly or developed shortly after LGLL diagnosis. Conversely, 14.2% of the patients had a documented previous B-cell lymphoproliferative disease, including diffuse large B-cell, Burkitt’s, and mantle cell lymphomas, chronic lymphocytic leukemia, multiple myeloma, and hairy cell leukemia.Citation15
Concerning the treatment choice in our patient, as far as we know, there is only one report of a patient with T-LGLL treated with bortezomib, and in the case reported the patient had a rapid complete hematological response.Citation16 Our patient had no response to bortezomib, but she responded rapidly to immunosuppressive doses of methotrexate. However, this does not demonstrate the inefficacy of bortezomib, and we find the hypothesis of bortezomib targeting the dysregulated pathways in LGLL as appealing.
In this group of diseases, there are multiple survival signaling pathways activated, such as the JAK/STAT3, sphingolipid, and Ras/MEK/ERK pathways, adding further complexity to treatment options.Citation17,Citation18 Because some patients are refractory to currently available treatments and none of these therapeutic modalities can cure T-LGLL, new therapeutic options are needed.Citation19 No standard therapy has yet been established because of the lack of large, prospective trials.
Conclusion
LGLL is a rare clonal disorder of T-lymphocytes with a complex biological and clinical appearance, which makes diagnosis a challenge. As seen in our case, circulating LGLs can be absent, as do LGL cytoplasmic azurophilic granules. Therefore, unexplained neutropenia should be further investigated for a possible LGLL.
There are no curative therapeutic modalities, and it is fairly well accepted that the treatment of choice is immunosuppression. All the molecular pathways recently recognized to be involved in this disease have opened perspectives on new potential therapeutic targets. Bortezomib and splenectomy are options worth considering. The rarity of the disease, however, makes prospective trials difficult to implement.
Acknowledgments
Drs CB and MZ are supported by a grant from the Romanian National Authority for Scientific Research, CNCS – UEFISCDI (Project number PN-II-RU-TE-2014-4-2074). The patient gave written consent for the use of her anonymized data for scientific purposes.
Disclosure
The authors report no conflicts of interest in this work.
References
- LoughranTPJrClonal diseases of large granular lymphocytesBlood19938211148324214
- WattersRJLiuXLoughranTPJrT-cell and natural killer-cell large granular lymphocyte leukemia neoplasiasLeuk Lymphoma201152122217222521749307
- ZhangDLoughranTPJrLarge granular lymphocytic leukemia: molecular pathogenesis, clinical manifestations, and treatmentHematology Am Soc Hematol Educ Program2012201265265923233648
- LamyTLoughranTPJrHow i treat LGL leukemiaBlood2011117102764277421190991
- LazaroEDuffauPChaigne DelalandeSGreibCPellegrinJLViallardJFLarge granular lymphocyte leukemia: clinical and pathogenic aspectsRev Med Interne201334555356023928096
- DeardenCLarge granular lymphocytic leukaemia pathogenesis and managementBr J Haematol2011152327328321158751
- LeblancFZhangDLiuXLoughranTPLarge granular lymphocyte leukemia: from dysregulated pathways to therapeutic targetsFuture Oncol20128778780122830400
- BockornyBDasanuCAAutoimmune manifestations in large granular lymphocyte leukemiaClin Lymphoma Myeloma Leuk201212640040522999943
- Autrel-MoignetALamyTAutoimmune neutropeniaPresse Med2014434 Pt 2e105e11824680423
- LoughranTPJrZicklLOlsonTLImmunosuppressive therapy of LGL leukemia: prospective multicenter phase II study by the Eastern Cooperative Oncology Group (E5998)Leukemia201529488689425306898
- StarkebaumGLoughranTPJrGaurLKDavisPNepomBSImmunogenetic similarities between patients with Felty’s syndrome and those with clonal expansions of large granular lymphocytes in rheumatoid arthritisArthritis Rheum19974046246269125243
- BurksEJLoughranTPJrPathogenesis of neutropenia in large granular lymphocyte leukemia and Felty syndromeBlood Rev200620524526616530306
- SokolLLoughranTPJrLarge granular lymphocyte leukemiaOncologist200611326327316549811
- BowmanSJBhavnaniMGeddesGCLarge granular lymphocyte expansions in patients with Felty’s syndrome: analysis using anti-T cell receptor V beta-specific monoclonal antibodiesClin Exp Immunol199510111824
- SkarbnikAPPortellCAMaciejewskiJPAssociation of large granular lymphocytic leukemia (LGL) with B-cell lymphoproliferative disordersBlood20131221387
- PellicciaSDi NapoliANasoVAlmaERebecchiniCCoxMCVery long-lasting remission of refractory T-large granular lymphocytes leukemia and myeloma by lenalidomide treatmentEur J Haematol201391218318623692265
- SteinwaySNLeBlancFLoughranTPJrThe pathogenesis and treatment of large granular lymphocyte leukemiaBlood Rev2014283879424679833
- RajalaHLPorkkaKMaciejewskiJPLoughranTPJrMustjokiSUncovering the pathogenesis of large granular lymphocytic leukemia-novel STAT3 and STAT5b mutationsAnn Med201446311412224512550
- KoskelaHLEldforsSEllonenPSomatic STAT3 mutations in large granular lymphocytic leukemiaN Engl J Med2012366201905191322591296