
Abstract
Prostate cancer (PCa), a multifocal clinically heterogeneous disease, is the most commonly diagnosed non-cutaneous neoplasm in men worldwide. The epigenome of PCa is a typical representation of catastrophic model of epigenetic alterations during tumorigenesis and its progression. Alterations in methylation patterns in tumor suppressors, cell cycle, oncogenes and metabolism-related genes are the most commonly observed epigenetic alterations in PCa. In this study, we have developed a computational strategy to identify methylated biomarker signature panels as potential targets of PCa by screening >160 genes reported to be epigenetically dysregulated, and shortlisted 26 differentially methylated genes (DMGs) that significantly contribute to oncogenesis. The gene ontology and functional enrichment analysis were performed, which showed that identified DMGs contribute to cellular and metabolic processes such as cell communication, cell cycle, response to drugs, apoptosis and p53 signaling. The top hub genes AR, CDH13, CDKN2A, DAPK1, GSTP1, CD44 and RASSF1 identified from protein–protein interaction network construction using Search Tool for the Retrieval of Interacting Genes contributed to hormonal response, inflammatory response, cell cycle, reactive oxygen species activity and receptor kinase activity, which are related to hallmarks of cancer as revealed by their functional enrichment analysis by Cytoscape. These genes were further scrutinized for CpG islands, transcription start sites and positions of methylated cytosines to study their methylation profiles. Our analysis revealed high negative correlation values between methylation frequencies and gene expressions of the hub genes, namely, AR, CDH13, CDKN2A, DAPK1, CD44, GSTP1 and RASSF1, which can be used as potential diagnostic biomarkers for PCa.
Introduction
Prostate cancer (PCa) accounts for being the most commonly diagnosed non-cutaneous neoplasm and third leading cause of cancer mortalities in men worldwide.Citation1 Reports suggest that men aged >40 years are ~30-fold more susceptible to developing PCa.Citation2 Although a number of genetic, surface and intracellular markers have been identified to study cancer progression such as CD133, CD49f, Trop2, p63 and PSA, PSA is the only established biomarker for the detection of PCa.Citation3 However, PSA lacks in specificity and sensitivity;Citation3 thus, there exists an urgent need to identify biomarkers that are more specific.
Epigenetic alterations in the form of DNA methylation and histone modifications have emerged as a major focus area for cancer treatment considering their contribution in modulation of crucial gene expressions in various malignancies.Citation4 Tumor suppressor genes and oncogenes play crucial roles in cancer by regulating functions such as cell cycle control, DNA repair, cell adhesion and apoptosis and are known to be differentially methylated in their promoter regions in several types of cancers.Citation5,Citation6 However till date, there is a limited knowledge of the specific functional mechanisms at the genome level that are being altered by epigenetic dysregulation during tumor progression in PCa.Citation7 Given their key role in various stages of carcinogenesis by causing abnormal gene expression of tumor suppressor genes and oncogenes, histone modifications and hypermethylation status of genes are being explored as potential biomarkers of cancer progression and prognosis.Citation8 Nowadays with the advancement of chromatin immunoprecipitation (ChIP) technology along with microarray profiling, scientists can screen thousands of differentially expressed genes (DEGs), analyze their mRNA profiles and methylation profiles computationally and subsequently scrutinize their involvement in various molecular and biological regulatory functions during various stages of tumor initiation and progression.Citation9
The present study aims to explore the contribution of epigenetic modulation of genes involved in progression of PCa using computational approach. We screened >160 differentially methylated genes (DMGs), which are reported to be epigenetically dysregulated, and identified 26 genes that significantly contribute to oncogenesis in prostate gland. This was followed by screening the identified DMGs for gene ontology (GO) and their pathway enrichment. The protein–protein interaction (PPI) and functional enrichment analysis further shed light on the crucial roles played by the identified DMGs during tumor initiation and progression. Our analysis showed a high negative correlation value between methylation frequencies and gene expressions of identified hub genes, which have the potential to act as early prognostic and diagnostic biomarkers of PCa.
Methods
Identification of DMGs
A comprehensive literature survey was performed using PubMed and Google Scholar. The keywords used were “Prostate cancer” and “DNA methylation”. The literature survey was limited to full text articles available in English. A preliminary abstract review was performed to decide relevance of these research articles to our methylation study. Once the literature survey was updated, an extensive data extraction exercise was carried out to retrieve genes that are differentially methylated in PCa using online databases. The Cancer Genome Atlas (TCGA) (cancergenome.nih.gov/),Citation10 Cancer Genetics Web (www.cancerindex.org/geneweb/)Citation11 and MethyCancer (methycancer.psych.ac.cn/)Citation12 were used for identifying the DMGs in PCa.
GO and functional enrichment analysis
Databases such as GeneCodisCitation13 and PantherCitation14 were used for functional enrichment analysis. GO of identified DMGs included categories such as cellular component (CC), biological process (BP) and molecular function (MF) terms. The parameters were set as p-value <0.01 for considering the results that were statistically significant. The enriched pathways were identified using Kyoto Encyclopedia of Genes and Genomes (KEGG)Citation15 analysis. To further analyze the interrelationship of identified hub genes in regulating cellular pathways, gene set enrichment analysis module of Cytoscape softwareCitation16 was used.
PPI network construction by search tool for the retrieval of interacting genes (STRING)
The interactive relationships between DMGs were identified using STRING database.Citation17 A combined score of >0.7 (high) of only experimentally validated interactions was considered to be statistically significant.
Epigenetic analysis
The FASTA sequences of identified differentially methylated hub genes in PCa were retrieved from nucleotide database of National Center for Biotechnology Information (www.ncbi.nlm.nih.gov/). CpGProD (doua.prabi.fr/software/cpgprod)Citation18 and EMBOSS CpGPlot (www.ebi.ac.uk/Tools/seqstats/emboss_cpgplot)Citation19 were used to monitor the presence of CpG islands if any. Information related to GC%, G + C skew, A + T skew, etc. was also retrieved from these online programs. Methylation and Expression database of Normal and Tumor tissues (MENT; mgrc.kribb.re.kr:8080/MENT/)Citation20 is an online integrative database to study DNA methylation and its correlation with gene expression in paired samples gathered from TCGA and Gene Expression Omnibus (GEO). MENT was used to identify the effect of methylation on the gene expression of the identified DMGs in PCa. Analysis of DNA methylation with exon–intron structures, genomic loci of epigenetic changes, positions of methylated cytosines, CpG islands and transcription start sites (TSS) using a graphical description was carried out using Prostate Epigenetic Database (PEpiD; wukong.tongji.edu.cn/pepid).Citation21 The identified hub genes after epigenetic analysis were further classified using extensive literature survey based on their specificity and sensitivity for determining their performance as prognostic and diagnostic biomarkers for PCa. The literature survey covered published reports in which the specificity and sensitivity of DMGs were determined in cell lines and also bodily samples such as serum, ejaculates, post-massage urine and biopsy samples using methylation-specific polymerase chain reaction (MSP-PCR), ChIP and microarray analysis.Citation22–Citation45
Results
Identification of DMGs involved in PCa initiation and progression
A consolidated list of >160 genes from literature survey and online databases with evidences of differential methylation during PCa progression was prepared (Table S1). We short-listed 26 candidate DMGs that were associated with metastatic and recurrent stages of the disease with at least two publications and were used for further analysis ().
Table 1 Differentially methylated genes (DMGs) shortlisted for analysis
Functional annotation and pathway enrichment analysis
The altered biological function and MF of the identified DMGs were then analyzed through GO using GeneCodis and Panther. A p-value of <0.01 was considered as significant during the analysis. The enrichment GO terms were categorized into MF, BP and CC (). During the MF analysis, we found that majority of DMGs contributed to protein binding (16 genes), zinc ion binding (seven genes), metal ion binding (eight genes), DNA binding (six genes) and receptor activity (six genes). Other regulated MFs included enzyme binding, sequence-specific DNA binding and calcium ion binding. The CC analysis revealed that majority of differentially methylated candidate genes were associated with cytoplasm (20 genes), nucleus (16 genes), plasma membrane (10 genes), nucleoplasm (seven genes) and cytosol (six genes). In the BP ontology, the most significantly regulated processes included response to drugs (six genes), negative regulation of cell proliferation (six genes), signal transduction (seven genes) and negative regulation of apoptotic process (five genes). The other regulated processes included response to estradiol stimulus, cell cycle arrest and negative regulation of MAPK cascade. Thus, the most significantly enriched pathways identified using KEGG analysis include pathways in cancer (10 genes), cell cycle (five genes), p53 signaling (three genes) and PCa (three genes; ). Functional gene set enrichment analysis using gene set enrichment analysis (GSEA) module of Cytoscape showed that the identified DMGs most commonly contributed in hormonal response, inflammatory response, cell cycle, reactive oxygen species (ROS) activity and receptor kinase activity, which are all related to hallmarks of oncogenesis (Table S2).
Table 2 Gene ontology (GO) analysis of candidate differentially methylated genes (DMGs) associated with prostate cancer (PCa)
PPI network
The PPI networks between DMGs were investigated using STRING server. In total, 26 nodes with 90 edges and 6.92 as average node degree were analyzed ( and ). In the PPI network, seven node proteins, namely, GSTP1, CDH13, CDKN2A, RASFF1, CD44, AR and DAPK1, showed maximum degree of association with the other proteins (>5), thus indicating that these genes (proteins) have higher hub degrees. Thus, these hub genes (proteins) may play a crucial role in PCa initiation and progression.
Figure 1 Protein–protein interaction (PPI) network of identified differentially methylated genes (DMGs).
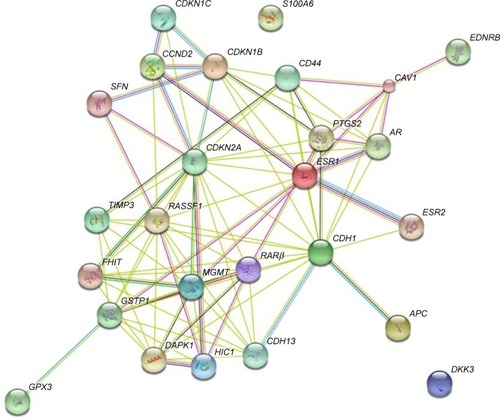
Table 3 Associated genes of identified seven differentially methylated hub genes
Epigenetic analysis
Evidences have shown that the cytosines in the CpG dinucleotides are often methylated to 5-methylcytosines leading to change in gene expression.Citation46 Almost 70%–80% cytosines in CpG mammals are seen to be methylated in mammals and are often located near the promoter regions.Citation47 CpGProd and EMBOSS CpGPlot were used to analyze positions of CpG islands, CpGo/e ratio, G + C skew and A + T skew (Table S3). The positions of methylated cytosines along with positions of TSS of the identified hub DMGs were also retrieved using PEpID (a prostate epigenetic database in mammals; Table S4). Once the CpG islands and the methylated cytosines positions were retrieved, MENT database was used to scrutinize the correlation between methylation frequencies and gene expressions. The MENT launched in 2012 is an integrated database of DNA methylation and gene expressions. Datasets containing information regarding DNA methylation and gene expression in paired samples were retrieved from TCGA and GEO. We opted for gene search tool to predict the correlation between methylation values and gene expressions of our hub genes. The Illumina methylation datasets consists of Human Methylation27 and Golden Gate Methylation Cancer Panel I, which is composed of 125 prostate tumor samples and 92 normal prostate samples for analysis. Based on the correlation values between mean methylation frequencies and gene expression, it was observed that expressions of hub genes GSTP1, CDH13, CDKN2A, RASFF1, AR, CD44 and DAPK1 were seen to be significantly affected by their hypermethylation during PCa progression due to an observed negative correlation of as high as −0.7 as shown in .
Table 4 Methylation and Expression database of Normal and Tumor tissues (MENT) data showing correlation between mean methylation values and gene expressions
These identified hub genes, ie, AR, CDH13, CDKN2A, DAPK1, GSTP1, CD44 and RASSF1, have been reported by various other research groups to show high specificity and sensitivity as independent biomarkers in prostatic intraepithelial neoplasia and different stages of PCa, including primary and metastatic tumors (). These genes have been reported to be differentially regulated due to hypermethylation in PCa cells, ie, PC3, DU145 and LNCaP, as well as bodily samples, including serum, ejaculates, post-massage urine and biopsy samples.Citation22–Citation45
Table 5 Specificity and sensitivity of hub genes identified in our study that are frequently hypermethylated as potential biomarkers for prostate cancer (PCa)
Discussion
Alterations in methylation patterns in tumor suppressor genes, cell cycle genes, oncogenes and metabolism-related genes are the most commonly observed epigenetic alterations in PCa. With the advancement in high-throughput technologies, including microarray and ChIP array, it has become feasible to analyze not only the expression levels of several genes simultaneously but also their methylation frequencies that can eventually lead to discovery of genes that can act as potential diagnostic, prognostic and therapeutic biomarkers. In the present study, we evaluated a panel of 26 DMGs using computational approaches to study their differential methylation and gene annotation profiles that are regulated during different stages of PCa. Consequently, their interrelationships were analyzed using PPI server.
Using thorough literature survey and online databases such as TCGA, Cancer Genetics Web and MethyCancer, we identified 26 DMGs in PCa. Here, we hypothesized that DMGs may act as potential diagnostic and prognostic biomarkers for PCa. However, to better understand the interrelationship and contribution of DMGs in cancer progression, we further performed GO and KEGG pathway enrichment analysis. The GO analysis showed that the DMGs significantly contributed in MFs such as protein binding, DNA binding, zinc ion binding and metal ion binding, which may contribute to carcinogenesis. Our hypothesis was further strengthened by the BP analysis which showed that these 26 DMGs significantly contributed to signal transduction, negative regulation of cell proliferation and negative regulation of apoptosis. These identified BPs pointed toward the involvement of the identified DMGs in major hallmarks of cancer. CDKN2A, CDKN1B and CDKN1C among the identified genes are known to be critical regulators of cell cycle, acting as checkpoints at various stages of cell cycle. Aberrantly, methylated cyclin-dependent kinase genes lead to carcinogenesis and progression of cancer due to uncontrolled cell growth.Citation48 CDKN2A has been seen to be frequently methylated in PCa progression.Citation48–Citation50 CCND2 is another important identified cell cycle regulator required for cell cycle G1/S transition.Citation51 Other cell cycle genes such as CD44,Citation52 HIC1Citation53 and PTGS2Citation54 identified in our analysis have been reported to be frequently methylated during PCa progression. Another important DMG identified was stratifin, which is known to inhibit the cyclin B1–CD2 complex from entering the nucleus by enforcing a G2/M arrest. The loss of stratifin due to promoter hypermethylation during tumor progression has been seen in various cancers, including PCa.Citation55 Death-associated protein kinase (DAPK) and fragile histidine triad (FHIT) among the identified DMGs are involved in apoptosis and metastasis and are reported to be frequently hypermethylated in various cancers, including PCa.Citation56,Citation57 Our analysis also identified endothelin B receptor type B, protein encoded by the EDNRB gene, which is known to induce apoptosis and inhibit tumor progression by interacting with endothelins. Several studies have reported promoter hypermethylation of EDNRB in higher frequencies in prostate tumors as compared to normal tissues.Citation2,Citation58
KEGG analysis showed that the major enriched pathways included pathways in cancer, cell cycle, p53 signaling, focal adhesion and Wnt signaling (). Wnt signaling is reported to play integral roles in cell fate decisions, proliferation, neural patterning, migration, differentiation, tumor aggression and stem cell renewal during many malignancies including PCa.Citation59,Citation60 In addition, differential regulation of p53 signaling as regulated by CCND2, SFN and CDKN2A has been reported in various cases of PCa.Citation61 Reports have suggested that expressions of AR and p53, which are balanced during androgen-dependent stages of PCa, eventually get disturbed during the progression of the disease.Citation62 p53 signaling also plays a pivotal role in cell cycle progression by regulating G1/S-, S- and G2/M-phase checkpoints and also transcription-dependent and -independent cell death program, DNA repair and apoptosis.Citation63 The functional enrichment analysis demonstrated that the identified genes contributed to hormonal response, cell cycle, ROS and inflammatory responses that are directly involved with cancer initiation and progression (Table S2). This further supported our hypothesis that the identified 26 DMGs played crucial roles in PCa development.
The PPI network constructed with identified DMGs led to the identification of high-degree hub genes ( and ) in the local network, which included AR, CDH13, CDKN2A, DAPK1, CD44, GSTP1 and RASSF1 that exhibited the highest degree of connectivity. Androgens that are responsible for stimulating growth of PCa cells mediate their effect through androgen receptors (ARs). Silencing of AR can lead to decreased growth and induce apoptosis.Citation42–Citation45 AR promoter hypermethylation has been reported by various groups to be between 8% and 39% in PCa tumor tissues.Citation2 The results of our analysis were in concordance with several published research papers that have reported the downregulated expression of AR due to hypermethylation during progression of PCa from androgen-dependent to androgen-independent stages.Citation42–Citation45,Citation64–Citation69 Thereby, this has led to emergence of androgen deprivation as one of the most effective treatment for advanced PCa, in addition to prostatectomy and radiation therapy.Citation42–Citation45 T-cadherin (CDH13), another identified hub gene located on 16q24, is a tumor suppressor gene known to play a critical role in cell–cell adhesion.Citation70 CDH13 expression can restrain the invasive potential and proliferation rate of tumor cells. Anomalous expression of CDH13 due to promoter methylation has been reported in various malignancies, including breast cancer, lung cancer and bladder cancer.Citation70 Yet, another gene identified in our analysis was glutathione S-transferase Pi 1 (GSTP1), which is known to be the most frequently epigenetically altered metabolic gene in PCa. DNA hypermethylation of GSTP1 is reported to be as high as 90% in prostate cancerous samples and 70% in prostatic interepithelial neoplasia.Citation22–Citation34 GSTP1 located on chromosome 11q3 eliminates foreign chemicals and detoxifies electrophilic toxic carcinogens and oxidants. Methylation in GSTP1 has been detected with high frequencies in PCa cell lines, urine samples, patient ejaculates and blood samples as compared to low or no methylation detected in healthy control patients.Citation71 Ras association domain family protein 1 isoform A (RASSF1A), another gene identified, was found to be associated with more than eight identified DMGs. RASSF1A is a tumor suppressor gene usually associated with DNA repair and apoptosis.Citation72 Evidences have shown that aberrant methylation of RASSF1A may hamper cell cycle control and DNA repair in tumors.Citation72 Silencing of RASSF1A has been observed in various cancers such as non-small-cell lung cancer, colorectal cancer, breast cancer and PCa. In fact, some researchers have shown the frequency of hypermethylation of RASSF1A to be correlated with PCa progression and aggressiveness.Citation72
Conclusion
We identified seven DMGs, namely, AR, CDH13, CDKN2A, DAPK1, GSTP1, CD44 and RASSF1, with high negative correlation (~0.7), which depicts that the increase in methylation directly leads to decrease in the expression of these genes. Biological and functional annotation showed that this panel of selected candidate genes played a crucial part in cell proliferation, binding, Wnt regulation, enzyme regulator activities and drug response. Although nonspecific epigenetic targeting might have short-term undesirable effects, gene-specific demethylation approaches offer promising prognostic and therapeutic targets for PCa diagnosis. Thus, results of the present study may be used for basic and translational treatment; however, further in vitro and in vivo investigations are needed to confirm our hypothesis.
Acknowledgments
This work was supported by grants from the Symbiosis Centre for Research and Innovation (SCRI) and Symbiosis School of Biological Sciences (SSBS); Symbiosis International University (SIU), Lavale, Pune, India and Junior Research INSPIRE Fellowship (JRF) from the Department of Science and Technology, Government of India, to Miss Anshika Nikita Singh.
Disclosure
The authors report no conflicts of interest in this work.
References
- TorreLABrayFSiegelRLFerlayJLortet-tieulentJJemalAGlobal cancer statistics, 2012CA Cancer J Clin20156528710825651787
- YangMParkJYDNA methylation in promoter region as biomarkers in prostate cancerMethods Mol Biol20128636710922359288
- VelonasVMWooHHdos RemediosCGAssinderSJCurrent status of biomarkers for prostate cancerInt J Mol Sci2013146110341106023708103
- SarkarSHornGMoultonKCancer development, progression, and therapy: an epigenetic overviewInt J Mol Sci20131410210872111324152442
- SharmaSKellyTKJonesPAEpigenetics in cancerCarcinogenesis2009311273619752007
- TamWLWeinbergRAThe epigenetics of epithelial-mesenchymal plasticity in cancerNat Med20081445724732
- FeinbergAPKoldobskiyMAGöndörAEpigenetic modulators, modifiers and mediators in cancer aetiology and progressionNat Rev Genet201617528429926972587
- JonesPAIssaJ-PJBaylinSTargeting the cancer epigenome for therapyNat Rev Genet2016171063064127629931
- ZhangLHuangYZhuoWZhuYZhuBChenZIdentification and characterization of biomarkers and their functions for Lapatinib-resistant breast cancerMed Oncol2017345
- WeinsteinJNCollissonEAMillsGBThe cancer genome Atlas Pan-Cancer Analysis Project JohnNat Genet2014451011131120
- CotterillSJ homepage on the InternetCancer Genetics Web Available from: http://www.cancer-genetics.org/index.htmAccessed October 8, 2016
- HeXChangSZhangJMethyCancer: the database of human DNA methylation and cancerNucleic Acids Res200836suppl 1836841
- Tabas-madridDNogales-cadenasRPascual-montanoAGeneCodis3: a non-redundant and modular enrichment analysis tool for functional genomicsNucleic Acids Res201240478483
- MiHPoudelSMuruganujanACasagrandeJTThomasPDPANTHER version 10: expanded protein families and functions, and analysis toolsNucleic Acids Res201644D1D336D34226578592
- KanehisaMGotoSKEGG: Kyoto Encyclopedia of Genes and GenomesNucleic Acids Res2000281273010592173
- LopesCTFranzMKaziFDonaldsonSLMorrisQBaderGDCytoscape Web: an interactive web-based network browserBioinformatics2011271323472348
- JensenLJKuhnMStarkMSTRING 8 – a global view on proteins and their functional interactions in 630 organismsNucleic Acids Res200937suppl 1412416
- PongerLMouchiroudDCpGProD: identifying CpG islands associated with transcription start sites in large genomic mammalian sequencesBioinformatics200218463163312016061
- LiWCowleyAUludagMThe EMBL-EBI bioinformatics web and programmatic tools frameworkNucleic Acids Res201543580584
- BaekSYangSKangTParkSSungYKimSMENT: methylation and expression database of normal and tumor tissuesGene2013518119420023219992
- ShiJHuJZhouQDuYJiangCPEpiD: a prostate epigenetic database in mammalsPLoS One201385e6428923696878
- AhmedHCappelloFRodolicoVEvidence of heavy methylation in the galectin 3 promoter in early stages of prostate adenocarcinoma: development and validation of a methylated marker for early diagnosis of prostate cancer 1Transl Oncol20092314615619701499
- SuhCIShanafeltTMayDJComparison of telomerase activity and GSTP1 promoter methylation in ejaculate as potential screening tests for prostate cancerMol Cell Probes200014421121710970725
- GoesslCKrauseHMuMSchraderMSachsingerJAdvances in brief fluorescent methylation-specific polymerase chain reaction for DNA-based detection of prostate cancer in bodily fluidsCancer Res20001559415945
- HardenSVGuoZEpsteinJISidranskyDQuantitative GSTP1 methylation clearly distinguishes benign prostatic tissue and limited prostate adenocarcinomaJ Urol200316931138114212576869
- HardenSVGoodmanSNPartinAAWPatrickCEpsteinJIQuantitative GSTP1 methylation and the detection of prostate adenocarcinoma in sextant biopsiesJ Natl Cancer Inst200395211634163714600096
- GoesslCMuMKrauseHSchostakMStraubBMethylation-specific PCR for detection of neoplastic DNA in biopsy washingsJ Pathol2002196333133411857497
- TokumaruYHardenSVSunDYamashitaKEpsteinJISidranskyDOptimal use of a panel of methylation markers with GSTP1 hypermethylation in the diagnosis of prostate adenocarcinomaClin Cancer Res2004105518552215328191
- CairnsPEstellerMHermanJGMolecular detection of prostate cancer in urine by GSTP1 hypermethylation molecular detection of prostate cancer in urine by GSTP1Clin Cancer Res2001792727273011555585
- EllingerJBastianPJJurganTCpG island hypermethylation at multiple gene sites in diagnosis and prognosis of prostate cancerUrology200871116116718242387
- SunamiEShinozakiMHiganoCSMultimarker circulating DNA assay for assessing blood of prostate cancer patients methodsClin Chem2009567559567
- GonzalgoMLPavlovichCPLeeSMNelsonWGJamesTBradyBProstate cancer detection by GSTP1 methylation analysis of postbiopsy urine specimensClin Cancer Res2003972673267712855646
- RogersCGGonzalgoMLYanGHigh concordance of gene methylation in post-digital rectal examination and post-biopsy urine samples for prostate cancer detectionJ Urol20061762280228417070312
- HoqueMOTopalogluOBegumSHenriqueRRosenbaumEQuantitative methylation-specific polymerase chain reaction gene patterns in urine sediment distinguish prostate cancer patients from control subjectsJ Clin Oncol2015232765696575
- RouprêtMHupertanVYatesDRMolecular detection of localized prostate cancer using quantitative methylation-specific PCR on urinary cells obtained following prostate massageClin Cancer Res20071361720172517363525
- MaruyamaRToyookaSToyookaKOAberrant promoter methylation profile of prostate cancers and its relationship to clinicopathological features aberrant promoter methylation profile of prostate cancers and its relationship to clinicopathological featuresClin Cancer Res20028251451911839671
- KangGHLeeSLeeHJHwangKSAberrant CpG island hypermethylation of multiple genes in prostate cancer and prostatic intraepithelial neoplasiaJ Pathol200420223324014743506
- YamanakaMWatanabeMYamadaYAltered methylation of multiple genes in carcinogenesis of the prostateInt J Cancer2003106338238712845678
- GinsburgGSWilliardHFGenomic and Personalized MedicineAmsterdamAcademia Press2012
- JanGDecreased expression Of CD44 in metastatic prostate cancerInt J Cancer1999483478483
- AlumkalJJZhangZHumphreysEBBiomarkers and risk factors effect of DNA methylation on identification of aggressive prostate cancerUrology20087261234123918387661
- JarrardDFKinoshitaHShiYMethylation of the androgen receptor promoter CpG island is associated with loss of androgen receptor expression in prostate cancer cellsCancer Res19985823531053159850055
- KinoshitaHShiYSandefurCMethylation of the androgen receptor minimal promoter silences transcription in human prostate cancerCancer Res200060133623363010910077
- NakayamaTWatanabeMSuzukiHEpigenetic regulation of androgen receptor gene expression in human prostate cancersLab Invest200080121789179611140692
- SasakiMTanakaYPerincheryGMethylation and inactivation of estrogen, progesterone, and androgen receptors in prostate cancerJ Natl Cancer Inst200294538439011880477
- JinBLiYRobertsonKDDNA methylation: superior or subordinate in the epigenetic hierarchy?Genes Cancer20112660761721941617
- LawJAJacobsenSEEstablishing, maintaining and modifying DNA methylation patterns in plants and animals JulieNat Rev Genet2011113204220
- BonelliPTuccilloFMBorrelliASchiattarellaABuonaguroFMCDK/CCN and CDKI alterations for cancer prognosis and therapeutic predictivityBiomed Res Int2014201436102024605326
- NguyenTTNguyenCTGonzalesFANicholsPWYuMCJonesPAAnalysis of cyclin-dependent kinase inhibitor expression and methylation patterns in human prostate cancersProstate200043323324210797499
- HermanGMerloAMaoLInactivation of the CDKN2/p16/MTSJ gene is frequently associated with aberrant DNA methylation in all common human cancers’Cancer Res19957145254530
- MusgroveEACaldonCEBarracloughJStoneASutherlandRLCyclin D as a therapeutic target in cancerNat Rev Cancer201111855857221734724
- VerkaikNSvan SteenbruggeGJvan WeerdenWMBussemakersMJvan der KwastTHSilencing of CD44 expression in prostate cancer by hypermethylation of the CD44 promoter regionLab Invest20008081291129810950120
- ZhengJXiongDSunXSignification of hypermethylated in cancer 1 (HIC1) as tumor suppressor gene in tumor progressionCancer Microenviron20121285293
- WoodsonKO’ReillyKJWardDECD44 and PTGS2 methylation are independent prognostic markers for biochemical recurrence among prostate cancer patients with clinically localized diseaseEpigenetics20061418318617998819
- LodyginDHermekingHThe role of epigenetic inactivation of 14-3-3 σ in human cancerCell Res200515423724615857578
- MichieAMMccaigAMNakagawaRVukovicMDeath-associated protein kinase (DAPK) and signal transduction: regulation in cancerFEBS J20102771748019878310
- FournierGCussenotOPescheSLatilABieILidereauRMolecular analysis of the FHIT gene in human prostate cancerOncogene19981614186318689583683
- PaoMMTsutsumiMLiangGUzvolgyiEGonzalesFAJonesPAThe endothelin receptor B (EDNRB) promoter displays heterogeneous, site specific methylation patterns in normal and tumor cellsHum Mol Genet200110990391011309363
- MohammadiNBabashahSInterplay between microRNAs and WNT/b -catenin signalling pathway regulates epithelial – mesenchymal transition in cancerEur J Cancer201551121638164926025765
- ZhanTRindtorffNBoutrosMWnt signaling in cancerOncogene201736111461147327617575
- OsmanIDrobnjakMFazzariMFerraraJScherHICordon-CardoCInactivation of the p53 pathway in prostate cancer: impact on tumor progressionClin Cancer Res1999582082208810473090
- CronauerMVSchulzWABurchardtTAckermannRBurchardtMInhibition of p53 function diminishes androgen receptor-mediated signaling in prostate cancer cell linesOncogene200423203541354915077179
- SteghAHTargeting the p53 signaling pathway in cancer therapyExpert Opin Ther Targets2012161678322239435
- MassieCEMillsIGLynchAGThe importance of DNA methylation in prostate cancer developmentJ Steroid Biochem Mol Biol201616611527117390
- TanHSoodARahimiHARb loss is characteristic of prostatic small cell neuroendocrine carcinoma Rb loss is characteristic of prostatic small cellClin Cancer Res201420489090324323898
- KinoshitaHShiYSandefurCMethylation of the androgen receptor minimal promoter silences transcription in human prostate cancerCancer Res200060133623363010910077
- GravinaGLMaramponFDi StasoM5-azacitidine restores and amplifies the bicalutamide response on preclinical models of androgen receptor expressing or deficient Prostate tumorsProstate2010117811661178
- GravinaGLFestucciaCMillimaggiDChronic azacitidine treatment results in differentiating effects, sensitizes against bicalutamide in androgen-independent prostate cancer cellsProstate200868779380118324645
- ZornCSWojnoKJMccabeMTKueferRGschwendJEDayMLCancer therapy: preclinical 5-Aza-2′-deoxycytidine delays androgen-independent disease and improves survival in the transgenic adenocarcinoma of the mouse prostate mouse model of prostate cancerClin Cancer Res20071372136214417404097
- AndreevaAVKutuzovMACadherin 13 in cancerGenes Chromosomes Cancer201049977579020607704
- MartignanoFGurioliGSalviSGSTP1 methylation and protein expression in prostate cancer: diagnostic implicationsDis Markers2016201616
- LiuLYoonJDammannRPfeiferGPFrequent hypermethylation of the RASSF1A gene in prostate cancerOncogene2002216835684012360410