
Abstract
The relapse and resistance to cytarabine (Ara-C) therapy is still a dominating obstacle to the successful clinical treatment of acute myeloid leukemia (AML). Recent studies have shown that dysregulation of miRNAs might modulate the resistance of cancer cells to anticancer drugs; yet, the mechanism is not fully understood. In this study, we showed a significant downregulation of miR-134 in human multidrug-resistant leukemia cells and relapsed/refractory AML patient samples. Overexpression of miR-134 sensitized K562/A02 and HL-60/ADM cells to Ara-C, inhibited cell colony formation, and enhanced the ability of Ara-C to induce apoptosis. Mechanistic analyses revealed that Mnks was a putative target of miR-134, which was inversely correlated with miR-134 expression in human multidrug-resistant leukemia cells and relapsed/refractory AML patient samples. Further investigation showed that miR-134 increased the anti-tumor effects of Ara-C through inhibiting phosphorylation of eukaryotic initiation factor 4E and downregulating Mcl-1 and bcl2, which was independent of p38 and Erk1/2 activation. Taken together, our results demonstrate that miR-134 plays a pivotal role in AML Ara-C resistance through increasing cell sensitivity to Ara-C and promoting apoptosis by targeting Mnks.
Introduction
Acute myeloid leukemia (AML) is a genetically heterogeneous clonal disorder leading to proliferation and accumulation of leukemic blasts in blood, marrow, and other organs.Citation1 Despite significant remission rates of chemotherapy, about 70% of AML patients will relapse and are associated with resistance and toxicity, resulting in dismal long-term survival rates and poor quality of life.Citation2,Citation3 The need for novel therapeutic approaches is urgent and of high clinical importance.
Multiple signaling pathways that promote leukemic cell survival and proliferation are constitutively activated in AML cells, providing potential therapeutic targets. Among them, the Ras/Raf/MAPK and the PI3K/Akt/mTOR signaling pathways are two vital cascades involved in the poor prognosis of AML and its development of resistance to cytarabine (Ara-C) treatment.Citation4–Citation7 Activation of these pathways has been associated with most AML cases. The oncogenic activity of the eukaryotic initiation factor 4E (eIF4E) driven by the Mnk kinases is a node of convergence of the two cascades.Citation8,Citation9
Mnks belong to a serine/threonine kinases family and two Mnk human genes, Mnk1 and Mnk2, have been identified. eIF4E is phosphorylated usually at Ser209 by Mnk kinases in response to mitogens and stress signals, and it subsequently promotes the synthesis of tumorigenic proteins.Citation9,Citation10 eIF4E is essential for cap-dependent mRNA translation and represents a key regulator in the control of mRNA translation and protein expression.Citation11,Citation12 Even moderate overexpression of eIF4E could cause dysregulated proliferation and tumorigenic transformation in immortalized cell lines including AML.Citation12,Citation13 Induction of antiapoptotic proteins was believed to play an important role for the relapse and resistance to Ara-C therapy. Those studies support a critical role for Mnk/eIF4E axis in the development of AML and indicate that targeting the Mnk/eIF4E axis may be a promising therapeutic strategy for the treatment of AML. However, efforts have been limited by the lack of Mnk inhibitor compounds with the potential for clinical development.
miRNAs are a class of short, noncoding RNA molecules with a significant regulatory capacity on gene expression and play an important role in various physiologic and pathologic processes, such as apoptosis, cell proliferation and differentiation.Citation14 miR-134 has recently been shown to be downregulated in a variety of diseases including cancers. For example, Sun et al found that miR-134 suppresses non-small cell lung cancer development through downregulation of oncogenic CCND1.Citation15 In addition, miR-134 is frequently downregulated in renal cell carcinoma cells and inhibits cell proliferation and epithelial-to-mesenchymal transition by targeting KRAS.Citation16 However, the role of miR-134 in AML has not been reported to the best of our knowledge. The aim of this study is to explore the biologic functions of miR-134 in AML and the underlying mechanisms. Here, we reported that miR-134 is downregulated in relapsed/refractory AML patient samples and human leukemia AML multidrug-resistant cell lines, and found that miR-134 sensitized K562/A02 and HL-60/ADM cells to Ara-C and promoted cell apoptosis by targeting the 3′ untranslated region (3′UTR) of Mnks.
Materials and methods
Patients and bone marrow samples
Forty-one newly diagnosed AML patients, 25 relapsed/refractory AML patients, and 27 healthy controls were included in this study. Diagnosis of AML was established according to clinical presentation and morphologic criteria of the French–American–British Classification. Leukemic mononuclear cells were isolated from the bone marrow by density-gradient centrifugation. Among these AML patients, six matched-pair bone marrow samples were available both at the diagnosis time prior to treatment and the relapsed/refractory state. An informed written consent in compliance with the Helsinki declaration was obtained from all the patients. The study was approved by the Ethics Committee of Huai’an First People’s Hospital.
Cell culture and transfection
The human leukemia cell lines, K562 and HL-60, and their multidrug-resistant counterparts, K562/A02 and HL-60/ADM, were purchased from the Department of Pharmacology, the Institute of Hematology of Chinese Academy of Medical Sciences (Tianjin, China). The cells were maintained in DMEM at 37°C/5% CO2. The cells were cultured for 2 weeks in drug-free medium prior to their use in the experiments. miRNA mimics for miR-134, as well as the negative control, were chemically synthesized from GE Dharmacon. Transfections were performed with Lipofectamine 2000 Reagent (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s instructions.
RNA isolation and quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from cell lines and tissues using the Trizol reagent (Thermo Fisher Scientific). To detect miR-134 expression, cDNA was reverse transcribed using TaqMan miRNA RT Kit (Life Technologies) and U6 was used as endogenous control. To detect Mnks expression, cDNA was reverse transcribed using a high-capacity cDNA reverse transcription kit (Thermo Fisher Scientific) and GAPDH was used as endogenous control. The qRT-PCR assays were analyzed by the Step One Plus RT-PCR system (Thermo Fisher Scientific) using SYBR PrimeScript RT-PCR Kit (Takara Bio, Shiga, Japan).
CCK-8 assay
After 24 h transfection, 5×103 K562/A02 and HL-60/ADM cells were seeded into 96-well plates. Ara-C was prepared freshly at various concentrations and then added. After 48 h incubation, cell viability was assessed by CCK-8 assay. Absorbance value at 490 nm was then measured. The concentration at which each drug produced 50% inhibition of growth (IC50) was estimated by the relative survival curve. At least three independent experiments were performed in quadruplicate.
Colony formation assay
Cells were seeded for the colony forming assay on six-well culture plates at 200 cells per well, in triplicate. After 14 days of incubation at 37°C with 5% CO2, colonies were fixed with acetic acid–methanol (1:4) and stained with dilute crystal violet (1:30) prior to being manually counted. Colonies containing 50 cells or more were counted as survivors.
Luciferase reporter assay
A fragment of the wild or mutated 3′UTR of Mnk1/2 that contained the putative miR-134 binding sites was cloned into downstream of the luciferase gene in the pGL3-REPORT luciferase vector (Thermo Fisher Scientific) at the HindIII and SpelI sites. For the luciferase assay, 293T cells were seeded in 24-well plates 24 h before transfection. The cells were co-transfected with pGL3-3′UTR and the control reporter plasmid, and miR-134 mimics or miRNA negative control. After 48 h, the luciferase activity was measured with a dual luciferase reporter assay system (Promega, Madison, WI, USA). Luciferase activity was normalized to Renilla luciferase activity for each transfected well.
Cell apoptosis analysis
K562/A02 and HL-60/ADM were plated in six-well plates (6×105 cells/well). After 24 h transfection, K562/A02 and HL-60/ADM cells were treated with Ara-C at a final concentration of 5 μM. After 48 h of treatment with Ara-C, the cells were stained with Annexin V and propidium iodide (BD Biosciences, San Jose, CA, USA) and then analyzed through flow cytometry (BD FACSAria™ Fusion).
Western blot
Antibodies directed against bcl2, Mcl-1 (Mcl-1L), Mnk1, Mnk2, eIF4E, and phosphorylated eIF4E at Ser209 (p-eIF4E), obtained from Abcam, were used to determine protein level by Western blot. β-actin (Abcam) was used as a loading control.
Statistical analysis
All data from three independent experiments were expressed as mean ± SD. Statistical analysis was performed using the GraphPad Prism Software. Statistical comparisons were performed using Student’s t-test, and p-values <0.05 were considered significant.
Results
Downregulation of miR-134 expression in bone marrow blasts from relapsed/refractory patients and human leukemia AML multidrug-resistant cell lines
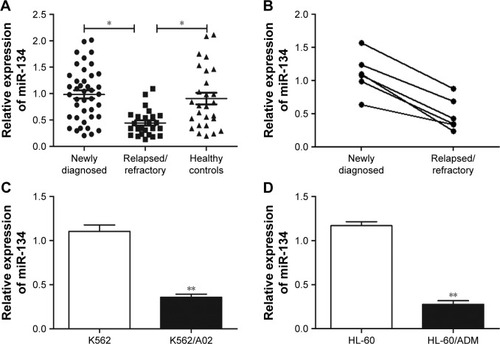
To investigate whether miRNA participated in drug resistance in AML, the expression of miR-134 was first assessed in bone marrow blasts from AML patients using qRT-PCR. Results revealed that miR-134 expression was significantly (p<0.05) reduced in relapsed/refractory patients in comparison to newly diagnosed AML patients (). However, no difference in miR-134 was observed between newly diagnosed AML patients and healthy controls. In six patients, matched-pair bone marrow samples were available both at diagnosis time prior to treatment and the relapsed/refractory state. We found a high level of miR-134 at diagnosis, whereas an obvious decrease of miR-134 expression was observed in the relapsed/refractory state (). Next, we examined miR-134 expression in human leukemia AML multidrug-resistant cell lines, and results demonstrated a lower expression of miR-134 in K562/A02 and HL-60/ADM cell lines, in comparison to that of their matched controls (K562 and HL-60), as shown in .
Figure 1 qRT-PCR analysis of miR-134 expression in relapsed/refractory AML patients and human leukemia AML multidrug-resistant cell lines.
Abbreviations: AML, acute myeloid leukemia; qRT-PCR, quantitative real-time PCR.
miR-134 enhances Ara-C sensitivity and induces apoptosis in K562/A02 and HL-60/ADM cells
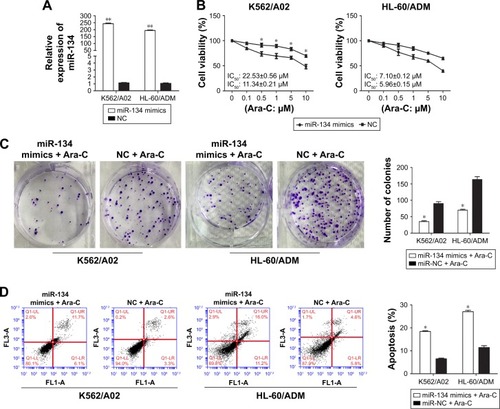
To further confirm the correlation between miR-134 and Ara-C drug resistance in AML, K562/A02 and HL-60/ADM cells were transiently transfected with miR-134 mimics or negative control. qRT-PCR assays demonstrated that miR-134 was successfully upregulated 210.72±7.02 and 179.81±5.65 fold in K562/A02 and HL-60/ADM cells transfected with miR-134 mimics, respectively (p<0.01; ). In both K562/A02 and HL-60/ADM cells, CCK-8 assay verified that those transfected with miR-134 mimics exhibited greatly enhanced sensitivity to Ara-C compared with the miRNA mimics control-transfected cells, respectively (). Colony formation assay also showed that overexpression of miR-134 inhibited the K562/A02 and HL-60/ADM cell colony formation (). To test whether miR-134 could mediate cell apoptosis in regulation of Ara-C resistance, we analyzed Ara-C-induced apoptosis after transfection of K562/A02 and HL-60/ADM cells with the miR-134 mimics by flow cytometry. In both K562/A02 and HL-60/ADM cells, overexpression of miR-134 resulted in a notable increase in cell apoptosis after Ara-C treatment compared with the negative control, respectively ().
Figure 2 Overexpression of miR-134 enhances Ara-C sensitivity and induces apoptosis in K562/A02 and HL-60/ADM cells.
Abbreviations: Ara-C, cytarabine; IC50, half maximal inhibitory concentration; NC, normal control.
miR-134 negatively regulates Mnk1/2
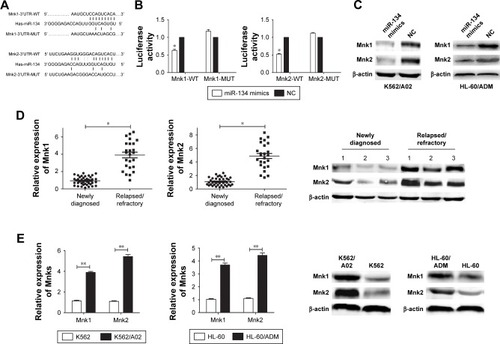
To explore the potential molecular mechanism of chemoresistance and apoptosis regulated by miR-134 in AML cells, we searched different databases (TargetScan and microRNA. org) for its potential targets involved in chemoresistance and apoptosis, and results demonstrated that Mnks was a predicted target of miR-134 (). Luciferase reporter assay results showed that miR-134 dramatically inhibited luciferase activity in 293T cells when the reporter plasmid carried the wild-type Mnks 3′UTR, but no significant inhibition was observed when the reporter plasmid carried a mutation (). Western blot results demonstrated that an obvious decrease of Mnk1/2 protein level was observed in transfected miR-134 mimics cells compared with the negative control cells in both K562/A02 and HL-60/ADM cells, respectively (). The expression of Mnks in bone marrow blasts from relapsed/refractory AML patients and human leukemia AML multidrug-resistant cell lines was also determined by qRT-PCR and Western blot ().
Figure 3 miR-134 negatively regulates Mnk1/2.
Abbreviations: 3′UTR, 3′ untranslated region; AML, acute myeloid leukemia; eIF4E, eukaryotic initiation factor 4E; MUT, mutant type; NC, normal control; WT, wild type.
miR-134 inhibits phosphorylation of eIF4E and downregulates Mcl-1 and bcl2
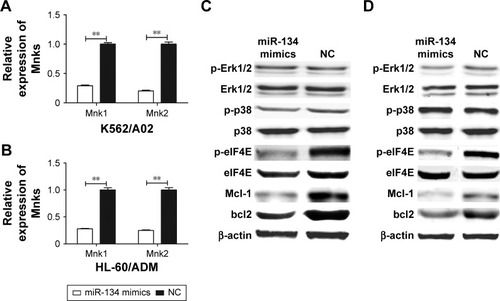
To investigate the effect of miR-134 on MAPK-mediated eIF4E pathways, Western blot analysis was performed after transfection with miR-134 mimics or miRNA mimic negative control in K562/A02 and HL-60/ADM cells. As expected, restoration of miR-134 in both K562/A02 and HL-60/ADM cells reduced the levels of respective Mnk1/2 mRNA expression by ~70%–85% (). Phosphorylation of Erk1/2 and p38 was not affected, while phosphorylation of eIF4E, Mcl-1, and bcl2 in miR-134-transfected cells was significantly reduced compared to miRNA mimic negative control transfected cells ().
Figure 4 miR-134 inhibits phosphorylation of eIF4E and downregulates Mcl-1 and bcl2.
Abbreviations: eIF4E, eukaryotic initiation factor 4E; NC, normal control; p-eIF4E, phosphorylation-eIF4E; p-Erk1/2, phosphorylation-Erk1/2; p-p38, phosphorylation-p38.
Discussion
Ara-C has been one of the cornerstone drugs in the treatment of AML for more than three decades.Citation17 Both intrinsic and acquired chemotherapeutic resistance develops in patients after prolonged treatment with Ara-C, which is a major limitation in the successful treatment of AML.Citation18 Evidence suggests that drug resistance mechanisms are involved in downregulated expression of hENT1, reduced intracellular concentrations of Ara-CTP and the active triphosphorylated form of Ara-C.Citation18–Citation20 In this work, we studied the functional role of miR-134 and the mechanism underlying its control of Ara-C-induced resistance in AML.
This study revealed that miR-134 expression was down-regulated in bone marrow blasts from relapsed/refractory AML patients and human leukemia AML multidrug-resistant cell lines compared with newly diagnosed AML patients and parallel drug-sensitive cell lines, respectively. Meanwhile, Ara-C is a primary drug for AML treatment in clinical practice. These results indicate that miR-134 might be involved in Ara-C-induced resistance. To check this hypothesis, CCK-8 assay and flow cytometry analysis were employed in K562/A02 and HL-60/ADM cell lines, and we found that miR-134 enhances Ara-C sensitivity and promotes cell apoptosis. This finding compliments previous observations that miR-134 was associated with gefitinib resistance in lung adenocarcinoma cells by targeting MAGI2, paclitaxel resistance in human ovarian cancer cells by regulating Pak2, and cisplatin resistance in triple-negative breast cancer through controlling STAT5B.Citation21–Citation23
Recent studies have found that the activated PI3K/Akt/mTOR and MAPK signaling pathways are involved in Ara-C resistance.Citation24–Citation26 Mnk/eIF4E axis is a specific and critical regulator of these two pathways. In this study, Mnks was confirmed to be a direct target of miR-134 by Western blot and luciferase reporter assay, and there was a negative correlation between the expression of miR-134 and Mnks in both relapsed/refractory AML patients and AML multidrug-resistant cell lines (). Further investigations of the effects of miR-134 on Mnk/eIF4E axis showed that miR-134 inhibits phosphorylation of eIF4E and downregulates Mcl-1 and bcl2, independent of p38 and Erk1/2 (). Taken together, these findings suggest that miR-134 sensitizes Ara-C-resistant AML cells and promotes cell apoptosis through targeting Mnk/eIF4E axis, and subsequently reduces Mcl-1 and bcl2 expression.
To summarize, we have shown that miR-134 can promote cell apoptosis and increase sensitivity to Ara-C by targeting Mnks. The miR-134–Mnk–eIF4E axis may provide new insights into the mechanisms of AML chemoresistance, and the restoration of miR-134 expression may offer a new strategy for the treatment of AML in the future.
Acknowledgments
We gratefully thank Dr Yufeng Li from the Department of Hematology, Huai’an First People’s Hospital for his helpful suggestions and advice during this project. Grants were provided by the Huai’an Science and Technology Plan (No HAP201423) and the Huai’an New Research Project of Clinical Diagnosis and Treatment (No HAS2014008, HAS2015026 and HAS201608).
Disclosure
The authors report no conflicts of interest in this work.
References
- EsteyEDöhnerHAcute myeloid leukemiaLancet200636895501894190717126723
- DavilaJSlotkinERenaudTRelapsed and refractory pediatric acute myeloid leukemia: current and emerging treatmentsPaediatr Drugs201416215116824158739
- EsteyEHTreatment of acute myeloid leukemiaHaematologica2009941101619118375
- PlataniasLCMap kinase signaling pathways and hematologic malignanciesBlood2003101124667467912623839
- MartelliAMEvangelistiCChiariniFGrimaldiCManzoliLMcCubreyJATargeting the PI3K/AKT/mTOR signaling network in acute myelogenous leukemiaExpert Opin Investig Drugs200918913331349
- NishiokaCIkezoeTYangJYokoyamaAInhibition of MEK signaling enhances the ability of cytarabine to induce growth arrest and apoptosis of acute myelogenous leukemia cellsApoptosis20091491108112019548087
- KhokharNZAltmanJKPlataniasLCEmerging roles for mammalian target of rapamycin inhibitors in the treatment of solid tumors and hematological malignanciesCurr Opin Oncol201123657858621892085
- WangXYuePChanCBInhibition of mammalian target of rapamycin induces phosphatidylinositol 3-kinase-dependent and Mnk-mediated eukaryotic translation initiation factor 4E phosphorylationMol Cell Biol200727217405741317724079
- HouJLamFProudCWangSTargeting Mnks for cancer therapyOncotarget20123211813122392765
- DiabSKumarasiriMYuMMAP kinase-interacting kinases–emerging targets against cancerChem Biol201421444145224613018
- SonenbergNGingrasACThe mRNA 5′cap-binding protein eIF4E and control of cell growthCurr Opin Cell Biol19981022682759561852
- TopisirovicIGuzmanMLMcConnellMJAberrant eukaryotic translation initiation factor 4E-dependent mRNA transport impedes hematopoietic differentiation and contributes to leukemogenesisMol Cell Biol200323248992900214645512
- AltmanJKSzilardAKonicekBWInhibition of Mnk kinase activity by cercosporamide and suppressive effects on acute myeloid leukemia precursorsBlood2013121183675368123509154
- BartelDPMicroRNAs: genomics, biogenesis, mechanism, and functionCell2004116228129714744438
- SunCCLiSJLiDJHsa-miR-134 suppresses non-small cell lung cancer (NSCLC) development through down-regulation of CCND1Oncotarget2016724359603597827166267
- LiuYZhangMQianJmiR-134 functions as a tumor suppressor in cell proliferation and epithelial-to-mesenchymal transition by targeting KRAS in renal cell carcinoma cellsDNA Cell Biol201534642943625811077
- MooreASKearnsPRKnapperSPearsonADZwaanCMNovel therapies for children with acute myeloid leukemiaLeukemia20132771451146023563239
- GalmariniCMThomasXCalvoFIn vivo mechanisms of resistance to cytarabine in acute myeloid leukemiaBr J Haematol2002117486086812060121
- KannoSHiuraTOhtakeTCharacterization of resistance to cytosine arabinoside (Ara-C) in NALM-6 human B leukemia cellsClin Chim Acta20073771–214414917097625
- SarkarMHanTDamarajuVCarpenterPCassCEAgarwalRPCytosine arabinoside affects multiple cellular factors and induces drug resistance in human lymphoid cellsBiochem Pharmacol200570342643215950950
- KitamuraKSeikeMOkanoTMiR-134/487b/655 cluster regulates TGF-β-induced epithelial-mesenchymal transition and drug resistance to gefitinib by targeting MAGI2 in lung adenocarcinoma cellsMol Cancer Ther201413244445324258346
- ShuangTWangMShiCZhouYWangDDown-regulated expression of miR-134 contributes to paclitaxel resistance in human ovarian cancer cellsFEBS Lett201558920 Pt B3154316426363097
- O’BrienKLowryMCCorcoranCmiR-134 in extracellular vesicles reduces triple-negative breast cancer aggression and increases drug sensitivityOncotarget2015632327743278926416415
- MartelliAMEvangelistiCChiariniFMcCubreyJAThe phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patientsOncotarget2010128910320671809
- MartelliAMTazzariPLEvangelistiCTargeting the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin module for acute myelogenous leukemia therapy: from bench to bedsideCurr Med Chem200714192009202317691943
- LiPDiabSYuMInhibition of Mnk enhances apoptotic activity of cytarabine in acute myeloid leukemia cellsOncotarget2016735568115682527462781