
Abstract
The Fms-like tyrosine kinase-3 (FLT3; fetal liver kinase-2; human stem cell tyrosine kinase-1; CD135) is a class III receptor tyrosine kinase that is normally involved in regulating the proliferation, differentiation, and survival of both hematopoietic cells and dendritic cells. Mutations leading it to be constitutively activated make it an oncogenic driver in ~30% of acute myeloid leukemia (AML) patients where it is associated with poor prognosis. The prevalence of oncogenic FLT3 and the dependency on its constitutively activated kinase activity for leukemia growth make this protein an attractive target for therapeutic intervention. Of the numerous small molecule inhibitors under clinical investigation for the treatment of oncogenic FLT3-positive AML, the N-benzoyl-staurosporine, midostaurin (CGP41251; PKC412; Rydapt®; Novartis Pharma AG, Basel, Switzerland), is the first to be approved by the US Food and Drug Administration for the treatment, in combination with standard chemotherapy, of newly diagnosed adult AML patients who harbor mutations in FLT3. Here, we describe the early design of midostaurin, the preclinical discovery of its activity against oncogenic FLT3, and its subsequent clinical development as a therapeutic agent for FLT3 mutant-positive AML.
Design of midostaurin as a kinase inhibitor
Midostaurin (structure shown in ), an N-indolocarbazole originally encoded “CGP 41251”, was discovered in 1986 as part of a drug discovery program aimed toward optimizing the inhibitory activity of staurosporine, a natural product isolated in 1977 from Streptomyces staurosporeus, against protein kinase C (PKC).Citation1 PKC was believed to play an important role in a number of pathologies, such as diabetes, inflammatory diseases, cancer, and others.Citation2 The role of activated or overexpressed PKC subtypes in cellular transformation, its activation by oncogenes, and augmentation of the transforming potential of oncogenes have been widely studied.Citation3 Midostaurin was found to be a reversible inhibitor of a panel of PKC subtypes in a purified enzyme assay, including cPKC-α, cPKC-β1, cPKC-β2, and cPKC-γ with IC50 values ranging from 0.02 to 0.03 μM.Citation4 Other PKC subtypes, including nPKC-δ, n-PKC-η, and nPKC-ε, were less potently inhibited by midostaurin (IC50 values ranging from 0.16 to 1.25 μM), with PKC-ζ showing insensitivity to the compound. Midostaurin progressed into clinical trials in patients with solid tumors; however, despite a well-tolerated dosing regimen being identified and implemented, the anti-tumor activity of the drug was insufficient to support further development.Citation4
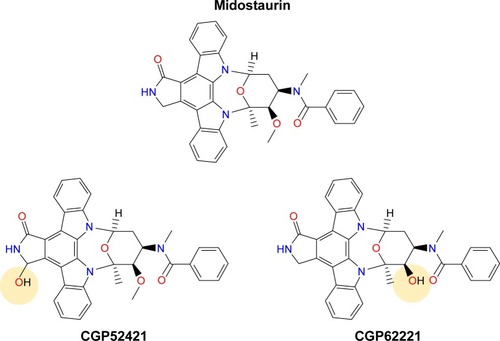
Figure 1 Chemical structures of midostaurin and its major metabolites CGP52421 (a mixture of two epimers) and CGP62221.
Around the same time, efforts to identify other kinase targets of midostaurin revealed that it was a potent inhibitor of vascular endothelial growth factor 2 (VEGFR-2; kinase insert domain receptor [KDR]; IC50=0.086 μM), KIT (CD117; IC50=0.086 μM), and platelet-derived growth factor (PDGFR; ).Citation4,Citation5 Like staurosporine, midostaurin belongs to the adenosine triphosphate (ATP)-competitive type I class of inhibitors, which bind to the catalytically active “DFG-in” conformation of the kinase to compete with ATP. The inhibitory activity of binding within the catalytic site of the enzymes in a type I or “DFG-in” binding mode involves direct competition with ATP in the ATP binding site. As such, midostaurin differs from type II or “DFG-out” ATP-competitive inhibitors, which engage a hydrophobic binding site that neighbors the ATP binding site.Citation6 The broad spectrum inhibitory capability of midostaurin is likely the reason underlying its extensive antiproliferative activity in vitro against a wide range of malignant cell types, including cancer of the breast, colon, bladder, and lung, as well as melanoma, glioblastoma, and epithelial carcinoma.Citation4
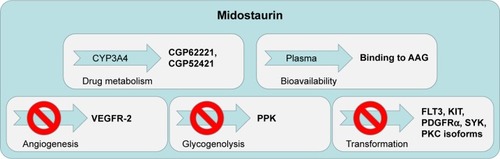
Figure 2 Schematic showing the multitargeted inhibitory effects and human protein binding properties of midostaurin and its major metabolites.
Discovery and development of midostaurin as a therapeutic agent for the treatment of mutant FLT3-positive acute myeloid leukemia (AML)
With over 20,000 new cases diagnosed in the United States annually, AML is the most common acute leukemia type in adults.Citation7 The disease is characterized by a partial blockage in differentiation and abnormal growth of myeloid progenitor cells.Citation8 This results in infiltration of the peripheral blood and bone marrow with primitive blasts, which leads to symptoms such as anemia, infection, and bleeding.Citation8
AML is a heterogeneous malignancy as it can be driven by many genetic and epigenetic lesions.Citation9 Approximately 30% of AML patients express activating mutations in Fms-like tyrosine kinase-3 (FLT3),Citation10 a gene normally involved in regulating hematopoiesis. The most prevalent form of mutant FLT3 is characterized by internal tandem duplications (ITD) in the juxtamembrane domain and is associated with a higher risk of relapse, a lower incidence of complete remission, and a lower survival rate.Citation11–Citation14 FLT3–ITD is found in ~20%–25% of AML patients and <5% of myelodysplastic syndrome (MDS) patients.Citation15–Citation21 Point mutations within the “activation loop” of FLT3 (ie, FLT3-D835Y, FLT3-D835V, FLT3-D835H, FLT3-D835E, FLT3-D835A, FLT3-D835N, FLT3-Y842C, among others) have been identified in 7% of AML patients.Citation22–Citation24 Patients may also have more than one mutation in FLT3, presumably present in different subclones of leukemia. Given that FLT3 mutations confer a poor prognosis, development of effective therapies aimed at blocking the function of FLT3 is urgently needed for patients who express this oncoprotein.
In 2001, as part of the collaboration between the Dana-Farber Cancer Institute and Novartis Pharmaceuticals, a drug screening effort was carried out that was geared toward identifying small molecule inhibitors of oncogenic FLT3-driven AML. In this drug screening of a panel of tyrosine kinase inhibitors, midostaurin was identified to be capable of inhibiting the activity of FLT3 ligand-stimulated wild-type (wt) FLT3 autophosphorylation in interleukin-3-dependent murine pro-B Ba/F3 cells.Citation22 It was also shown for the first time to inhibit the activity of mutated FLT3.Citation22 Specifically, midostaurin was observed to induce programmed cell death of Ba/F3 cells transduced with the human FLT3–ITD oncogene through direct suppression of FLT3 kinase activity. There was a correlative relationship between inhibition of FLT3 in vitro kinase activity and inhibition of growth of FLT3–ITD- and FLT3-D835Y-expressing Ba/F3 cells (IC50 of ≤10 nM) by midostaurin.Citation25 Midostaurin prolonged survival in vivo in Balb/c mice transplanted with bone marrow transduced with an FLT3–ITD-containing retrovirus, and FLT3 target engagement was supported by the fact that cells made resistant to midostaurin overexpressed mutant FLT3.Citation25 Later, studies were conducted with midostaurin, which demonstrated its ability to inhibit FLT3 activity in human FLT3–ITD-positive cell lines and primary cells.Citation26,Citation27
Clinical investigation of midostaurin as a treatment for FLT3 mutant-positive AML
Drug resistance and drug intolerance due to drug toxicity have been the limiting factors in the success of available treatments for AML. Use of standard chemotherapy as a stand-alone treatment is beneficial in that it leads to a lower incidence of treatment-associated mortality; however, the emergence of drug resistance results in an increased risk of disease recurrence and clinical relapse.Citation28 For allogeneic bone marrow transplantation (alloBMT), the opposite is true with patients having less frequent relapse but suffering a higher mortality rate due to treatment toxicity.Citation28 A major limiting factor for alloBMT is that it is most effective in patients under the age of 65 years.Citation29 However, the majority of AML patients tend to be older and thus are not good candidates for alloBMT.Citation29 There has unfortunately been little progress in the treatment of AML for the past four decades, and the disease is marked by a high rate of relapse, with a 5-year remission rate of only 40% in younger patients (<60 years) and a mere 10%–20% in older patients.Citation30 AML has an annual death rate of over 10,000.Citation7
Midostaurin entered clinical trials for mutant FLT3-positive AML at doses shown to be tolerated in earlier clinical trials. When previously tested in advanced solid tumor patients, midostaurin was found to be able to be administered orally across a broad range of doses.Citation31 The binding of midostaurin to α-1 acidic glycoprotein in human serum is thought to reduce its bioavailabilityCitation5 (); daily dosing with midostaurin leads to maximum serum concentrations after around 1 week of treatment, and this is followed by a decline in drug levels even with continuation of treatment.Citation32 Two predominant metabolites of midostaurin, CGP52421 (a mixture of two epimers), which is a less potent inhibitor of PKC but equipotent to midostaurin with respect to FLT3 inhibition,Citation25 and CGP62221, which displays potency similar to midostaurin with respect to PKC as a target, are generated in the liver by hepatic CYP3A4 ().Citation5 In light of this, to avoid metabolism-related complications, treatments that block or stimulate CYP3A4 are avoided in the clinical testing of midostaurin.Citation33
Midostaurin was found to be generally well-tolerated when administered as a single agent to relapsed patients (oral dosing at 100–225 mg); early clinical studies suggested that clinical efficacy could be improved when midostaurin was administered with standard chemotherapy (daily dosing of 100–200 mg midostaurin combined with other agents).Citation32,Citation34–Citation36 However, dose-limiting toxicities leading to discontinuation of treatment for some patients were associated with combined treatment with midostaurin (at higher doses, 50–100 mg twice daily) plus cytarabine and daunorubicin.Citation37 Sequential administration of midostaurin and standard chemotherapy was determined to be superior to simultaneous administration in terms of unwanted side effects and treatment tolerance.
In the first of two Phase IIB clinical trials, midostaurin was tested as a single agent (75 mg three times a day) in newly diagnosed mutant FLT3-positive patients who were not eligible for treatment with standard chemotherapy and mutant FLT3-positive relapsed or refractory AML patients or patients with high-risk MDS.Citation34,Citation35 Midostaurin, as a single agent, was generally well-tolerated, with nausea and vomiting being the major adverse side effects of treatment. Therapy with midostaurin led to transient effects, with a decline of blasts by at least 50% in the peripheral blood of 70% (14/20) of patients and in the bone marrow of 30% (6/20) of patients.Citation34,Citation35
In the second of the two Phase IIB clinical trials involving 95 AML and high-risk MDS patients as well as wt FLT3-expressing patients (35 mutant FLT3-expressing patients and 60 wt FLT3-expressing patients), midostaurin was administered orally twice a day at either 50 or 100 mg dose randomly to both wt and mutant FLT3-expressing patients.Citation36 As in the other Phase II trial, midostaurin as a single agent was generally well-tolerated with nausea and vomiting as adverse side effects of treatment. Midostaurin led to a decline of blasts in the peripheral blood or bone marrow by at least 50% in 71% of mutant FLT3-positive patients and in 42% of wt FLT3-expressing patients.Citation36 One partial response was reported for one patient harboring FLT3–ITD, who had received the 100 mg dose regimen.
A Phase IB clinical trial was carried out that tested the combination of midostaurin and standard chemotherapy (daunorubicin and cytarabine) in a group of 69 younger and newly diagnosed, wt or mutant FLT3-expressing patients (≥18 years but <60 years of age).Citation37 It was discovered that a dose of 50 mg twice a day was better tolerated than a dose schedule of 100 mg twice a day, with a lower discontinuation rate for the lower dosing regimen as well as a lack of grade 3/4 nausea and vomiting.Citation37 Treatment with midostaurin was carried out at the same time as chemotherapy or following chemotherapy.Citation37 Those patients receiving lower doses in this study achieved a better clinical response than those patients receiving higher doses. This was believed to be due to dose-limiting toxicity, leading to attenuation of treatment. In this trial, 35% and 74% of wt FLT3-positive patients in the high- and low-dose groups, respectively, showed a complete remission rate compared with 83% and 92% of mutant FLT3-positive patients in the high- and low-dose groups, respectively. What is particularly encouraging about this study is that a significant number of midostaurin-treated patients were able to move forward and undergo hematopoietic stem cell transplantation (HSCT).
Following these encouraging early phase studies, midostaurin was tested in combination with conventional induction and consolidation treatments in an international, randomized, double-blind Phase III study (RATIFY [CALGB 10603]) focusing on FLT3 mutant-expressing AML patients <60 years of age.Citation38 This trial enrolled 717 AML patients with FLT3 mutations, randomized between midostaurin and placebo, both given with standard induction chemotherapy. Overall survival was increased in the midostaurin arm compared to the placebo arm (74.7 vs 26.0 months, p=0.007).Citation38 In this study, although the p-value showing a survival advantage with midostaurin is significant, it is important to note that the large difference between groups may be related to the inflection point in the Kaplan–Meier survival curves, but the HR is 0.78, which is likely a more accurate reflection of the benefit obtained from midostaurin.
This study supports the notion that inhibition of FLT3 is important in patients with mutations in the FLT3 gene and also highlights the clinical effectiveness of midostaurin as a therapeutic for AML. Importantly, it should be noted that, as opposed to other small molecule FLT3 inhibitors, midostaurin offers the benefit of targeting a wide range of FLT3 activation loop mutations in addition to inhibiting FLT3–ITD. The efficacy of midostaurin in combination with standard chemotherapy in FLT3-mutated AML patients led to the eventual approval of midostaurin by Health Authority (Rydapt®).Citation39
Clinical investigation of midostaurin as a treatment for KIT mutant-positive systemic mastocytosis (SM)
KIT is a class III receptor tyrosine kinaseCitation40 that is expressed in mast cells and hematopoietic stem cells, as well as melanocytes, germ cells, and Cajal cells of the gastrointestinal tract.Citation41 Gain-of-function mutations in KIT have been identified in hematologic malignancies, including acute leukemia and mastocytosis, and non-hematologic malignancies, including germ cell tumor and gastrointestinal stromal cell tumor.Citation41
A mutated form of c-Kit, KITD816V, was first identified in the blood mononuclear cells of patients with SM, a rare disorder that is characterized by excessive production of mast cells that build up in the skin and internal organs and tissues.Citation42 There are limited choices of treatment for patients with advanced SM, for example, SM with an associated hematologic neoplasm (SM-AHN), mast cell leukemia (MCL), and aggressive SM (ASM), and the prognosis is generally poor.
Midostaurin is one of several tyrosine kinase inhibitors, including imatinib, nilotinib, and dasatinib, that has been investigated as an inhibitor of c-KitCitation43 and found in vitro to potently inhibit D816V- and D816Y-mutated KIT receptor tyrosine kinase,Citation44,Citation45 while being substantially less active against the non-mutated enzyme. Based on this profile, midostaurin has been studied for activity in SM. When administered at 100 mg twice daily to 15 SM patients, over half of whom harbored the D816V, midostaurin led to clinical responses, including reduced ascites and pleural effusion and an increase in hemoglobin as well as a decrease in bone marrow mast cell burden.Citation43 In a Phase II trial conducted with 26 advanced SM patients administered 100 mg midostaurin twice daily, a 50% reduction in mast cell burden in the bone marrow was achieved in 68% of patients.Citation46 In addition, a decrease in serum tryptase levels was observed in 46% of patients, with a median overall survival of 40 months for the whole patient population.Citation46 In a larger study testing the effects of orally administered midostaurin in 116 patients with advanced SM, clinical responses were achieved with a median overall survival of 28.7 months and a median progression-free survival of 14.1 months.Citation47 Currently, Phase II clinical trials are ongoing with midostaurin for ASM and MCL.
Discussion
Midostaurin was approved by US Food and Drug Administration for the treatment of newly diagnosed, mutant FLT3-expressing adult AML patients, in combination with standard chemotherapy (cytarabine and daunorubicin induction and cytarabine consolidation), on April 28, 2017.Citation39 Oncogenic FLT3 mutations as biomarkers are identified in a companion diagnostic, the LeukoStrat CDx FLT3 Mutation Assay. On the same day, midostaurin was approved for adult patients with SM-AHN, MCL, and ASM.
Midostaurin appears to possess a different clinical profile in AML patients compared to other FLT3 inhibitors.Citation38 It is currently speculated that this might be the result of the drug, together with its major circulating metabolites, inhibiting a number of kinases important for the viability of leukemic cells (). Indeed, the clinical responsiveness to midostaurin of AML patients expressing wt FLT3 supports the notion of midostaurin likely having beneficial off-target effects on kinases other than mutated FLT3. However, since midostaurin has been shown to inhibit wt FLT3 in vitro,Citation25 it cannot be ruled out that effects of midostaurin in wt FLT3-expressing patients may be due to elevated expression of wt FLT3 in those drug-responsive patients. As AML is a heterogeneous disease marked by significant variability in mechanism of leukemogenesis and phenotype, there is potential benefit for midostaurin being able to inhibit multiple signaling molecules that play an integral role in cancer progression. There is also benefit to be gained from the ability of midostaurin to block the activity of kinases that are mutated or hyperactivated and contribute to transformation and/or resistance to targeted therapy. For instance, studies with midostaurin have demonstrated its ability to inhibit angiogenesis in vivo, presumably due to its inhibitory effects on the VEGF receptor ().Citation5,Citation48 This may have a significant impact on the clinical responsiveness of AML patients as angiogenesis has been reported to be increased in AML.Citation49 However, it should be noted that three fatalities associated with pulmonary toxicity were reported in early clinical trials, and the VEGFR inhibitory activity of midostaurin and antiangiogenesis activity of the drug are believed to have possibly played a role in two of the cases.Citation35
As mentioned previously, PKC is a primary target of midostaurin. The inhibition of PKCs involved in transformationCitation3 may also add to the anticancer activity of midostaurin ().Citation4 In addition to the potential clinical benefit is the ability of midostaurin to target proteins such as KIT, which has been found to be mutated and while insufficient by itself for tumorigenesis does play an important role in cancer occurrence ().Citation50
Another inhibited target of midostaurin, as mentioned above, is PDGFR (IC50=0.08 μM in a purified enzyme assay).Citation4 PDGFRA and PDGFRB activating mutations are associated with myeloproliferative neoplasms ().Citation43 Somatic mutations lead to chimeric proteins involving PDGFR-alpha, such as the well-characterized, imatinib-sensitive activating mutation, FIP1L1–PDGFRA, which causes hypereosinophilic syndrome (HES).Citation51 A mutation in FIP1L1–PDGFRA, T674I, which is analogous to the imatinib-resistant T315I “gatekeeper” mutation in BCR–ABL, renders FIP1L1–PDGFRA similarly resistant to imatinib.Citation51 Midostaurin was shown to override this resistance.Citation52
Midostaurin also potently inhibits an enzyme associated with glycogen metabolism, polyphosphate kinase (PPK) (IC50=0.038 μM in a purified enzyme assay).Citation4 As glycogen metabolism is upregulated in a number of tumor types and contributes to cancer cell pathophysiology, inhibition of PPK by midostaurin may contribute to midostaurin’s anticancer activity ().
Spleen tyrosine kinase (Syk) was found to be an additional target of midostaurin (IC50=0.095 μM in a purified enzyme assay; ).Citation4 Syk has been characterized as a driver oncogene for hematologic malignancies including chronic lymphocytic leukemia, mantle cell lymphoma, and B-cell lymphoma.Citation53–Citation55 The product of Syk and ITK (IL-2-inducible T cell kinase) fusion, ITK–Syk, has been identified as a recurrent translocation in 17% of unspecified peripheral T-cell lymphoma patients.Citation56,Citation57 Activated Syk has been implicated in AML,Citation58,Citation59 which was found to cause a B-acute lymphocytic leukemia (ALL)-like disease in vivo,Citation60 and was detected in a patient with an atypical MDS with leukemic transformation.Citation61,Citation62
Recently, midostaurin was validated in cell-based assays as a dual inhibitor of FLT3 and, constitutively, active Syk.Citation63 Highly activated Syk was discovered to be enriched in FLT3–ITD-positive patients, and Syk has also been shown to be important for transformation and necessary for the development of myeloproliferative disease (MPD).Citation64 In addition, Syk overexpression confers resistance to FLT3 inhibitors.Citation64 The ability of midostaurin to inhibit Syk is of potential clinical significance given the reported role of Syk in FLT3 kinase inhibitor resistance, transformation, and maintenance of AML.
Conclusion
There is a pressing need for new and improved treatments that are well-tolerated and that will prolong AML patient survival. Midostaurin was one of a number of targeted FLT3 kinase inhibitors tested in clinical trials, and it exhibited sufficient efficacy and a favorable enough toxicity profile to move from testing as a single agent to testing in combination with standard therapies. In general, the transient and partial responses observed in AML patients treated with FLT3 inhibitors as single agents raise the question of the extent to which mutated FLT3 is an effective stand-alone target for therapy. Indeed, a continuation of phosphorylation of downstream signaling molecules following FLT3–ITD inhibition in some primary AML cells and occasional lack of correlation between inhibition of FLT3–ITD autophosphorylation and induction of cell death may explain the limited effectiveness of FLT3 inhibitors as a monotherapy in the clinic. It has also been proposed that the inability of FLT3 inhibitors to cause sustained and complete clinical responses in AML patients may account for their limited utility.Citation25,Citation65,Citation66
Importantly, full genome sequencing results suggest that AML is initiated by as many as 20 distinct mutations, and the detection of FLT3 mutations in only a subset of AML cells suggests that they may be disease promoting as opposed to initiating. As such, aberrant signaling independent of mutant FLT3-mediated signaling may contribute to leukemic cell survival even in the presence of an FLT3 inhibitor. As there is significant cross talk between the main signaling pathways downstream of FLT3, this may provide a survival advantage to mutant FLT3-expressing leukemic cells and requires a multi-targeted therapy to override this. The polypharmacology that characterizes midostaurin may uniquely work in its favor in more effectively suppressing those factors that enable leukemic cells to thrive in the presence of small molecule FLT3 inhibition and may explain, at least in part, midostaurin’s efficacy in patients.
Disclosure
PWM is an employee of Novartis Pharma AG. JDG and PWM have a financial interest with Novartis Pharma AG. The authors report no other conflicts of interest in this work.
References
- CaravattiGMeyerTFredenhagenAInhibitory activity and selectivity of staurosporine derivatives towards protein kinase CBioorg Med Chem Lett199443399404
- Mochly-RosenDDasKGrimesKVProtein kinase C, an elusive therapeutic target?Nat Rev Drug Discov2012111293795723197040
- BlobeGCObeidLMHannunYARegulation of protein kinase C and role in cancer biologyCancer Metastasis Rev1994133–44114317712599
- FabbroDBuchdungerEWoodJInhibitors of protein kinases: CGP 41251, a protein kinase inhibitor with potential as an anticancer agentPharmacol Ther199982229330110454207
- FabbroDRuetzSBodisSPKC412- a protein kinase inhibitor with a broad therapeutic potentialAnticancer Drug Des2000151172810888033
- LiuYGrayNSRational design of inhibitors that bind to inactive kinase conformationsNat Chem Biol20062735836416783341
- SiegelRLMillerKDJemalACancer statistics, 2015CA Cancer J Clin201565152925559415
- McKenzieSBAdvances in understanding the biology and genetics of acute myelocytic leukemiaClin Lab Sci2005181283715747784
- IslamMMohamedZAssenovYDifferential analysis of genetic, epigenetic, and cytogenetic abnormalities in AMLInt J Genomics20172017291364828713819
- Cancer Genome Atlas Research NetworkGenomic and epigenomic landscapes of adult de novo acute myeloid leukemiaN Engl J Med2013201336820592074
- WhitmanSPArcherKJFengLAbsence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a cancer and leukemia group B studyCancer Res200161197233723911585760
- SchlenkRFDohnerKKrauterJMutations and treatment outcome in cytogenetically normal acute myeloid leukemiaN Engl J Med2008358181909192818450602
- KayserSSchlenkRFLondonoMCGerman-Austrian AML Study Group (AMLSG). Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcomeBlood2009114122386239219602710
- PatelJPGonenMFigueroaMEPrognostic relevance of integrated genetic profiling in acute myeloid leukemiaN Engl J Med2012366121079108922417203
- NakaoMYokotaSIwaiTInternal tandem duplication of the FLT3 gene found in acute myeloid leukemiaLeukemia19961012191119188946930
- HoriikeSYokotaSNakaoMTandem duplications of the FLT3 receptor gene are associated with leukemic transformation of myelodysplasiaLeukemia1997119144214469305595
- KiyoiHTowatariMYokotaS1998. Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the productLeukemia1998129133313379737679
- KondoMHoribeKTakahashiYPrognostic value of internal tandem duplication of the FLT3 gene in childhood acute myelogenous leukemiaMed Pediatr Oncol199933652552910573574
- RomboutsWJBloklandILowenbergBBiological characteristics and prognosis of adult acute myeloid leukemia with internal tandem duplications in the FLT3 geneLeukemia200014467568310764154
- KellyLMYuJCBoultonCLCT53518, a novel selective FLT3 antagonist for the treatment of acute myelogenous leukemia (AML)Cancer Cell20021542143212124172
- MattisonRJOstlerKRLockeFLImplications of FLT3 mutations in the therapy of acute myeloid leukemiaRev Recent Clin Trials20072213514118473998
- YamamotoYKiyoiHNakanoYActivating mutation of D835 within the activation loop of FLT3 in human hematologic malignanciesBlood20019782434243911290608
- JiangJPaezJGLeeJC2004. Identifying and characterizing a novel activating mutation of the FLT3 tyrosine kinase in AMLBlood200410461855185815178581
- KindlerTBreitenbuecherFKasperSIdentification of a novel activating mutation (Y842C) within the activation loop of FLT3 in patients with acute myeloid leukemia (AML)Blood2005105133534015345593
- WeisbergEBoultonCKellyLInhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412Cancer Cell20021543344312124173
- PratzKWSatoTMurphyKMFLT3-mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AMLBlood201011571425143220007803
- WilliamsCBKambhampatiSFiskusWPreclinical and phase I results of decitabine in combination with midostaurin (PKC412) for newly diagnosed elderly or relapsed/refractory adult patients with acute myeloid leukemiaPharmacotherapy201333121341135223798029
- MathewsVDiPersioJFStem cell transplantation in acute myelogenous leukemia in first remission: what are the options?Curr Hematol Rep20043423524115217552
- WitherspoonRPDeegHJAllogeneic bone marrow transplantation for secondary leukemia or myelodysplasiaHaematologica199984121085108710586209
- SanzMBurnettALo-CocoFLowenbergBFLT3 inhibition as a targeted therapy for acute myeloid leukemiaCurr Opin Oncol200921659460019684517
- McDonaldACPopperDKingDPhase I and pharmacokinetic study of CGP 41521, an inhibitor of protein kinase (abstract)Proc Am Soc Clin Oncol199715212a
- PropperDJMcDonaldACManAPhase I and pharmacokinetic study of PKC412, an inhibitor of protein kinase CJ Clin Oncol20011951485149211230495
- DutreixCMunariniFLorenzoSInvestigation into CYP3A4-mediated drug-drug interactions on midostaurin in healthy volunteersCancer Chemother Pharmacol20137261223123424085261
- StoneRMDe AngeloJGalinskyIPKC412 FLT3 inhibitor therapy in AML: results of a phase II trialAnn Hematol200483Suppl 1S89S9015124689
- StoneRMDeAngeloDJKlimekVPatients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412Blood20051051546015345597
- FischerTStoneRMDeangeloDJPhase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3J Clin Oncol201028284339434520733134
- StoneRMFischerTPaquetteRPhase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemiaLeukemia20122692061206822627678
- StoneRMMandrekarSJSanfordBLMidostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutationN Engl J Med2017377545446428644114
- Commissioner, Office of the “Press Announcements – FDA approves new combination treatment for acute myeloid leukemia”2017 Available from: www.fda.govAccessed September 28, 2017
- YardenYKuangWJYang-FengTHuman protooncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligandEMBO J1987611334133512448137
- MiettinenMLasotaJKIT (CD117): a review on expression in normal and neoplastic tissues, and mutations and their clinicopathologic correlationAppl Immunohistochem Mol Morphol200513320522016082245
- NagataHWorobecASOhCKIdentification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorderProc Natl Acad Sci U S A1995922310560105647479840
- TefferiAMolecular drug targets in myeloproliferative neoplasms: mutant ABL1, JAK2, MPL, KIT, PDGFRA, PDGFRB and FGFR1J Cell Mol Med200913221523719175693
- GrowneyJDClarkJJAdelspergerJActivation mutations of human c-KIT resistant to imatinib mesylate are sensitive to the tyrosine kinase inhibitor PKC412Blood2005106272172415790786
- GleixnerKVMayerhoferMAichbergerKJPKC412 inhibits in vitro growth of neo-plastic human mast cells expressing the D816V-mutated variant of KIT: comparison with AMN107, imatinib, and cladribine (2CdA) and evaluation of cooperative drug effectsBlood200610752752779
- DeAngeloDJGeorgeTILinderAEfficacy and safety of midostaurin in patients with advanced systemic mastocytosis: 10-year median follow-up of a phase II trialLeukemia2017
- GotlibJKluin-NelemansHCGeorgeTIEfficacy and safety of midostaurin in advanced systemic mastocytosisN Engl J Med2016374262530254127355533
- FerraraNChenHDavis-SmithTVascular endothelial growth factor is essential for corpus luteum angiogenesisNature Med1998433363409500609
- PadroTRuizSBiekerRIncreased angiogenesis in the bone marrow of patients with acute myeloid leukemiaBlood20009582637264410753845
- AbbaspourBMKamalidehghanBSaleemMReceptor tyrosine kinase (c-Kit) inhibitors: a potential therapeutic target in cancer cellsDrug Des Dev Ther20161024432459
- CoolsJDeAngeloDJGotlibJA tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndromeN Engl J Med2003348131201121412660384
- CoolsJStoverEHBoultonCLPKC412 overcomes resistance to imatinib in a murine model of FIP1L1-PDGFRα-induced myeloproliferative diseaseCancer Cell20033545946912781364
- RinaldiAKweeITaborelliMGenomic and expression profiling identifies the B-cell associated tyrosine kinase Syk as a possible therapeutic target in mantle cell lymphomaBr J Haematol2006132330331616409295
- ChenLMontiSJuszczynskiPSYK-dependent tonic B-cell receptor signaling is a rational treatment target in diffuse large B-cell lymphomaBlood200811142230223718006696
- BuchnerMFuchsSPrinzGSpleen tyrosine kinase is over-expressed and represents a potential therapeutic target in chronic lymphocytic leukemiaCancer Res200969135424543219549911
- PechloffKHolchJFerchUThe fusion kinase ITK-SYK mimics a T cell receptor signal and drives oncogenesis in conditional mouse models of peripheral T cell lymphomaJ Exp Med201020751031104420439541
- StreubelBVinatzerUWillheimMNovel t(5;9)(q33;q22) fuses ITK to SYK in unspecified peripheral T-cell lymphomaLeukemia200620231331816341044
- HahnCKBerchuckJERossKNProteomic and genetic approaches identify Syk as an AML targetCancer Cell200916428129419800574
- SprisslerCBelenkiDMaurerHDepletion of STAT5 blocks TEL-SYK-induced APMF-type leukemia with myelofibrosis and myelodysplasia in miceBlood Cancer J201448e24025148222
- WossningTHerzogSKohlerFDeregulated Syk inhibits differentiation and induces growth factor-independent proliferation of pre-B cellsJ Exp Med2006203132829284017130299
- KunoYAbeAEmiNAn atypical myelodysplastic syndrome with t(9;12)(q22;p12) and TEL gene rearrangementBr J Haematol1999106257057110460625
- KunoYAbeAEmiNConstitutive kinase activation of the TEL-Syk fusion gene in myelodysplastic syndrome with t(9;12)(q22;p12)Blood20019741050105511159536
- WeisbergELPuissantAStoneRCharacterization of midostaurin as a dual inhibitor of FLT3 and SYK and potentiation of FLT3 inhibition against FLT3-ITD-driven leukemia harboring activated SYK kinaseOncotarget2017832520265204428881711
- PuissantAFenouilleNAlexeGSYK is a critical regulator of FLT3 in acute myeloid leukemiaCancer Cell201425222624224525236
- SiendonesEBarbarrojaNTorresLAInhibition of Flt3-activating mutations does not prevent constitutive activation of ERK/Akt/STAT pathways in some AML cells: a possible cause for the limited effectiveness of monotherapy with small-molecule inhibitorsHematol Oncol2007251303717128418
- ChuSJSmallDMechanisms of resistance to FLT3 inhibitorsDrug Resist Updat2009121–281619162530