
Abstract
Anaplastic lymphoma kinase (ALK) rearrangement responds to ALK tyrosine kinase inhibitors (TKIs) in lung cancer. Many cases ultimately acquire resistance to crizotinib. Resistance, including ALK-dominant or ALK non-dominant, mechanisms have been described. Transformation to small-cell lung cancer is rare. Herein, we report a 49-year-old man diagnosed with adenocarcinoma, who was negative for EGFR and ALK genes as detected by reverse transcription polymerase chain reaction, and was treated with crizotinib. A new biopsy showed a small-cell lung cancer after disease progression. Then, next-generation sequencing (NGS) was carried out and detected a TP53 gene mutation, an ALK rearrangement, and no loss of the retinoblastoma gene (RB). Although a regimen for small-cell lung cancer may be one treatment option, a heterogeneous tumor may exist at the time of diagnosis and manifest during the course of disease.
Introduction
Anaplastic lymphoma kinase (ALK) and echinoderm microtubule-associated protein-like 4 (EML4) gene rearrangements occur in 2%–7% of patients with non-small cell lung cancer (NSCLC).Citation1 Crizotinib is the standard treatment for advanced ALK-positive NSCLC and has been shown to have impressive single-agent activity in this type of patient.Citation2,Citation3 While most patients respond to crizotinib, every patient will ultimately experience disease progression within 1–2 years.Citation2 Drug resistance has not only become the major limitation of clinical efforts, but also is the most urgent issue in need of resolution for prolonging life in patients with NSCLC. It is encouraging that molecular acquired resistance mechanisms to tyrosine kinase inhibitors (TKIs) have been identified. With in-depth research, new therapeutic strategies to overcome resistance have been used to prolong survival of patients with advanced NSCLC. Mechanisms of acquired resistance to crizotinib that have been explored include on-target genetic alterations or off-target mechanisms of resistance;Citation4 however, other complex resistance mechanisms, such as mechanisms of resistance to EGFR-TKIs, still exist.Citation5 Histologic transformation into small-cell lung cancer (SCLC) has been reported in some ALK fusion adenocarcinomas; however, there are no established treatment strategies to manage such transformations.
Here, we report a patient who developed acquired resistance to crizotinib with transformation to SCLC. Moreover, we have summarized some published reports which have improved our understanding of the transformation to SCLC in ALK fusion adenocarcinoma.
Case report
A 49-year-old man, a never smoker, presented to our hospital with a 1-month history of cough and blood-stained sputum in October 2012. CT scans revealed a mass in the right upper lung, right hilar lymph node enlargement, and multiple nodules in the right lung. Cerebral and lumbar magnetic resonance imaging (MRI) revealed a nodule in the right cerebellum and a centrum tumor at the L2 level (T4N1M1b stage IV). A pathologic diagnosis of adenocarcinoma cells was established based on a bronchoscopic biopsy (). Immunohistochemical (IHC) analysis demonstrated positivity for TTF-1 and Napsin A, and negativity for cytokeratin (CK) 5/6 and P63 ( and ). Tumor tissue was shown to be wild-type of epidermal growth factor receptor variants by ARMS (AmoyDx, Xiamen, People’s Republic of China), and EML4-ALK fusion was shown by reverse transcription polymerase chain reaction (RT-PCR; AmoyDx; ). The patient received five cycles of first-line chemotherapy with cisplatin and paclitaxel from October 2012 to January 2013. The best tumor response was a partial response according to RECIST criteria, and the progression-free survival (PFS) was 7.0 months. In May 2013, the tumor progressed (right lower lobe nodules and brain metastases). The patient underwent crizotinib treatment (250 mg/bid, orally) from May 2013 to October 2014. He received whole-brain radiotherapy (2 Gy per fraction, 1 fraction per day ×20 days; total radiation dose 50 Gy). The curative effect of crizotinib treatment was stable disease (SD). After progression on crizotinib, the patient underwent multiple cycles of cytotoxic chemotherapy (gemcitabine, docetaxel, and bevacizumab; ). A secondary biopsy of the enlarging mass in the right lung was performed. Histologic examination of the biopsy specimen revealed SCLC without adenocarcinoma components (). IHC analysis demonstrated positivity for Syn and CD56, negativity for CgA, and a Ki-67 index of 98% (). To search for new therapeutic strategies, additional gene detection was performed on the tissue sample by next-generation sequencing (Gene plus, Beijing, People’s Republic of China), which showed a TP53 gene mutation (p.R248W) and ALK rearrangement accompanied by ERBB3 p.P554Q, NOTCH1 p.P3S, IL7R p.P417R, and IL6ST p.A896V, but no loss of the retinoblastoma gene (RB). The next generation sequencing (NGS) assay was performed using the HiSEquation 4000 (Illumina, San Diego, CA, USA). Then, the patient underwent etoposide monotherapy. After two cycles, the effect was SD. The patient remains alive at the time of writing this article. We have listed small-cell transformations following treatment with ALK-TKIs, including crizotinib/alectinib/ceritinib/lorlatinib in . The authors confirm that written informed consent for publication of case details and any accompanying images has been provided by the patient.
Figure 1 Hematoxylin–eosin staining and immunohistochemistry in adenocarcinoma before crizotinib treatment.
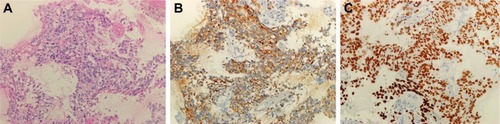
Figure 2 Amplification plot of EML4-ALK-positive by amplification refractory mutation system method.
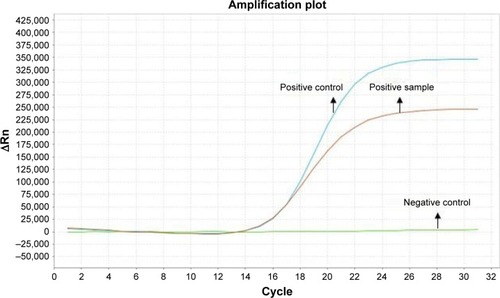
Figure 3 Hematoxylin–eosin staining and immunohistochemistry in small-cell cancer after crizotinib treatment.
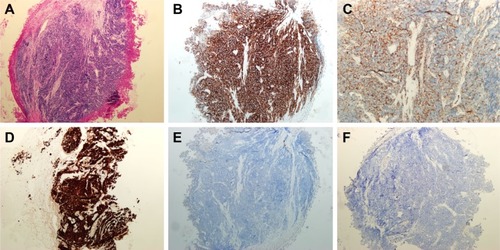
Table 1 Primary antibodies used for immunhistochemical staining
Table 2 Details of the treatment process
Table 3 List of reported small-cell transformations resistant to crizotinib/alectinib/ceritinib/lorlatinib
Discussion
With the rapid development of molecular diagnostic technology, targeted therapy is effective for patients with advanced NSCLC and associated driver genes; however, a majority of patients will eventually acquire resistance to the targeted drug and experience disease progression. Therefore, some ALK-positive patients also develop acquired resistance to ALK-TKIs. Known acquired mechanisms that cause resistance to ALK-TKIs have been described, and can be divided into two types (ALK dominant and ALK non-dominant).Citation13 ALK-dominant mechanisms include ALK secondary mutations and ALK amplification, whereas ALK non-dominant mechanisms include bypassing downstream signaling, such as the epidermal growth factor receptor (EGFR), KRAS, KIT, MET, and insulin-like growth factor 1 receptor pathways. Approximately 25% of acquired mechanisms of resistance are mediated by unknown mechanisms.Citation14 With respect to EGFR-TKIs, 3%–15% of the patients with EGFR-mutated lung cancer develop acquired resistance to EGFR-TKIs by histologic transformation to SCLC.Citation15,Citation16 Similarly, a mechanism underlying SCLC transformation in ALK-positive tumors has been reported in some cases.Citation6–Citation12
In the present case in which the re-biopsied SCLC tissue was examined, ALK rearrangement was still detected. In addition, chemotherapy and/or anti-angiogenic drug treatment-induced change in initial tumor morphology should, therefore, be considered. If the transformation had occurred before crizotinib treatment, a rapid growth of the primary lesion during crizotinib therapy would have been observed. Moreover, the primary lesion did not show an increase in size during albumin paclitaxel or docetaxel combination with bevacizumab therapy, and only limited agent activity on SCLC was reported with these agents.Citation17 These findings imply that the transformation to SCLC during crizotinib treatment was the main cause of acquired resistance in this case. The pathophysiologic mechanism underlying transfor-mation to SCLC following ALK-TKI treatment is not well understood. According to the mechanism of EGFR-TKIs, two possibilities have been stated, including a phenotype switch from NSCLC to SCLC, and SCLC and adenocarcinoma may coexist at baseline, with SCLC becoming dominant during disease progression after EGFR-TKI therapy.Citation5,Citation18 We thought the mechanism of transformation to SCLC in ALK-TKIs was similar to that with EGFR-TKIs. Although the pathologic features of the present case were re-evaluated, SCLC was not identified in the primary biopsy specimen. Because of the initial bronchial biopsy, there was limited availability of the tissue sample.
In contrast, although the cause of transformation into SCLC is unclear, inactivation of tumor suppressor genes (RB and TP53) seems important and constitutes an initial event in the tumorigenic process.Citation16 Some reports have revealed the role of RB gene loss and/or TP53 mutation in ALK-positive NSCLC transformed into SCLCCitation11,Citation15,Citation16; however, in our case, there was no loss of RB, but a TP53 gene mutation occurred. We reasoned that RB gene loss or TP53 gene mutation could participate in the transformation of adenocarcinoma to SCLC, but the two genes were not always concurrent as the transforming event. Furthermore, we demonstrated a NOTCH1 gene mutation, and this constituted the first reported gene involving transformation to SCLC. It has been reported that approximately 25% of patients with SCLC have a NOTCH1 gene family abnormality identified on next-generation sequencing.Citation19 We hypothesize that, if inactivation of the RB1 or P53 or NOTCH1 gene is identified in the primary tumor tissue, the tumor may be at higher risk of SCLC transformation following target drug therapy.
The present case supports attempts to re-biopsy the tumor for selection of treatment after acquired resistance. There are no established treatment strategies to manage patients with transformation to SCLC. In agreement with reported cases, Fujita et alCitation8 reported treatment with a combination of irinotecan and alectinib, resulting in an ongoing partial response to the primary lesion with a maintained partial response to the other lesions. Miyamoto et alCitation9 showed that after two cycles of cisplatin–irinotecan treatment, there was a reduction in the size of mediastinal lymph nodes, whereas there was an increase in the size and number of nodes in both lung fields. Ou et alCitation12 suggest that the patient switch to a third-generation ALK-TKI within 2 months of treatment and disease progression. The patient underwent etoposide monotherapy in our case. After two cycles, the effect was SD. Nevertheless, as the heterogeneity of response to different therapeutic strategies of the reported case suggests, the re-biopsy site only represents a partial pathology of the resistance, and the mechanism of resistance may differ from one site to another. Given the heterogeneity and the variety of acquired resistance mechanisms to target drugs, re-biopsy of only one site might not always be appropriate. We think detection of gene variations in blood samples with homogeneity in ctDNA by NGS may be a supplemental method with re-biopsy for analysis, in some cases. It is clinically challenging to choose the treatment mode when transformation to SCLC occurs. More research on optimal treatment methods are needed.
Conclusion
Oncologists should realize the possibility of transformation to SCLC after patients acquire resistance to ALK-TKI therapy. A re-biopsy should be performed to facilitate histologic and molecular analysis. Transformation to SCLC is an important consideration for choosing appropriate therapy due to the potential efficacy of standard SCLC treatments or a combination of next-generation AKL-TKIs.
Acknowledgments
This work was supported by the Zhejiang Province of China (2013KYB051), Zhejiang Administration of Traditional Chinese Medicine Foundation (2013ZQ005), Science and Technology Planning Project of Zhejiang Province (2015C33194), and the National Clinical Key Specialty Construction Program (2013).
Disclosure
The authors report no conflicts of interest in this work.
References
- EttingerDSWoodDEAkerleyWNational Comprehensive Cancer NetworkNon-small cell lung cancer, version 6. 2015J Natl Compr Canc Netw201513551552425964637
- SolomonBJMokTKimDWPROFILE 1014 InvestigatorsFirst-line crizotinib versus chemotherapy in ALK-positive lung cancerN Engl J Med2014371232167217725470694
- ShawATKimDWNakagawaKCrizotinib versus chemotherapy in advanced ALK-positive lung cancerN Engl J Med2013368252385239423724913
- MatikasAKentepozidisNGeorgouliasVKotsakisAManagement of resistance to crizotinib in anaplastic lymphoma kinase-positive non-small-cell lung cancerClin Lung Cancer201617647448227341790
- YuHAArcilaMERekhtmanNAnalysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancersClin Cancer Res20131982240224723470965
- ChaYJChoBCKimHRLeeHJShimHSA case of ALK-rearranged adenocarcinoma with small cell carcinoma-like transformation and resistance to crizotinibJ Thorac Oncol2015115e55e5826752677
- TakegawaNHayashiHIizukaNTransformation of ALK rearrangement-positive adenocarcinoma to small cell lung cancer in association with acquired resistance to alectinibAnn Oncol20162752532
- FujitaSMasagoKKatakamiNYatabeYTransformation to SCLC after treatment with the ALK inhibitor AlectinibJ Thorac Oncol2016116e67e7226751586
- MiyamotoSIkushimaSOnoRTransformation to small-cell lung cancer as a mechanism of acquired resistance to crizotinib and alectinibJpn J Clin Oncol201646217017326613679
- CaumontCVeillonRGrosALaharanneEBégueretHMerlioJPNeuroendocrine phenotype as an acquired resistance mechanism in ALK-rearranged lung adenocarcinomaLung Cancer201692151826775590
- LevacqDD’HaeneNde WindRRemmelinkMBerghmansTHistological transformation of ALK rearranged adenocarcinoma into small cell lung cancer: a new mechanism of resistance to ALK inhibitorsLung Cancer2016102384127987586
- OuSILeeTKYoungLDual occurrence of ALK G1202R solvent front mutation and small cell lung cancer transformation as resistance mechanisms to second generation ALK inhibitors without prior exposure to crizotinib. Pitfall of solely relying on liquid re-biopsy?Lung Cancer201710611011428285684
- CamidgeDRDoebeleRCTreating ALK-positive lung cancer-early success and future challengesNat Rev Clin Oncol20129526827722473102
- IsozakiHTakigawaNKiuraKMechanisms of acquired resistance to ALK inhibitors and the rationale for treating ALK-positive lung cancerCancers (Basel)20157276378325941796
- NorkowskiEGhignaMRLacroixLSmall-cell carcinoma in the setting of pulmonary adenocarcinoma: new insights in the era of molecular pathologyJ Thorac Oncol20138101265127124457237
- OserMGNiederstMJSequistLVEngelmanJATransformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of originLancet Oncol2015164e165e17225846096
- KalemkerianGPAkerleyWBognerPSmall cell lung cancerJ Natl Compr Canc Netw2013111789823307984
- SequistLVWaltmanBADias-SantagataDGenotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitorsSci Transl Med201137575ra26
- KikuchiHSakakibara-KonishiJFurutaMExpression of Notch1 and Numb in small cell lung cancerOncotarget201786103481035828060745