Abstract
Background
Activating point mutation of the RAS gene has been generally accepted as an oncogenic event in a variety of malignancies. It represents one of the most common genetic alterations in acute myeloid leukemia (AML). However, little is known about its clinical relevance in the treatment outcome for this leukemia.
Objective
This study aimed to clarify the biologic and prognostic impact of K-RAS mutations in relation to the dose of cytarabine (ara-C) used in postinduction consolidation chemotherapy in adult AML patients.
Patients and methods
The study comprised of 71 de novo AML patients with male/ female ratio 1.4:1; their ages ranged from 21–59 years with a median of 37 years. They were subjected to full clinical evaluation, routine laboratory investigations, cytogenetic studies by G-banding (Giemsa staining), and K-RAS mutation detection using real-time polymerase chain reaction. The patients were randomized into two groups according to the ara-C dose used in consolidation treatment, the high the dose ara-C (HDAC) group receiving 400 mg ara-C and-low-dose ara-C (LDAC) group receiving 100 mg ara-C; they were followed over a period of five years.
Results
Mutations in the K-RAS gene (mutRAS) were detected in 23 patients (32%) with the remaining 48 patients (68%) having wild-type RAS (wtRAS). The percent of blast cells was significantly lower in mutRAS compared to wtRAS patients (P ≤ 0.001) while M4 subtype of AML and Inv(16) frequencies were significantly higher in mutRAS compared to wtRAS patients (P = 0.015) and (P = 0.003), respectively. The patients were followed up for a median of 43 months (range 11–57 months). There was no significant difference in overall survival (OS) between mutRAS and wtRAS (P = 0.326). Within the mutRAS patients treated with HDAC, cumulative OS was significantly higher than those treated with LDAC (P = 0.001). This was not the case in the wtRAS group (P = 0.285). There was no significant difference in disease-free survival (DFS) between mutRAS and wtRAS groups (P = 0.923). mutRAS patients treated with HDAC had a statistically higher cumulative DFS than mutRAS patients treated with LDAC (P = 0.001). Patients with wtRAS also benefited from HDAC, but to a lesser extent. Among patients with wtRAS, those treated with HDAC showed higher cumulative and median DFS than patients treated with LDAC (P = 0.031).
Conclusion
It was concluded that adult AML patients carrying mutations in the K-RAS gene benefit from higher ara-C doses more than wtRAS patients, so pretreatment mutation detection could be an important predictor for treatment strategy and survival of adult AML patients. These findings counter the prevailing bias that oncogene mutations lead to more aggressive behavior in human malignancies.
Introduction
Activating point mutations of RAS genes have been generally accepted as oncogenic events in the tumorigenesis of a variety of malignancies. The mechanisms by which mutant RAS (mutRAS) is transforming affected cells have been extensively studied. In general, RAS mutations lead to a resistance of the RAS proteins to signaling provided by guanosine triphosphatase (GTPase)-activating proteins thus remaining locked in the active GTP-bound state.Citation1,Citation2 This constitutive activity is furthermore translated via a cascade of key signaling events that contribute to the regulation of proliferation, apoptosis, and differentiation. One example for such a signaling cascade is RAS-GTP recruitment of RAF proteins to the plasma membrane which leads to activating phosphorylation of mitogen-activated protein kinase (MAPK) and consequently extracellular signal-regulated kinases (ERK). ERK activity has been linked to proliferative stimulation and activation of antiapoptotic pathways.Citation3,Citation4
In acute myelogenous leukemia (AML), activating point mutations could be shown to affect almost exclusively N-RAS and K-RAS at codons 12, 13, and 61. This alteration was detected with varying frequencies (25%–40%) and represents one of the most common genetic alterations detected in AML.Citation5
Given the potential effect of activating point mutations on cellular physiology, one might expect in AML patients an association between RAS mutations and an aggressive course of the disease. However, under certain conditions, RAS activity may even give rise to increased senescence and proapoptotic signaling.Citation6,Citation7 Recently, some studies showed that there may be interactions between genetic alterations and therapeutic modalities. For example, RAS mutations harbor poor prognosis in lung cancer without adjuvant chemotherapy, whereas they are associated with better outcome after chemotherapy, which may be caused by a differential sensitivity of RAS mutated cells towards cytarabine- (ara-C) containing chemotherapy.Citation8,Citation9 Other studies revealed conflicting data and did not show an independent effect of RAS mutations on therapy outcome.Citation10,Citation11 Some studies have analyzed the association of RAS mutation with specific French– American–British (FAB) subtypes, karyotypes, or blast cell tumor load in AML, but the results were conflicting.
This study aimed to clarify the biologic and prognostic impact of K-RAS mutations in relation to high- and low-dose ara-C used in postinduction consolidation therapy of de novo adult AML patients.
Material and methods
Patients
Eighty-nine consecutive patients with de novo AML attending the Hematology and Oncology Unit, Zagazig University Hospital, Egypt were enrolled in this study cohort for the biologic and prognostic impact of RAS mutations over a period of five years. The age requirement for eligibility was greater than 18 years with an upper limit of 60 years. Patients having acute promyelocytic leukemia were not enrolled in the study. Only patients who achieved a complete remission (CR) after one or two courses of induction chemotherapy were eligible for inclusion in this analysis of postremission therapy. Patients with a prior history of myelodysplasia, other antecedent hematologic malignancies, prior nonsteroidal cytotoxic chemotherapy or radiation therapy, pre-existing liver disease, or uncontrolled infection were not included in the study. Written informed consent was obtained from all patients.
Treatments
Patients received induction chemotherapy of daunorubicin 45 mg/m2/day intravenously for three days and cytarabine 200 mg/m2/day as a continuous infusion for seven days. Those who attained a CR after one or two courses of induction therapy (n = 74) were randomly assigned to one of two postinduction arms that differed in dose-intensity of ara-C. These arms included four cycles of (a) high-dose in ara-C (HDAC) at 400 mg/m2 as a continuous infusion for five days or (b) low-dose ara-C (LDAC) at 100 mg/m2 as a continuous infusion for five days. In each case, this was followed by maintenance treatment consisting of four monthly treatments with ara-C (100 mg/m2 every 12 hours) for five days by subcutaneous injection and daunorubicin 45 mg/m2 on the first treatment day. Thereafter, patients were followed up with bone marrow (BM) examination every three months for one year, every six months for two years, and then every year for two additional years.
Methods
All patients were subjected to the following: full clinical assessment including history taking, clinical examination and abdominal ultra sonography, liver and kidney function tests, complete blood count and BM aspiration with examination of well prepared films by Leishman and peroxidase stains, flowcytometric immunophenotypic analysis of peripheral blood (PB) and/or BM aspirates, conventional cytogenetic analysis by G-banding (Giemsa staining), and K-RAS mutation detection using real-time polymerase chain reaction (PCR). Lab investigations were carried out at the Clinical Pathology Department of Zagazig University Hospital.
Cytogenetic analysis
Cultivation and harvesting
All reagents were supplied by GIBCO-BRL (Invitrogen Carlsbad, CA). BM was cultured (two tubes for each patient) on RPMI-1640 (Sigma-Aldrich, St Louis, MO) to which fetal calf serum, L-glutamine, and penicillin/streptomycin were added; the tubes were then incubated at 37°C in a CO2 incubator for 24 and 48 hours. Colcemid was added to arrest mitosis followed by hypotonic treatment of the cell pellets and subsequent fixation with methanol–acetic acid solution.Citation12
Slide preparation and banding
Fixed cells were then dropped onto a frosted, alcohol-cleaned slide. Aging of slides for 24 hours in an incubator at 37°C was done for proper banding. At least five slides were prepared for each patient and evaluated under the phase contrast microscope for metaphases. Banding with trypsin solution and counterstaining with Giemsa was performed. The slides were examined microscopically using an oil emersion lens. At least 20 metaphases were subjected to analysis; we used an Imstar image analyzer for karyotyping (Paris, France).
DNA extraction and RAS mutations detection
All molecular analyses were conducted in a blinded fashion on DNA extracted from cryopreserved cells taken at the time of diagnosis. Seventy-four patients were submitted for analysis; however, three (4%) were deemed ineligible because of defective DNA samples. Screening for RAS mutations was performed using real-time PCR in 71 patients.
DNA extraction
PB mononuclear cells were isolated on a Ficoll gradient. DNA was isolated using standard procedure and extracted promptly using DNA extraction and purification kit (High Pure PCR Template Preparation Kit; Roche Diagnostics, Mannheim, Germany) according to manufacturer’s instructions. The eluted purified DNA was stored at −80°C until used.
Real-time assay
The method was carried out on the LightCycler instrument (Roche Diagnostics), comprising of amplification of a 171 bp fragment of the K-RAS gene using specific primers, together with hybridization probes labeled with LightCycler Red 640. The detection depends on the fact that the amplification of the wild type K-RAS codons 12/13 DNA is suppressed by a competitor so that its melting peak at 64.7°C was not detectable, whereas mutants of K-RAS codons 12/13 DNA show melting peaks of 61.0°C in channel 640/530.
PCR protocol
PCR was performed in glass capillary tubes using 1 μL of genomic DNA and LightMix Kit K-RAS codons 12/13 (Roche Diagnostics) with final volume of 20 μL. The PCR program started with incubation at 95°C for 10 minutes, followed by 45 cycles of PCR amplification. Each cycle consists of three segments with three different temperatures: denaturation at 95°C for 10 seconds, annealing at 58°C for 10 seconds, and extension at 72°C for 25 seconds. The temperature was then increased at a rate of 0.1°C/second up to 85°C with continuous fluorescence monitoring. The software provided with the equipment gives the temperature of melting; the mutation detection was achieved by melting curve analysis. In samples with wild-type K-RAS, no melting curve will be displayed.
Response to therapy
CR was defined as the presence of morphologically normal BM in patients with at least 1.5 × 109/L granulocytes and 100 × 109/L platelets in the blood. Relapse was defined as >5% leukemic blasts in a BM aspirate or new extramedullary leukemia. Overall survival (OS) was measured from the protocol on-study date until the date of death regardless of cause, censoring for those alive at last follow-up. Disease-free survival (DFS) was estimated from the time of first CR to relapse or death in CR. Patients who were still alive and disease free were censored at the date of last follow up.
Statistical methods
Statistical analysis of data was performed with SPSS computer program (version 16.0;SPSS Inc., Chicago, IL). Data are presented as median and ranges. Student’s t-test, Mann– hitney U-test, chi-square, and Fisher’s exact test were used for comparison between groups. Kaplan–Meier method was used to estimate survival, and the differences between groups were analyzed using logrank test. P < 0.05 was considered significant.
Results
Eighty-nine patients participated in this study. Fifteen were excluded after induction chemotherapy (four died and 11 failed to attain remission). Another three patients were excluded because of defective DNA samples. K-RAS mutation detection and statistical analyses were carried out for the remaining 71 patients, who comprised 42 males and 29 females with male/female ratio 1.4:1; their ages ranged from 21 to 59 years with a median of 37 years.
Mutations in K-RAS were detected in leukemic cells from 23 patients (32%), with the remaining 48 patients (68%) having wtRAS alleles. shows comparison of clinical and hematological features of AML patients with and without K-RAS mutations. No statistically significant differences were found for age, hepatomegaly, splenomegaly or lymphadenopathy, hemoglobin level, platelet count, or total lymphocyte count. The median of BM and PB blasts percent was significantly higher in mutRAS compared to wtRAS patients (P < 0.001 and P = 0.025, respectively).
Table 1 Comparison of clinical and hematological features of AML patients with and without RAS mutations
Thirty-nine patients were assigned to HDAC therapy (13 mutant and 26 wild RAS), while 32 patients were assigned to receive LDAC (10 mutant and 22 wild), with nonstatistical difference in the percentage of patients with respect to the mutational status in each group (P = 0.852).
K-RAS mutation demonstrated significant heterogeneity among FAB subgroups, being more common in M4 (P = 0.015) (). Also K-RAS mutation varied between karyotypic subtypes, with evident overrepresentation in inv(16) (p13; q22) compared to othercytogenetic groups. Inv(16) was significantly higher in mutRAS cases than in wtRAS cases (P = 0.003) ().
Table 2 Relationship between FAB subtypes and RAS mutation
Table 3 Relationship between cytogenetic groups and RAS mutation
The patients were followed up for a median of 43 months (range 11–57 months). There was no significant difference in OS between mutRAS and wtRAS (P = 0.326) (). Within the mutRAS group, cumulative OS at four years in patients treated with HDAC was significantly higher than those treated with LDAC (P = 0.001) (). This was not the case in the wtRAS group (P = 0.285) ().
Figure 1 Overall survival of AML patients according to RAS mutation status (mutRAS and wtRAS).
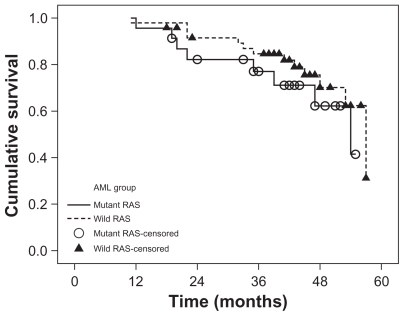
Figure 2 Overall survival of AML patients with mutant RAS according to consolidation therapy (HDAC and LDAC).
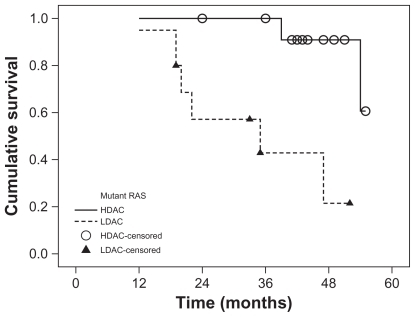
Figure 3 Overall survival of AML patients with wild RAS according to consolidation therapy (HDAC and LDAC).
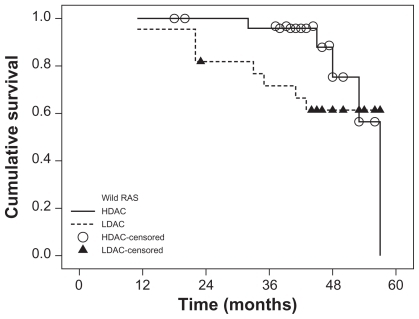
There was no significant difference in DFS between mutRAS and wtRAS groups (P = 0.923). When both the RAS status and consolidation therapy were taken into account, mutRAS patients treated with HDAC had a statistically higher cumulative survival than mutRAS patients treated with LDAC (P = 0.001) ().
Table 4 Overall survival at four years and disease-free survival at three years of the AML patients and their relationship to RAS mutational state and consolidation therapy
Patients with wtRAS also benefited from HDAC, but to a lesser extent. Among patients with wtRAS, those treated with HDAC showed higher cumulative and median DFS than patients treated with LDAC (P = 0.031).
Discussion
Mutations in the RAS proto-oncogenes have been implicated in the genesis of AML and have been described in approximately 25% of cases.Citation13 This frequency makes aberrations in RAS the most common molecular abnormality in this heterogeneous disease. Despite the prevalence of RAS mutations, the clinical significance of such molecular aberrations in AML is unclear. To assess the impact of K-RAS mutations on this disease, we sought to analyze a relatively uniform cohort of de novo AML patients who were diagnosed in a standard fashion and treated with different dose intensity chemotherapeutic regimens.
The frequency of K-RAS mutation in our small cohort of AML patients was comparable with that reported in the literature (12%–44%).Citation5,Citation14 The largest study described data from 232 patients with (28%) RAS mutations.Citation15
Unexpected findings emerged from our study. A significant association was gleaned between the presence of a K-RAS mutation and a lower fraction of blasts in the BM. Currently, we have no biologic explanation for this result. However, a strong RAS activation may be associated with reduced proliferation and enhanced maturation and differentiation of leukemic cells;Citation16 this is a possible explanation.
We demonstrated an association between K-RAS mutation frequency and M4 subtype of AML. This finding may provide indirect evidence in implicating K-RAS mutation as an important functional pathologic event in selected cases of AML. Selection and expansion of K-RAS mutant clones may provide a differentiative stimulus toward the monocytic lineage, given that K-RAS mutation was overrepresented in FAB subtypes M4 and M5. In vitro data also suggest that mutant RAS promotes a myeloid maturation defect, with relative sparing of the monocyte/macrophage lineage.Citation17
Evaluation of K-RAS mutation in association with karyotype revealed a high frequency of inv(16) in patients with K-RAS mutation compared to patients with wtRAS. This is consistent with the finding of Valk et al who proposed that signal transduction pathway mutations are common with RAS mutation.Citation18 Our sample size for inv(16) was small, and independent confirmation of the high K-RAS mutation frequency is required from larger cohorts. However, this finding is in harmony with the higher prevalence of K-RAS mutation in M4 as inv(16) is most frequently encountered in this group.
Earlier studies of the prognostic significance of mutations of K-RAS in AML showed contradictory results. Some studies reported that patients with RAS mutations had improved OS,Citation16,Citation19 whereas others found that these patients had worse complete remission rate and OS.Citation20 In most of these studies, the type of postremission treatment was not taken in account in the analysis of clinical outcome.Citation21,Citation22
However, when we considered the K-RAS status and consolidation therapy, statistical analysis revealed that the impact of K-RAS mutations on OS and DFS in our AML patients depends on the type of postremission chemotherapy. Kaplan–Meier analysis of our data revealed that the probability of OS at four years for mutRAS and wtRAS groups is comparable, whereas the estimated four years OS among patients with mutRAS assigned to HDAC was significantly higher than in those subjected to LDAC, an observation which was not found in patients with wtRAS. Although therapy with HDAC resulted in a higher DFS both in patients with and without RAS mutations, its benefit was much more pronounced in patients with mutRAS. In vitro data showed that mutations in K-RAS render tumor cell lines derived from AML, nonsmall-cell lung cancer, and colon carcinoma more sensitive to certain cytotoxic drugs, such as ara-C or topoisomerase II inhibitors.Citation23
Koo et al have demonstrated that cells harboring an activated RAS oncogene fail to arrest in the S-phase of the cell cycle in response to cytarabine treatment and that this results in their apoptotic death. In contrast, tumor cells with wtRAS genes undergo marked S-phase growth arrest on exposure to ara-C that is reversible once the drug is removed. The authors concluded that the presence of a RAS mutation may change cellular response to ara-C from cytostatic to cytotoxic, most likely because of altered cellular checkpoint functions in response to ara-C.Citation24 Other studies provide experimental evidence that mutated RAS not only induces proliferation, apoptosis, senescence, or differentiation (depending on the cellular context in which it is expressed), but it may also induce a DNA damage checkpoint response.Citation25–Citation27 These results provide biologic plausibility to our clinical observations.
Conclusion
This study evaluating AML patients with mutant K-RAS suggests that HDAC consolidation chemotherapy is critical for treating such patients. It is also possible that alternative treatment approaches that use more intensive induction therapy will be equally effective in treating this mutant subtype of AML. If our findings are confirmed, testing for K-RAS mutations could become crucial, in addition to abnormal cytogenetic AML detection, for risk adapted stratification to HDAC postremission treatment in adults with de novo AML. As new parameters with powerful prognostic significance are rapidly evolving, considering all variables in one comprehensive model is likely to provide more exact quantitative estimations of the prognosis. These prognostic distinctions are likely to provide the elementary foundations for treatment choice in the near future.
Disclosure
The authors report no conflicts of interest in this work.
References
- ChanITKatoJLWilliamslRConditional expression of oncogenic K-ras from its endogenous promoter induces a myeloproliferative diseaseJ Clin Invest2004113452853814966562
- DownwardJTargeting RAS signaling pathways in cancer therapyNat Rev Cancer200331112212509763
- HaferlachTMolecular genetic pathways as therapeutic targets in acute myeloid leukemiaHematology Am Soc Hematol Educ Program200840041119074117
- ChangFSteelmanLSLeeJTSignal transduction mediated by the Ras/Raf, MEK, and ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic interventionLeukemia20031771263129312835716
- IllmerTThiedeCFredersdorfAActivation of the RAS pathway for a chemosensitive phenotype of acute myelogenous leukemia blastsClin Cancer Res20051193217322415867216
- BarlettaEGoriniGVineisPRAS gene mutations in patients with acute myeloid leukemia and exposure to chemical agentsCarcinogenesis200425574975514688017
- BenantiJAGallowayDAThe normal response to RAS: senescence or transformation?Cell Cycle20043671571715153805
- MilellaMKomblauSMEstrovZTherapeutic targeting of the MEK/MAPK signal transduction module in acute myeloid leukemiaJ Clin Invest2001108685185911560954
- RodenhuisSBoerrigterLTopBMutational activation of the K-ras oncogene and the effect of chemotherapy in advanced adenocarcinoma of the lung: a prospective studyJ Clin Oncol19971512852918996154
- KiyoiHNaoeTNakanoYPrognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemiaBlood19999393074308010216104
- StirewaltDLKopeckyKJMeshinchiSFLT3, RAS, and TP53 mutations in elderly patients with acute myeloid leukemiaBlood200197113589359511369655
- SeabrightMA rapid banding technique for human chromosomesLancet1971277319719724107917
- CoghlanDWMorleyAAMatthewsJPBishopJFThe incidence and prognostic significance of mutations in codon 13 of the N-ras gene in acute myeloid leukemiaLeukemia1994810168216877934163
- TynerJWErichsonHDeiningerMWHigh-throughput sequencing screen reveals novel transforming RAS mutations in myeloid leukemia patientsBlood200911381749175519075190
- RitterMKimTDLisskePThiedeCSchaichMNeubauerAPrognostic significance of N-RAS and K-RAS mutations in 232 patients with acute myeloid leukemiaHaematologica200489111397139915531466
- NeubauerADodgeRKGeorgeSLPrognostic importance of mutations in the ras proto-oncogenes in de novo acute myeloid leukemiaBlood1994836160316118123851
- BowenDTFrewMEHillsRRAS mutation in acute myeloid leukemia is associated with distinct cytogenetic subgroups but does not influence outcome in patients younger than 60 yearsBlood200510662113211915951308
- ValkPJBowenDTFrewMEGoodeveACLowenbergBReillyJTSecond hit mutations in the RTK/RAS signaling pathway in acute myeloid leukemia with inv(16)Haematologica200489110614754614
- SchaichMRitterMIllmerTMutations in ras proto-oncogenes are associated with lower mdr1 gene expression in adult acute myeloid leukemiaBr J Haematol2001112230030711167822
- MeshinchiSStirewaltDLAlonzoTAActivating mutations of RTK/RAS signal transduction pathway in pediatric acute myeloid leukemiaBlood200310241474147912702504
- BraunBSTuvesonDAKongNSomatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorderProc Natl Acad Sci U S A2004101259760214699048
- BacherUHaferlachTSchochCKernWSchnittgerSImplications of NRAS mutations in AML: a study of 2502 patientsBlood2006107103847385316434492
- KooHMMonksAMikheevAEnhanced sensitivity to 1-beta-D-arabinofuranosylcytosine and topoisomerase II inhibitors in tumor cell lines harboring activated ras oncogenesCancer Res19965622521152168912859
- KooHMMcWilliamsMJAlvordWGVande WoudeGFRAS oncogene-induced sensitization to 1-beta-D-arabinofuranosylcytosineCancer Res199959246057606210626790
- Di MiccoRFumagalliMCicaleseAOncogene-induced senescence is a DNA damage response triggered by DNA hyper-replicationNature2006444711963864217136094
- KnaufJAOuyangBKnudsenESFukasawaKBabcockGFaginJAOncogenic RAS induces accelerated transition through G2/M and promotes defects in the G2 DNA damage and mitotic spindle checkpointsJ Biol Chem200628173800380916316983
- FikarisAJLewisAEAbulaitiATsygankovaOMMeinkothJLRas triggers ataxia-telangiectasia-mutated and Rad-3-related activation and apoptosis through sustained mitogenic signalingJ Biol Chem200628146347593476716968694