
Abstract
Anthracyclines, and doxorubicin in particular, remain a mainstay of sarcoma therapy. Despite modest activity and significant toxicities, no cytotoxic monotherapy has yet yielded superior overall survival over doxorubicin for therapy of advanced soft tissue sarcomas in a randomized trial. Similarly, combination regimens have also been unable to overcome doxorubicin in terms of overall survival. Strategies to ameliorate the most prominent side effect of doxorubicin, cardiotoxicity, are available, but their use in sarcoma patients has been limited. Aldoxorubicin is a prodrug consisting of doxorubicin with a covalent linker. It binds rapidly after intravenous infusion to cysteine-34 of human serum albumin. The drug–albumin conjugate is preferentially retained in tumor tissue, with uptake into tumoral cells. At physiologic pH, the complex is stable. Hydrolysis can occur under the acidic conditions of the endocytic lysosome, releasing doxorubicin. Doxorubicin then distributes to various cellular compartments, including Golgi, mitochondrion, and nucleus, with subsequent cytotoxic effects. Aldoxorubicin has demonstrated in vitro and in vivo activities in both cancer model systems and human xenografts. Preclinical models also support its decreased cardiac effects vs doxorubicin, although such promising results require formal comparison at efficacy equivalent doses of the two drugs. Phase I studies confirmed the tolerability of aldoxorubicin in humans. Clinical cardiotoxicity was not observed, but molecular and subclinical cardiac effects could be demonstrated. A Phase II study in treatment-naïve, advanced sarcoma patients demonstrated improved progression-free survival and response rate over doxorubicin, although no survival benefit was evident. A Phase III study of aldoxorubicin vs investigator’s choice from a panel of chemotherapy regimens in the salvage setting was unable to demonstrate a benefit in progression-free or overall survival in the entire population. Progression-free survival in L-sarcomas (leiomyosarcomas and liposarcomas) was documented. While evidence of subclinical cardiac effects was seen in a small proportion of aldoxorubicin-treated patients, data from both the Phase II and III studies indicated a favorable cardiotoxicity profile vs doxorubicin. Despite the negative results from this Phase III study, the importance of anthracycline therapy in sarcoma management merits further investigation of the potential role of aldoxorubicin in this indication. Other avenues for progress include identification of sensitive histologies and biomarkers of activity, exploration of clinical niches without proven standard therapies, and exploration of alternate dosing strategies.
Introduction
Sarcomas are a diverse group of mesenchymal neoplasms. At least 50 different subtypes exist, some of which are individually quite rare. In the United States in 2019, there will be an estimated 12,750 new cases and 5,270 deaths for soft-tissue sarcomas (STS) and 3,500 cases and 1,660 deaths for bone sarcomas.Citation1 The impact of these conditions is magnified by the relatively young age population being affected. The National Cancer Institute has recognized sarcomas as an area of unmet need in its Roadmap for Sarcoma Research.Citation2
Surgical resection remains the mainstay of primary treatment.Citation3 Radiation therapy frequently plays a role in the neoadjuvant, adjuvant, or palliative settings. Use of systemic therapy is primarily reserved for the advanced disease setting.Citation4,Citation5 For several subtypes primarily associated with the pediatric population (osteosarcoma, Ewing’s sarcoma, rhabdomyosarcoma), systemic therapy is an important component of the primary therapy.Citation4,Citation6,Citation7 Systemic neoadjuvant or adjuvant therapy may be used in the primary treatment of other sarcoma subtypes, although the benefit of such therapy is less certain.Citation4,Citation6,Citation8
Doses of aldoxorubicin are referred in primary studies in terms of both absolute dose of aldoxorubicin and doxorubicin equivalent (DE) doses. To avoid confusion, for all doses of aldoxorubicin discussed, the dose in the referenced publication is stated. If this is in reference to the DE dose, this is indicated by “DE” after the dose. If dosing in the publication is in absolute amounts of aldoxorubicin, this is stated, along with the DE indicated parenthetically thereafter. References to free doxorubicin concentrations and dosing are indicated in DE. References to amounts of epirubicin, an alternate anthracycline, or doxorubicinol, a metabolite of doxorubicin, are simply indicated in the absolute amounts of each moiety.
Doxorubicin (DOX) in the management of sarcomas
Since its introduction in the 1970s, DOX essentially remains either the backbone of multi-drug sarcoma treatment regimens or the primary monotherapy treatment for advanced sarcoma treatment (eg, see recent clinical trials assessing DOX-based combinations vs DOX monotherapy in STS).Citation9–Citation12 Consequently, DOX has been employed in investigations of sarcoma treatment on numerous occasions. Several studies are available to characterize its activity in sarcoma management. A large meta-analysis of anthracyclines in STS management estimated objective response rate (ORR) of 26% and median overall survival (OS) of 51 weeks/12.7 months.Citation13 Kaplan–Meier curves for overall survival (OS) were superimposable for all of the included treatment arms. The ORR estimate in this meta-analysis included a variety of clinical trials using older technologies to estimate ORR and thus may overestimate this parameter vs more recent trials. In addition, this study included studies of anthracycline-based combinations, which might be anticipated to yield a higher ORR than anthracycline monotherapy.
Enrolling patients from 2003 to 2010, Judson et al assigned DOX to 228 patients to the control arm of this randomized trial comparing DOX to DOX and ifosfamide (IFOS).Citation9 This trial remedied some of the methodological limitations of earlier trials: modern imaging assessment techniques were used; histologies non-responsive to chemotherapy were excluded; and the trial was adequately powered to address the scientific questions. Consequently, the control arm of this trial yielded high-quality estimates of DOX monotherapy activity in STS.
DOX (75 mg/m2 doxorubicin equivalent [DE] every 21 days) yielded estimated median overall survival (mOS) and median progression-free survival (mPFS) of 12.8 months (95% CI 10.5–14.3 months) and 4.6 months (95% CI 2.9–5.6 months), respectively. The ORR estimate, according to Response Evaluation Criteria in Solid Tumors (RECIST) 1.0 criteria, was 14% (95% CI 9.5%–19%). This is lower than that reported by Van Glabbeke et al, likely due more modern imaging assessment methods and inclusion of multi-agent, anthracycline-based combinations in the meta-analysis.Citation9,Citation13 Remarkably, the mOS for DOX monotherapy was virtually identical to that reported in the meta-analysis.
DOX has been studied extensively in combination regimens. Combinations with IFOS, alone or with other agents, have been the focus of efforts to improve the outcomes of anthracycline-based therapy. Multiple randomized clinical trials, reported over the course of >20 years, have failed to demonstrate a survival benefit in advanced STS for the DOX/IFOS combination, vs DOX alone.Citation9,Citation14–Citation16
In recent years, several novel agents have been assessed in large, randomized Phase III clinical trials to determine whether they enhance the clinical activity of DOX in STS. These include two novel derivatives of IFOS, palifosfamide and evofosfamide, and the humanized monoclonal antibody (mAb) olaratumab, targeting platelet-derived growth factor-α.Citation10,Citation11,Citation17,Citation18 Despite supportive preclinical data and strong Phase II clinical results, large, well-conducted Phase III studies of all three agents failed to demonstrate improvement in OS over DOX monotherapy. Thus, as of February 2019, DOX monotherapy remains a viable treatment option for advanced STS that new agents must either enhance or supersede.
Strategies to avoid DOX-mediated cardiotoxicity
The Achilles’ heel of anthracycline therapy, even when demonstrably effective, is dose-dependent cardiomyopathy. This has been recognized essentially since the introduction of anthracycline antineoplastics in the 1960s. The clinical condition appears to have acute (during treatment administration), early-chronic (up to 1 year after therapy completion), and late-chronic phases (>1 year after treatment completion).Citation19,Citation20 Estimates of the frequency of these effects vary, although <1%, 1.6%–2.1%, and 1.6%–5%, respectively, have been proposed. Late effects have been observed decades after treatment.
Increased risk is associated with a variety of treatment and clinical factors.Citation21 Patient factors include extremes of age, pre-existing heart disease, female sex, hypertension, and prior receipt of mediastinal irradiation. Treatment factors include rate of drug administration and, critically, cumulative anthracycline dose. Cumulative doses <300 mg/m2 DE have generally been associated with low rates of cardiotoxicity, representing a threshold value for the institution of cardio-protective maneuvers.Citation22
Several strategies have been explored to minimize cardiotoxicity. Limiting anthracycline dosing, possibly a viable solution in the pediatric setting, may not be viable in an adult demonstrating benefit for treatment of metastatic disease.Citation23 Altering the manner of drug administration from bolus to continuous intravenous infusion (CIV) is another strategy.Citation24 Several older randomized trials in breast cancer and sarcoma treatment have indicated that CIV may reduce anthracycline cardiotoxicity.Citation25–Citation27 A meta-analysis favored CIV to decrease cardiotoxicity.Citation28 Despite these results, uptake of this strategy has been limited in sarcoma treatment: a recently conducted clinical trial, in which investigators could administer DOX by either CIV or bolus, demonstrated use of CIV in only 84/640 (13%) of patients enrolled.Citation29
Dexrazoxane was designed to prevent cardiotoxicity associated with anthracycline use. It acts as a free radical scavenger, one of the postulated mechanisms of anthracycline cardiotoxicity.Citation30 Multiple randomized studies, primarily in breast cancer patients, but also in adult and pediatric sarcoma patients, indicate that dexrazoxane pretreatment can significantly decrease the incidence of anthracycline cardiotoxicity.Citation31–Citation34 These studies face methodological criticisms, including limited sample sizes and differing definitions and assessment methods for cardiotoxicity. In the pediatric population, dexrazoxane yields a 14.7- and 12.3-fold decreases in the incidence at 20 years for 300 and 400 mg/m2 DE cumulative DOX.Citation23
A final strategy to reduce cardiotoxicity has been the development of anthracycline derivatives with less cardio-toxicity. Epirubicin, a DOX analog with lower equidose cardiotoxicity, was not found to be a superior alternative to DOX in sarcoma treatment.Citation28,Citation35–Citation38 Liposomal forms of DOX have been proposed as a less cardiotoxic alternative. For example, a retrospective study in 141 heavily pretreated metastatic breast cancer patients showed no significant cardiotoxicity.Citation39 Two patients demonstrated abnormalities on electrocardiogram and one experienced a subclinical decrease in left ventricular ejection fraction (LVEF). A meta-analysis supported the observation of decreased clinical and subclinical cardiotoxicity of liposomal anthracycline preparations.Citation28
A number of clinical trials have explored liposomal DOX in sarcoma therapy.Citation40–Citation43 No clinical cardiac toxicity was reported. Clinical activity was modest. In the one randomized Phase II study, enrolling 94 patients, ORR was 10% for liposomal DOX and 9% for conventional DOX.Citation42 Inclusion of patients with gastrointestinal stromal tumors, a relatively chemotherapy-insensitive histology, may explain the low clinical activity observed.
Development of aldoxorubicin (ALDOX)
ALDOX ([6-maleimidocaproyl] hydrazone derivative of DOX) was originally derived to allow conjugation of DOX to thiolcontaining mAbs.Citation44 It has a long chemical half-life (T1/2 =158 hours) under neutral conditions (pH =7). Decomposition with the release of free DOX occurs much more rapidly (T1/2 =3.2 hours) under acidic conditions (pH =5) consistent with the endocytic lysosome.
Early studies of ALDOX used BR96, a mAb that binds a tumor antigen expressed on human carcinomas and closely related to Lewis Y.Citation44 After binding, BR96 is internalized into endocytic lysosomes. ALDOX-conjugated BR96 demonstrated a 50-fold improvement in the 50% Inhibitory Concentration (IC50) in vitro vs a non-binding control mAb in the L2987 lung carcinoma model.
Further experiments employing BR96 were conducted in xenograft models to assess the ALDOX-derivatized mAb in vivo.Citation45 Significant activity, including long-term (>1 year) cures in mice, was obtained after administration of the BR96–DOX conjugate in human lung, colon, and breast carcinoma xenografts.Citation45 Neither optimized doses of DOX, BR96 alone, mixtures of DOX with unconjugated BR96, nor ALDOX conjugates of control mAb yielded significant antitumor activity.
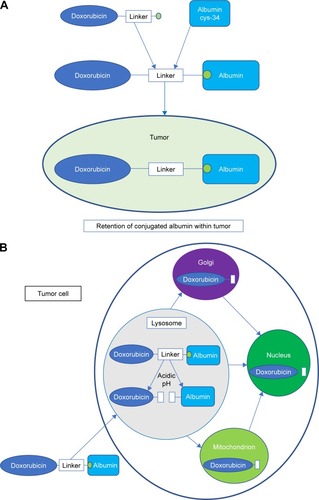
ALDOX conjugation to transferrin and albumin sought to target antineoplastic drugs more selectively to tumor tissues.Citation46,Citation47 The presumed mechanism of action is illustrated in . Preferential intratumoral accumulation of macromolecules, particularly albumin, has long been recognized ().Citation48–Citation51 Albumin has a number of additional advantages as a drug carrier, including its availability in pure form, relative stability, lack of immunogenicity, lack of toxicity, and biodegradability.Citation46
Figure 1 Mechanism of action of aldoxorubicin. (A) Intravenously administered aldoxorubicin forms a covalent bond with the free thiol of cysteine-34 in circulating human serum albumin. The drug–albumin conjugate is retained in tumors due to its size, poor tumoral blood flow, and absent tumoral lymphatic system.Citation46,Citation48–Citation52,Citation65 (B) At the level of the tumor cell, the drug–albumin conjugate is internalized into the endocytic lysosome. The acidic internal environment of the lysosome catalyzes release of free doxorubicin from the conjugate. This then traffics intracellularly to the Golgi, mitochondrion, and nucleus, where doxorubicin mediates cytotoxic effects.Citation53–Citation55
The ALDOX conjugate of albumin demonstrated in vitro activity comparable to free DOX in breast and leukemia cell lines.Citation46 Similar conjugates designed to be non-acid hydrolysable were inactive. This confirmed the importance of the acid hydrolysable linker in the activity of these compounds, allowing putative drug release in the acidic lysosome environment.
Using an albumin conjugate with a phenylacetyl linker, rather than the aliphatic linker of ALDOX, Kratz et al demonstrated in vitro binding of the DOX prodrug to human serum albumin.Citation52 Specifically, the resulting albumin conjugate was bound to the free cysteine at position 34 (cysteine-34) of human serum albumin (). This unusual functional group represents one of the few known free thiol groups in a circulating protein.Citation53
Further investigations characterized the activity of ALDOX in detail.Citation53 Binding to cysteine-34 of albumin was confirmed. Molecular modeling indicated that the spacer containing five methylene residues, the molecule originally identified by Willner et al, was optimal for binding at this site.Citation44 Binding was confirmed to both exogenous human serum albumin and endogenous human serum albumin in blood plasma.Citation53 Coupling of the ALDOX prodrug to albumin was complete after 2 minutes incubation in vitro and was associated almost entirely with the albumin component of plasma. In whole human blood, ~90% of ALDOX binds human albumin in the plasma phase, near the theoretical maximum amount that can be so distributed. In contrast, only 30% of free DOX is found in the plasma, consistent with sequestration of the majority of DOX in blood cells.
Incubation of ALDOX with MCF-7 tumor cells yielded gradually increasing amounts of free DOX, confirming intracellular release of free DOX. In vitro and in vivo antitumoral activities of ALDOX were also observed. Collectively, these data confirmed that ALDOX can be administered as a prodrug to bind its albumin target in whole blood, the ALDOX–albumin conjugate is taken up by tumor cells, free DOX is released from the tumor cells, and antitumoral activity is evident. Administration of the ALDOX prodrug is simpler to implement than preparation and administration of exogenously derivatized human albumin.
The intracellular distributions of DOX–protein conjugates, liposomal DOX, and free DOX in LXFL 529 lung carcinoma cell line were compared using confocal microscopy ().Citation54 This study used the albumin conjugate with a phenylacetylhydrazone linker, as opposed to ALDOX, for the acid-hydrolysable DOX–albumin construct. All tested preparations demonstrated cytotoxicity, apart from a non-acid hydrolysable conjugate of human albumin, again confirming the importance of acid hydrolysis for the activity of DOX–albumin conjugates.
Four hours after the addition of drug, the intracellular localizations of DOX were different. While free DOX was largely localized to the nucleus, DOX conjugates were primarily cytoplasmic. Interestingly, lysosomal localization was absent, indicating that this step in the proposed mechanism leads to rapid egress from the lysosome. By 24 hours, the intracellular distributions of free and hydrolysable albumin-bound preparations were similar, consistent with the liberation of free DOX from the conjugate. In contrast, the transferrin and non-hydrolysable albumin constructs were not found in the nucleus at 24 hours. Both of the latter preparations were also less active or inactive, respectively, in cytotoxicity assays vs free DOX or hydrolysable albumin-bound preparations.
These data are consistent with the proposed mechanism of albumin-bound DOX: the initial trafficking of the conjugate is different than free DOX, but becomes similar after acid hydrolysis and release of free DOX. Albumin as a carrier more closely approximates the intracellular management of free DOX after release than does transferrin. Efficient hydrolysis of the bound DOX is important for activity. Nuclear localization of DOX appears important for the cytotoxic activity. While activities targeted at DNA have been thought of as the dominant mode of anthracycline activity, other mechanisms of action involving other organelles have also been identified.Citation55 In LXFL 529, the nuclear activities may predominate.Citation54
Preclinical studies of ALDOX
Extensive preclinical studies have documented the activity of ALDOX and related derivatives in models of human cancer. Using a phenylacetyl spacer instead of the aliphatic spacer of ALDOX, acid-hydrolysable, albumin-bound DOX demonstrated IC50 approximating that of free DOX in clonogenic assays of 12 diverse human xenografts.Citation56 The drug–albumin conjugate also demonstrated superior activity to free DOX in the MDA-MB-435 human carcinoma model. This may have been due to reduced toxicity of the drug conjugate, allowing greater DE doses to be administered. Parenthetically, the MDA-MB-435 model, while initially thought to represent a breast carcinoma line, presently is thought to be melanoma.Citation57
Administration of ALDOX itself has been assessed in a variety of human xenografts. One of the challenges in these studies is the selection of doses for comparison, allowing a fair comparison of drugs. In xenograft studies of MDA-MB-435, ALDOX showed similar or better tumor growth inhibition vs equimolar concentrations of DOX.Citation53,Citation56 This included doses in which DOX exceeded the toxicity threshold in the murine xenograft system. ALDOX appears to have efficacy in this experimental system at least comparable to DOX, if not greater.
In 3366 breast carcinoma, A2780 ovarian carcinoma, and H209 small cell lung carcinoma, ALDOX achieves greater tumor inhibition than free DOX.Citation58 This may be due to its favorable toxicity profile, allowing a 3- to 4.5-fold increase in its DE dosing. Combinations of ALDOX and free DOX in the A2780 ovarian carcinoma and MIA PaCa-2 pancreatic carcinoma models may be even more effective.Citation59 In part, this may reflect a decreased toxicity of the combination regimen, with similar antineoplastic effects mediated by lower cumulative DOX doses. In addition, albumin-bound and free DOX may have somewhat different mechanisms of action, as hinted by the earlier confocal microscopy studies showing their different intracellular drug trafficking patterns.Citation54
Extensive studies have been conducted in myeloma.Citation60 Angiogenesis was inhibited by ALDOX in a concentration- and pH-dependent manner, along with correlated expression of Flk-1. Two xenograft models were established from myeloma patients. ALDOX demonstrated significant single-agent activity and moreover enhanced the activity of bortezomib. Uptake of albumin into the actual myeloma tumors was confirmed through visualization with Evans’ blue, an albumin-binding dye.
A major problem with DOX, highlighted earlier, is dose-dependent cardiomyopathy. ALDOX was designed to preferentially accumulate in tumors, hopefully sparing the myocardium. To assess this preclinically, rats received equimolar doses of free DOX or ALDOX (low-dose), or a dose of ALDOX equivalent to three times the equimolar dose (high-dose).Citation61 The DOX dose was selected based on its being equitoxic to that administered in the high-dose ALDOX group.Citation62
After seven weekly doses, assessment at 48 weeks of rats treated with free DOX demonstrated clinical, histopathological and molecular markers of severe myocardial damage.Citation61 Out of 15 rats treated, five died before completion of the study. In contrast, only one of 20 ALDOX-treated rats died before study completion, without gross pathologic changes. Clinical signs were comparable to the untreated control group. Heart weight in the low-dose ALDOX group was similar to control, while that of the high-dose ALDOX group was slightly higher than controls. Hearts in the DOX-treated rats were significantly increased in weight above all other groups. Pleural effusions, hepatomegaly, or macroscopic renal changes were not present in control or ALDOX-treated groups, in contrast to virtually every member of the free DOX-treated cohort.
Histologic and molecular markers also indicated decreased myocardial effects of ALDOX vs free DOX. However, detectable effects were not entirely absent. In the high-dose ALDOX group, histological markers of myocardial damage, depressed mitochondrial DNA copy number, and elevated superoxide levels were present, although to a significantly lesser extent than those treated with free DOX. Mitochondrial DNA deletions were present in both DOX- and ALDOX-treated rats, although to a lesser extent in the latter. As a control, the same markers of mitochondrial damage were assessed in skeletal muscle. This tissue did not demonstrate the same pattern of damage, indicating that changes were specific to the myocardium.
These data indicate that ALDOX has less cardiotoxicity than free DOX, on an equimolar basis, in this rat model. High-dose ALDOX still elevated some markers of mitochondrial damage in a dose-dependent manner, indicating that the drug is not free from cardiac effects. The dose selected for the high-dose ALDOX group, judged equitoxic to the selected free DOX dose, seems to have a lesser effect than free DOX on indices of cardiac damage. This is noted despite administration of a threefold greater absolute amount of DEs. One would hypothesize that an ALDOX dose could be identified yielding evidence for cardiotoxicity similar to the DOX dose selected. The authors did not report such higher dose studies, preventing clear delineation of the cardiotoxicity dose–response curve. In this experimental model, ALDOX does not eliminate anthracycline cardiotoxicity.
In anticipation of first-in-human studies, a preclinical toxicity study was conducted.Citation62 This was conducted in CD-1 mice, Sprague–Dawley rats, and Beagle dogs. In mice and rats, the minimum LD50 was 2- to >5-fold higher than for an equivalent dose of free DOX. A dose-dependent peripheral neuropathy occurred in mice and rats, about 1–3 weeks after administration, similar to a phenomenon observed in these species with DOX. Hematologic effects in rats were decreased threefold vs DOX in repeat dosing studies, although testicular/oligospermia were similar at equitoxic doses. In rats, the ALDOX no–observed-adverse-effect level (NOAEL) of 4 weekly doses of 2.5 mg/kg DE was identical to the maximum tolerated dose (MTD) of DOX.
In Beagle dogs, the NOAEL was 2 weekly ALDOX doses of 1.5 mg/kg DE, approximately twice the MTD of DOX. Some dogs experienced a histamine-like reactions within 3 hours of dosing. Temporary hematologic, urinary, and histopathologic effects were noted in animals receiving mid-range or high doses (3.0 and 4.5 mg/kg DE, respectively). The authors recommended a starting dose of 20 mg/m2 DE ALDOX for human Phase I studies.
Phase I clinical studies of ALDOX
The first-in-human Phase I study of ALDOX was conducted as a conventional 3+3 dose escalation, starting at a dose of 20 mg/m2 DE every 3 weeks.Citation63 Eligible patients could have received up to 350 DE or 700 mg/m2 of prior DOX or epirubicin therapy, respectively. The authors do not report the number of patients with such prior therapy. Patients with peripheral neuropathy greater than grade 2 were excluded, in consideration of preclinical data indicating dose-dependent peripheral neuropathy in mice and rats.Citation62
No toxicity attributable to ALDOX was observed up to 200 mg/m2 DE. At 260 mg/m2 DE, grade 2 anemia/thrombocytopenia, grade 3 neutropenia/leukocytopenia, and grade 2 mucositis were each observed in at least two patients. The next dose level, 340 mg/m2 DE, met the definition of MTD, with grade 4 neutropenia/leukocytopenia, grade 3 thrombocytopenia, grade 2 anemia, and grade 3 mucositis in at least two patients each. Based on the pre-specified definition, 260 mg/m2 DE every 3 weeks was designated as the recommended Phase II dose (RP2D) level. This represents a 3.5-fold increase in absolute amount of DOX dosed vs conventional DOX dosing at 75 mg/m2 DE per 3-week cycle.
Potential for cardiotoxicity was a major concern. Other than clinically asymptomatic electrocardiographic changes in three patients, no cardiac changes were observed. No echocardiographic (ECHO) changes suggestive of heart failure were identified. This was observed despite administration in some patients of ALDOX doses averaging 1,650 mg/m2 DE, significantly more than dosing of free DOX leading to heart failure in more than 50% of treated patients. These results confirmed the earlier preclinical studies indicating that ALDOX had a more favorable cardiac toxicity profile than DOX, assuming equipotency of the two drugs. Confirmation of this observation could only be undertaken in a formal comparison of the two agents.
Pharmacokinetic (PK) studies were conducted assuming a noncompartmental analysis, using a model requiring constant infusion. The analytical methods described in the report assessed total DOX within the plasma samples (albumin-bound + free). These studies indicated a terminal half-life of ALDOX of 17.6–38.2 hours, similar to that of free DOX. Mean area under curve (AUC) values of 234.5–2,547 h µmol/L of albumin-bound DOX were much higher than the value of 3 h µmol/L anticipated for DOX. This is consistent with the prolonged residence of albumin-bound DOX in the circulation, vs the rapid and extensive distribution of free DOX from plasma into tissues.Citation64
Corresponding to this elevated AUC is a lower rate of clearance of ALDOX from the circulation of 2.15–3.37 mL/min/m2, vs 580 mL/min/m2 for DOX. The volume of distribution (VD) was 2.14–7.28 L/m2 vs 700–1,000 L/m2 for DOX. Only 1.4%–7.7% of administered ALDOX was excreted in the urine, similar to the proportion of free DOX so eliminated.Citation64 Since prolonged circulation in the blood is necessary for enhanced tumoral accumulation of the albumin–drug conjugate, these human PK parameters are consistent with the design objectives of ALDOX: to enhance tumoral targeting of the chemotherapy warhead.Citation63,Citation65
Further characterization of the PK profile of ALDOX was undertaken in a study to assess a new formulation of ALDOX.Citation66 Here, 18 patients received either 230 mg/m2 (170 mg/m2 DE; n=11) or 350 mg/m2 (260 mg/m2 DE; n=7) of ALDOX every 3 weeks.Citation66 The higher dose level included the RP2D from the prior Phase I study.Citation63 Prior DOX dosing was allowed, although the number of such patients was not reported. Those with clinically significant heart disease were excluded. Eight patients had lung cancer, while the remainder represented a variety of malignancies. One STS patient was included.
A key methodological difference in this second PK study was inclusion of analyses specifically investigating free DOX and the metabolite doxorubicinol, as well as albumin-bound DOX. Doxorubicinol is proposed to play a major role in DOX cardiotoxicity.Citation67 After the initial dose of DOX, the time to peak plasma concentrations (tmax) of ALDOX and free DOX were similar (0.75 and 0.58 h for 230 mg/m2 [170 mg/m2 DE] and 1.00 and 0.68 h for 350 mg/m2 [260 mg/m2 DE], respectively). In contrast, tmax for doxorubicinol was 36.5 h (230 mg/m2, 170 mg/m2 DE) and 48.5 h (350 mg/m2, 260 mg/m2 DE). Peak plasma concentrations (Cmax) of albumin-bound DOX (mean 67,400–105,000 ng/mL DE) were 40- to 300-fold higher on an individual basis than Cmax of free DOX (mean 1,200–2,470 mg/mL DE). Mean Cmax of doxorubicinol levels were near the limits of detection (4.17–13.7 ng/mL). The latter finding may explain decreased or absent cardiotoxicity after ALDOX administration.Citation67 The mean circulating half-life of ALDOX was measured at 20.1–21.1 h. VD was 3.96–4.08 L/m2. Mean clearance was 0.136–0.152 L/h/m2 (2.27–2.53 mL/min/m2). In concordance with the prior Phase I study of Unger et al,Citation63 only small amounts of DOX were detectable in urine, the majority as unchanged drug. Doxorubicinol was present in only trace quantities in the urine. These data indicate that ALDOX is stable in the circulation without substantial accumulation in non-bloodstream compartments. In addition, the bulk of DOX circulating in the plasma remains bound to albumin, rather than free.
Grade 3 or 4 adverse events were observed on eight occasions in the lower dosing group and 14 occasions in the higher dosing group.Citation66 Assessment for cardiac adverse events was an important clinical objective of the study. Ten patients had prolongation of QTc >5% from baseline, and one patient had a QTc >500 ms. These were not considered clinically significant. One patient had a nonclinically significant decrease in LVEF of >10%. No other evidence of potential cardiotoxicity was noted.
A Phase Ib/II study was conducted to assess a new formulation of ALDOX.Citation68 It is unclear from the clinical report whether this represents the same formulation as described in the Phase I study of Mita et al.Citation66 Patients received 230, 350, or 450 mg/m2 (170, 260, or 335 mg/m2 DE, respectively) of this new preparation, administered to 5, 18, and 2 patients, respectively. Of the 25 enrolled patients, 13 had STS, with the remainder representing a broad range of neoplasms. Assessment of adverse events and determination of MTD were the primary objectives of the Phase Ib component.
The MTD was determined to be 350 mg/m2 (260 mg/m2 DE). At 450 mg/m2 (335 mg/m2 DE), both patients experienced grade 3–4 neutropenia, one with febrile neutropenia, leading to closure of the cohort. At MTD, myelosuppression, nausea/vomiting, fatigue, and stomatitis/mucositis were observed in at least three of the 18 patients. One patient died of septic shock due to urinary tract infection and neutropenia. From the cardiac perspective, no significant changes in LVEF were observed. One patient experienced QTc prolongation to >500 ms. Seven patients demonstrated elevated troponin levels judged to be clinically insignificant.
A Phase Ib/II study was reported in abstract/poster form in which patients with metastatic solid tumors received ALDOX and gemcitabine.Citation69 Primary endpoints were safety and identification of RP2D. ALDOX was administered on day 1 and gemcitabine was administered on days 1 and 8 of a 21-day cycle. Thrombocytopenia was the dose-limiting toxicity, with myelosuppression, infectious complications, and stomatitis also being prominent toxicities. No clinically significant cardiac toxicity was reported among the 27 enrolled patients, although 6/27 experienced at least a 10% decrease in LVEF. The RP2D was 200 mg/m2 ALDOX (150 mg/m2 DE) and 500 mg/m2 gemcitabine.
Efficacy studies of ALDOX in the management of STSs
STS has been a major focus of the efficacy assessments of ALDOX to date (). The Phase Ib/II study noted above also attempted to assess the treatment efficacy of ALDOX, especially in STS.Citation68 Among all patients enrolled in this trial, five (20%) experienced a PR, as assessed by RECIST 1.1 criteria, all among STS patients receiving at least 350 mg/m2 (260 mg/m2 DE) of ALDOX (liposarcoma, leiomyosarcoma, spindle cell sarcoma, pleomorphic sarcoma, malignant peripheral nerve sheath tumor; ORR for STS =29%; ORR =38% among those receiving at least 350 mg/m2 or 260 mg/m2 DE).
Table 1 Clinical trials of ALDOX enrolling soft tissue sarcoma patients
A multicenter, international randomized Phase II trial was conducted to compare ALDOX with DOX in advanced STS.Citation70 Patients were randomized 2:1 to treatment every 3 weeks with either ALDOX (350 mg/m2, 260 mg/m2 DE) or to conventional DOX at 75 mg/m2 DE. Dosing of both agents was planned for six cycles, with up to two additional cycles possible. Patients could have received up to 225 mg/m2 DE of prior DOX-based adjuvant therapy, but were treatment-naïve for advanced disease. Patients with sarcomas that would not be expected to respond to DOX or for which there existed other standard treatment approaches were excluded.
Primary endpoint of the study was PFS. Both local investigator and central review for PFS were undertaken. It is unclear from the report which of these two reviews served as the primary endpoint assessment, although both were statistically significant (see below). OS, ORR by RECIST 1.1, and safety/adverse events were also assessed.
Of 126 eligible patients, 86 were randomly assigned to ALDOX and 40 to DOX. Seven (6%) patients had received prior DOX. Three patients in the ALDOX group withdrew consent prior to treatment and were not evaluable. All DOX patients received assigned treatment. Patients were evaluable with respect to the final efficacy analysis if treated with at least one dose of study drug and evaluated by one imaging study beyond baseline (described as a “modified intent-to-treat” [ITT] analysis).
Whether by investigator or blinded central review, the study met its primary endpoint. PFS was improved in the ALDOX group (investigator: mPFS 8.3 months, 95% CI 6.4–9.7 months; central: mPFS 5.6 months, 95% CI 3.0–8.1 months) vs the DOX group (investigator: mPFS 4.6 months, 95% CI 2.7–5.9 months; central: mPFS 2.7 months, 95% CI 1.6–4.3 months; investigator: P=0.001; central: P=0.02). There was no statistically significant improvement in OS, a secondary study endpoint (ALDOX: mOS =15.8 months, 95% CI 13.0 months to not reached; DOX: mOS =14.3 months, 95% CI 8.6–20.6 months; HR =0.73, 95% CI 0.44–1.20, P=0.21). However, the study was not powered to assess the OS endpoint. ORR both by investigator (23% vs 5%, P=0.01) and central review (25% vs 0%, P=0.0007) favored ALDOX.
Adverse events in the ALDOX group mirrored those in the antecedent studies. Nausea, vomiting, stomatitis, alopecia, and decreased appetite were reported in >20% of patients. The frequency of these was greater among ALDOX-treated patients. Grade 3–4 toxicity was also more frequent in the ALDOX-treated patients (80% vs 58%, P=0.01).
From the cardiac standpoint, no patients experienced clinical cardiac dysfunction, based on clinical or imaging grounds, during treatment or in follow-up up to 11 months after treatment. LVEF decreased below 50% in three DOX-treated patients. During treatment, 12% of ALDOX- and 29% of DOX-treated patients experienced at least a 10% reduction in LVEF from baseline. Another measure of cardiac effects, serum troponin, was unchanged in ALDOX-treated patients from baseline, but was increased in DOX-treated patients until 5 months after treatment completion.
A Phase III study of ALDOX in STS was subsequently conducted.Citation71 Its design differed significantly from the antecedent randomized Phase II.Citation70 The Phase III study enrolled STS patients who had relapsed after, or were refractory to, initial systemic therapy. Patients were randomized to either ALDOX 350 mg/m2 (260 mg/m2 DE) every 3 weeks or investigator’s choice of one of three pre-selected regimens at a given participating institution. The control regimens included dacarbazine, pazopanib, gemcitabine/docetaxel, DOX, or IFOS, all administered according to institutional standards. The primary endpoint was PFS among the ITT population, as assessed by central radiology review. Additional pre-specified analyses included geographic outcomes for this international trial, histologic subtype (leiomyosarcoma/liposarcoma vs others), and receipt of prior DOX.
A total of 433 patients were randomized 1:1, with 218 assigned to ALDOX and 215 assigned to the control arm. Two thirds of the patients in each group received prior DOX. L-sarcomas comprised 55% of the ALDOX patients (42% leiomyosarcoma, 13% liposarcoma) and 59% of the control group (44% leiomyosarcoma, 15% liposarcoma). Among 213 ALDOX-treated patients who received planned therapy, cumulative median dosing of ALDOX was 1,400 mg/m2 (1,040 mg/m2 DE).
PFS, the primary endpoint, was not significantly improved (ALDOX: mPFS 4.11 months, 95% CI 2.79–5.06 months; control: mPFS 2.96 months, 95% CI 2.56–4.17 months; HR =0.81, 95% CI 0.64–1.03, P=0.087). For L-sarcomas, PFS was improved (ALDOX: mPFS 5.32 months, 95% CI 3.45–7.16 months; control: mPFS 2.96 months, 95% CI 2.10–4.37 months; HR =0.62, 95% CI 0.44–0.88, P=0.007). OS in the ITT population was not improved (ALDOX: mOS 12.88 months, 95% CI 10.05–15.11 months; control: mOS 12.16 months, 95% CI 10.38–13.31 months; HR =0.97, 95% CI 0.74–1.28, P=0.86). ORR was also unimproved in either the ITT population or the L-sarcoma subgroup. Adverse events were similar in both groups. However, treatment-related adverse events, those of grade 3–5, those resulting death, those that qualified as serious, and those resulting in drug discontinuation were all higher in the ALDOX-treated group.
Echocardiographic data were presented comparing the ALDOX-treated patients (n=213) with the subset of control patients treated with DOX (n=47). A decrease in LVEF of at least 20% from baseline was seen in 8/213 ALDOX-treated patients and 4/43 DOX-treated patients (P=0.116). A decrease in LVEF to <50% was observed in 9/213 ALDOX-treated patients and 9/47 receiving DOX (P<0.001). These groups are not necessarily entirely comparable. Two thirds of ALDOX-treated patients received prior DOX. One would presume that most or all patients receiving DOX had not received the agent previously. Comparison of only DOX-naïve patients in each group would perhaps give a more accurate picture. In addition, accounting for differential follow-up duration might lead to a more rigorous comparison. Nevertheless, in this study administering a 3.5-fold increase in the DE dose (260 mg/m2 DE of ALDOX vs 75 mg/m2 DE of DOX), there was no increase in clinical cardiotoxicity of ALDOX-treated patients vs those treated with DOX.
In another Phase II study, sarcoma patients received therapy with ALDOX at 170 mg/m2 (125 mg/m2 DE; n=7) or 250 mg/m2 (185 mg/m2 DE; n=38) on day 1 and IFOS at 1 g/m2/day on days 1–14 of a 28-day cycle.Citation72,Citation73 The combination was given up to six cycles, but ALDOX could continue thereafter as monotherapy. Prominent toxicities included myelosuppession, nausea/vomiting, stomatitis, and febrile neutropenia. No clinically significant cardiac toxicity occurred. Among the 44 evaluable patients at both dose levels, 16 (36%) demonstrated PR by RECIST 1.1 criteria.
Additional studies in other malignancies have been reported. In glioblastoma, a Phase II study of ALDOX in first relapse demonstrated that 3/21 (14%) patients with PR were evaluable.Citation74 Pseudoprogression was documented surgically in two patients who showed radiological evidence of progression. This suggests that ALDOX may penetrate the central nervous system in humans, as indicated preclinically.Citation75
Preliminary evidence of ALDOX activity in AIDS-associated Kaposi’s sarcoma has been reported.Citation76 Partial responses were observed in 10/15 enrolled patients. This was seen even in patients with visceral progressive disease with prior DOX-based therapy. Studies in small cell lung cancer vs topotecan (NCT 0200757) and as monotherapy in pancreatic cancer (NCT1580397) are reportedly in progress (see ClinicalTrials.gov).
Current status of ALDOX and directions for future study
Numerous clinical trials have attempted to supplant DOX in sarcoma treatment. None have yet succeeded. In some ways, this is surprising. First, DOX is not a highly active drug. Randomized trials of DOX, or any other anthracycline, vs placebo have not been conducted, and such studies will almost certainly not be. Thus, the impact of anthracycline therapy on survival in advanced sarcomas is unclear. However, ORR, probably best estimated by Judson et al at 14%, is modest.Citation9 This low rate may simply reflect the wide diversity of histologies that fall under the “sarcoma” label, with some histologies being more anthracycline-responsive than others. Perhaps it is unreasonable to expect that any cytotoxic monotherapy would achieve ORR in sarcomas markedly higher than this.
Of course, the second problem with DOX is toxicity, specifically its dose-dependent cardiotoxicity. Even when the drug is having a demonstrably beneficial effect, it must eventually be discontinued. Strategies, discussed earlier, have been developed to address anthracycline cardiotoxicity. Despite the great dependence of sarcoma treatment on anthracycline therapy, uptake of approaches to minimize cardiotoxicity has been surprisingly limited.Citation29
ALDOX comes upon this scene with several potential advantages. There is a strong preclinical data set supporting its use in this setting. Its novel mechanism may allow preferential targeting of the drug to tumors. Experimental systems and multiple in-human studies support the proposed mechanism. Preclinical studies suggest activity of ALDOX at least comparable to DOX, if not superior.
Assuming superiority, several mechanisms might be invoked to explain ALDOX superiority. These include preferential targeting of drug to tumors, allowance of greater DE doses to be administered, existence of an antineoplastic mechanism distinct from DOX, or any combination of these. Fundamental to clarification of these questions is definitive confirmation of superior efficacy of ALDOX vs DOX. Such evidence does not yet exist in humans.
A major argument in favor of ALDOX is decreased cardiac effects vs DOX. Data from both experimental systems and human studies suggest that cardiotoxicity is less prominent with ALDOX. Confirmation of this requires 1) comparing doses of ALDOX and DOX that are at least equipotent from an efficacy standpoint and 2) monitoring patients for cardio-toxicity after sufficient drug dosing and follow-up periods to allow meaningful comparison with data for DOX and other anthracyclines. Better characterization of the ALDOX dose– response relationship with regard to cardiotoxicity is needed to confirm decreased cardiotoxicity vs DOX. Critically, studies of ALDOX do not indicate absent cardiac effects, despite their being deemed subclinical in studies to date.Citation63,Citation66,Citation68–Citation71
The clinical experience in sarcomas at present is best captured by the randomized Phase II and III clinical trials discussed above.Citation70,Citation71 The Phase II study appears to have been a well-designed and conducted study assessing preliminarily the activity and safety of ALDOX vs DOX in STS. Two points in the trial’s design are important to consider while evaluating the Phase II results and interpreting the results of the subsequent Phase III trial.
First, this study enrolled patients who were naïve to systemic therapy for advanced disease. Only 13 of 123 evaluable patients received neoadjuvant or adjuvant chemotherapy. Presumably, most of this subgroup received DOX-based therapy. No significant work has yet been undertaken to define mechanisms of acquired resistance to ALDOX. It would not be surprising if resistance mechanisms were shared with conventional DOX. That 89% of the participants had not been exposed to prior chemotherapy minimized this problem. Exclusion of all patients with prior anthracycline therapy should have been considered.
Second, PFS was the primary endpoint.Citation77 This is a perfectly reasonable endpoint in this setting to assess whether a more rigorous Phase III assessment should be conducted. The trial achieved this target. OS was not significantly improved. Based on the size of the trial, the observed hazard ratio for OS of 0.73, and the 2:1 treatment allocation, the statistical power of the trial to detect such a difference was only 42%. With 2:1 treatment allocation, a trial of 351 evaluable patients would have been required to have 80% statistical power to detect such a difference.
Overall, the Phase II trial results give reasonably strong evidence to support undertaking a confirmatory Phase III trial. Design of Phase III trials from antecedent Phase II designs and outcomes is challenging.Citation78 A simple repeat of a Phase II on a larger scale is rarely adequate. Alteration in endpoints (such as defining a co-primary endpoint of PFS and OS), treatment, study population, or any other factor can alter the study enough to alter outcomes. The clinical trial size noted above (n=351), with some additional patients added to account for practical considerations such as drop-out, could reasonably have been conducted to confirm the OS result seen (HR =0.73 in the Phase II trial). Notably, this is approximately the degree of improvement in OS sought by three large recent trials in STS that used OS as primary endpoint.Citation9–Citation11 A positive trial would have had a major impact on the sarcoma field. Even a study that demonstrated improved PFS and decreased cardiac effects, without improvement in OS, could reasonably have led to regulatory approval. Further development did not proceed in this manner.
The Phase III study looked at a very different clinical scenario: salvage treatment in disease either relapsed after or refractory to initial therapy.Citation71 The decision to change the focus of ALDOX development in sarcomas may have been driven by other clinical trials being conducted or planned in parallel, such as the DOX combinations with palifosfamide, evofosfamide, or olaratumab.Citation10,Citation11,Citation18 As noted above, these trials ultimately showed no improvement over DOX, but that was unknown at the time the ALDOX Phase III trial was being designed and executed.
The primary endpoint of PFS was selected.Citation71 Several sarcoma drugs have been approved in the same setting with PFS as the primary endpoint and no OS improvement (eg, pazopanib, trabectedin).Citation79,Citation80 The size of the trial (n=433 randomized) would have been more than able to accommodate the design size noted above (n=351) to even detect a difference in OS similar to that seen in the Phase II study.Citation70
A critical factor was the fraction of patients with prior DOX exposure. While this amounted to 11% in the Phase II study, two thirds of the Phase III patients had prior DOX exposure and had progressed.Citation71 If acquired resistance to DOX can lead to some degree of ALDOX cross-resistance, then this high proportion of anthracycline-exposed patients will place ALDOX at a disadvantage.
Although the control population included 47/215 who received DOX therapy as assigned treatment, the remainder received agents that would not be expected to have a high degree of cross-resistance to DOX. At best, the high proportion of DOX-treated patients has no effect. More likely, prior DOX exposure and resistance impaired ALDOX responsiveness and had a lesser impact on the control treatments. This would tend to minimize differences between the two study arms.
Unfortunately, PFS was not improved in the overall population. There was improvement in PFS among L-sarcomas (leiomyosarcoma and liposarcoma), making up 57% of the study population. This was not coupled with additional outcome improvements (such as OS or ORR) that might have justified regulatory submission for approval in this subgroup. A similar outcome was seen in a trial of eribulin in L-sarcomas, where regulatory approval was ultimately granted only for liposarcoma treatment, despite an overall statistical improvement in OS for the entire study population.Citation81
So, where does this leave ALDOX at this point vis-à-vis sarcomas? Several trials are exploring combinations, such as with IFOS or gemcitabine, which might be pertinent. However, these do not address the real question: can ALDOX supplant DOX in sarcoma treatment? Concern about failure of a Phase III to “beat” DOX and concurrent clinical trials that might have established new DOX-based treatment regimens may have led to the Phase III design that was actually conducted. That Phase III trial neither answered the key question nor provided another route for approval.
Several routes for further development in sarcomas are evident. The obvious avenue is through a confirmatory Phase III study which closely approximates the Phase II study. The design should 1) focus on co-primary endpoints of PFS and cardiac toxicity with adequate power and follow-up, 2) exclude those with prior anthracycline therapy, 3) enroll STS patients who are treatment-naïve for advanced unresectable or metastatic disease, 4) include evaluation of activity in L-sarcomas as a planned PFS subgroup analysis, and 5) be adequately powered to also evaluate the OS endpoint, even if defined as secondary.
Alternatively, if there was concern that ALDOX may not be able to demonstrate superiority DOX on standard clinical criteria, then a non-inferiority trial could be undertaken, with decreased cardiotoxicity as a co-primary outcome. Here, the primary objective would be to demonstrate the ability of ALDOX to substitute for DOX, with the objective of demonstrating ALDOX superiority with respect to cardiotoxicity. Selection of dosing for such a trial is important, although the Phase III dosing of ALDOX (350 mg/m2 =260 mg/m2 DE) is probably reasonable, given its prior use suggesting lower cardiotoxicity than conventional doses of DOX (75 mg/m2 DE) and evidence of activity in the STS setting from the Phase II trial. Non-inferiority designs however face large patient requirements, based on the degree of non-inferiority that must be confirmed. Such a trial may or may not be practical.
A third strategy would be to look for niches into which ALDOX could fit. This could involve identification of sensitive histologies (eg, activity in L-sarcomas indicated by the Phase III, similar for the development of eribulin and trabectedinCitation80,Citation81) or biomarkers of response. Development in clinical settings for which no proven standard therapy yet exists could also be considered. Neoadjuvant STS therapy arguably falls into this category. Exploration of alternate dosing strategies, such as prolonged lower-dose administration, could also be productive. Any of these approaches would require generation of more preliminary data prior to a registration trial, and thus are more prolonged approaches than either of the strategies noted above based on the randomized Phase II trial results in the advanced/metastatic setting.
Conclusion
ALDOX has been developed to exploit novel biological observations, with the intent of improving the therapeutic index of DOX. STSs are one of the few classes of neoplasms where DOX monotherapy remains standard (as of February 2019), first-line therapy for advanced disease.Citation9,Citation18 The present time (February 2019) offers a window-of-opportunity for such a trial. Multiple recent Phase III trials to examine agents that would supplement DOX in STS have reported negative results, most recently a trial of olaratumab.Citation9–Citation11,Citation18 DOX mono-therapy remains a viable standard comparator therapy. The limitations of the previously conducted ALDOX Phase III study should not preclude further evaluation of ALDOX in sarcomas. The current data set suggests that ALDOX could be an important addition to the sarcoma treatment armamentarium, and one that would finally allow the sarcoma field to move forward, to a world beyond DOX.
Acknowledgments
This is to acknowledge Lauren Wood for her assistance in manuscript preparation. Funding was provided by CytRx, the manufacturer of ALDOX, to Dr Cranmer’s prior institution, the University of Arizona, for conducting a clinical study. Dr Cranmer’s relationship with the University of Arizona and CytRx ended in October 2015.
Disclosure
Dr Cranmer reports grants from Tracon, AADi, Eli Lilly, Merck, Advenchen, and BetaCat, outside the submitted work. Dr Cranmer also received grants and personal fees from BluePrint outside the submitted work. The author reports no other conflicts of interest in this work.
References
- SiegelRLMillerKDJemalACancer statistics, 2019CA Cancer J Clin Epub201918
- Sarcoma Progress Review GroupA Roadmap for Sarcoma ResearchNational Cancer Institute, U.S. Department of Health and Human Services2004 Available from: https://sarcomahelp.org/articles/sarcoma-research-roadmap.htmlAccessed October 4, 2018
- CorreaRGómez-MillánJLobatoMRadiotherapy in soft-tissue sarcoma of the extremitiesClin Transl Oncol20182091127113510.1007/s12094-018-1848-x29476322
- LiebnerDAThe indications and efficacy of conventional chemotherapy in primary and recurrent sarcomaJ Surg Oncol2015111562263110.1002/jso.2386625581912
- ShengJYMovvaSSystemic therapy for advanced soft tissue sarcomaSurg Clin North Am20169651141115610.1016/j.suc.2016.06.00627542647
- CanterRJChemotherapy: does neoadjuvant or adjuvant therapy improve outcomes?Surg Oncol Clin N Am201625486187210.1016/j.soc.2016.05.01327591503
- SangkhathatSCurrent management of pediatric soft tissue sarcomasWorld J Clin Pediatr2015449410510.5409/wjcp.v4.i4.9426566481
- GronchiAFerrariSQuagliuoloVHistotype-tailored neoadjuvant chemotherapy versus standard chemotherapy in patients with high-risk soft-tissue sarcomas (ISG-STS 1001): an international, open-label, randomised, controlled, phase 3, multicentre trialLancet Oncol201718681282210.1016/S1470-2045(17)30334-028499583
- JudsonIVerweijJGelderblomHDoxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trialLancet Oncol201415441542310.1016/S1470-2045(14)70063-424618336
- RyanCWMerimskyOAgulnikMPICASSO III: a phase III, placebo-controlled study of doxorubicin with or without palifosfamide in patients with metastatic soft tissue sarcomaJ Clin Oncol201634323898390510.1200/JCO.2016.67.668427621408
- TapWDPapaiZVan TineBADoxorubicin plus evofosfamide versus doxorubicin alone in locally advanced, unresectable or metastatic soft-tissue sarcoma (TH CR-406/SARC021): an international, multi-centre, open-label, randomised phase 3 trialLancet Oncol20171881089110310.1016/S1470-2045(17)30381-928651927
- Lartruvo® (Olaratumab) [package insert]Indianapolis, INEli Lilly and Company82018
- Van GlabbekeMvan OosteromATOosterhuisJWPrognostic factors for the outcome of chemotherapy in advanced soft tissue sarcoma: an analysis of 2,185 patients treated with anthracycline-containing first-line regimens – a European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group StudyJ Clin Oncol199917115015710.1200/JCO.1999.17.1.15010458228
- AntmanKCrowleyJBalcerzakSPAn intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomasJ Clin Oncol19931171276128510.1200/JCO.1993.11.7.12768315425
- EdmonsonJHRyanLMBlumRHRandomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomasJ Clin Oncol19931171269127510.1200/JCO.1993.11.7.12698315424
- SantoroATurszTMouridsenHDoxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first-line treatment of advanced soft tissue sarcomas: a randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma GroupJ Clin Oncol19951371537154510.1200/JCO.1995.13.7.15377602342
- TapWDJonesRLVan TineBAOlaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trialLancet201638810043488497 Erratum appears in Lancet. 2016 Jul 30; 388(10043):46410.1016/S0140-6736(16)30587-627291997
- Eli Lilly and CompanyLilly Reports Results of Phase 3 Soft Tissue Sarcoma Study of LARTRUVO2019 Available from: https://investor.lilly.com/news-releases/news-release-details/lilly-reports-results-phase-3-soft-tissue-sarcoma-studyAccessed February 6, 2019
- BabikerHMMcBrideANewtonMCardiotoxic effects of chemotherapy: a review of both cytotoxic and molecular targeted oncology therapies and their effect on the cardiovascular systemCrit Rev Oncol Hematol201812618620010.1016/j.critrevonc.2018.03.01429759560
- CuriglianoGCardinaleDSuterTCardiovascular toxicity induced by chemotherapy, targeted agents and radiotherapy: ESMO clinical practice guidelinesAnn Oncol201223Suppl 7vii155vii16610.1093/annonc/mds29322997448
- ShakirDKRasulKIChemotherapy induced cardiomyopathy: pathogenesis, monitoring and managementJ Clin Med Res20091181210.4021/jocmr2009.02.122522505958
- SeymourLBramwellVMoranLAUse of dexrazoxane as a cardio-protectant in patients receiving doxorubicin or epirubicin chemotherapy for the treatment of cancer. The provincial systemic treatment disease site groupCancer Prev Control19993214515910474762
- LiesseKHarrisJChanMSchmidtMLChiuBDexrazoxane significantly reduces anthracycline-induced cardiotoxicity in pediatric solid tumor patients: a systematic reviewJ Pediatr Hematol Oncol201840641742510.1097/MPH.000000000000111829432315
- BielackSSErttmannRWinklerKLandbeckGDoxorubicin: effect of different schedules on toxicity and anti-tumor efficacyEur J Cancer Clin Oncol198925587388210.1016/0277-5379(89)90135-12661240
- HortobagyiGNFryeDBuzdarAUDecreased cardiac toxicity of doxorubicin administered by continuous intravenous infusion in combination chemotherapy for metastatic breast carcinomaCancer198963137452910423
- ShapiraJGotfriedMLishnerMRavidMReduced cardiotoxicity of doxorubicin by a 6-hour infusion regimen. A prospective randomized evaluationCancer19906548708732297656
- ZalupskiMMetchBBalcerzakSPhase III comparison of doxorubicin and dacarbazine given by bolus versus infusion in patients with soft-tissue sarcomas: a Southwest Oncology Group StudyJ Natl Cancer Inst199183139269322067035
- SmithLACorneliusVRPlummerCJCardiotoxicity of anthracycline agents for the treatment of cancer: systematic review and meta-analysis of randomised controlled trialsBMC Cancer20101033710.1186/1471-2407-10-33720587042
- CranmerLDLyYBlammanKVLoggersETPollackSToxicity and efficacy of bolus (BOL) versus continuous intravenous (CIV) dosing of doxorubicin (DOX) in soft tissue sarcoma (STS): post hoc analysis of a prospective randomized trialJ Clin Oncol201735supplabstr.1102310.1200/JCO.2017.35.15_suppl.11023
- WoutersKAKremerLCMMillerTLHermanEHLipshultzSEProtecting against anthracycline-induced myocardial damage: a review of the most promising strategiesBr J Haematol2005131556157810.1111/j.1365-2141.2005.05759.x16351632
- WexlerLHDeLaneyTFTsokosMIfosfamide and etoposide plus vincristine, doxorubicin, and cyclophosphamide for newly diagnosed Ewing’s sarcoma family of tumorsCancer1996784901911 Erratum appears in Cancer 1997 Feb 15;79(4):86710.1002/(SICI)1097-0142(19960815)78:4<901::AID-CNCR30>3.0.CO;2-X8756388
- VenturiniMMichelottiADel MastroLMulticenter randomized controlled clinical trial to evaluate cardioprotection of dexrazoxane versus no cardioprotection in women receiving epirubicin chemotherapy for advanced breast cancerJ Clin Oncol199614123112312010.1200/JCO.1996.14.12.31128955656
- LopezMViciPDi LauroKRandomized prospective clinical trial of high-dose epirubicin and dexrazoxane in patients with advanced breast cancer and soft tissue sarcomasJ Clin Oncol1998161869210.1200/JCO.1998.16.1.869440727
- MartyMEspiéMLlombartAMonnierARapoportBLStahalovaVMulticenter randomized phase III study of the cardioprotective effect of dexrazoxane (Cardioxane) in advanced/metastatic breast cancer patients treated with anthracycline-based chemotherapyAnn Oncol200617461462210.1093/annonc/mdj13416423847
- HortobagyiGNYapHYKauSWA comparative study of doxorubicin and epirubicin in patients with metastatic breast cancerAm J Clin Oncol198912157622643296
- PerezDJHarveyVJRobinsonBAA randomized comparison of single-agent doxorubicin and epirubicin as first-line cytotoxic therapy in advanced breast cancerJ Clin Oncol19919122148215210.1200/JCO.1991.9.12.21481960557
- MouridsenHTBastholtLSomersRAdriamycin versus epirubicin in advanced soft tissue sarcomas. A randomized phase II/phase III study of the EORTC Soft Tissue and Bone Sarcoma GroupEur J Cancer Clin Oncol198723101477148310.1016/0277-5379(87)90089-73479329
- NielsenOSDombernowskyPMouridsenHHigh-dose epirubicin is not an alternative to standard-dose doxorubicin in the treatment of advanced soft tissue sarcomas. A study of the EORTC soft tissue and bone sarcoma groupBr J Cancer19987812163416399862576
- RomJBechsteinSDomschkeCEfficacy and toxicity profile of pegylated liposomal doxorubicin (Caelyx) in patients with advanced breast cancerAnticancer Drugs201425221922410.1097/CAD.000000000000003724247203
- ChidiacTBuddGTPelleyRPhase II trial of liposomal doxorubicin (Doxil) in advanced soft tissue sarcomasInvest New Drugs200018325325910958594
- GarciaAAKempfRARogersMMuggiaFMA phase II study of Doxil (liposomal doxorubicin): lack of activity in poor prognosis soft tissue sarcomasAnn Oncol1998910113111339834828
- JudsonIRadfordJAHarrisMRandomised phase II trial of pegylated liposomal doxorubicin (DOXIL/CAELYX) versus doxorubicin in the treatment of advanced or metastatic soft tissue sarcoma: a study by the EORTC soft tissue and bone sarcoma groupEur J Cancer200137787087711313175
- SkubitzKMPhase II trial of pegylated-liposomal doxorubicin (Doxil) in sarcomaCancer Invest200321216717612743981
- WillnerDTrailPAHofsteadSJ(6-Maleimidocaproyl)hydrazone of doxorubicin–a new derivative for the preparation of immunoconjugates of doxorubicinBioconjug Chem1993465215277508268
- TrailPAWillnerDLaschSJCure of xenografted human carcinomas by BR96-doxorubicin immunoconjugatesScience (New York, NY)1993261511821221510.1126/science.8327892
- KratzFBeyerUColleryPPreparation, characterization and in vitro efficacy of albumin conjugates of doxorubicinBiol Pharm Bull199821156619477169
- KratzFBeyerURothTTransferrin conjugates of doxorubicin: synthesis, characterization, cellular uptake, and in vitro efficacyJ Pharm Sci199887333834610.1021/js970246a9523988
- MaedaHWuJSawaTMatsumuraYHoriKTumor vascular permeability and the EPR effect in macromolecular therapeutics: a reviewJ Control Release2000651–227128410699287
- MatsumuraYMaedaHA new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancsCancer Res19864612 Pt 1638763922946403
- SchillingUFriedrichEASinnHSchrenkHHCloriusJHMaier-BorstWDesign of compounds having enhanced tumour uptake, using serum albumin as a carrier–part II. In vivo studiesInt J Rad Appl Instrum B19921966856951522023
- SinnHSchrenkHHFriedrichEASchillingUMaier-BorstWDesign of compounds having an enhanced tumour uptake, using serum albumin as a carrier. Part IInt J Rad Appl Instrum B19901788198272079429
- KratzFMuller-DriverRHofmannIDrevsJUngerCA novel macromolecular prodrug concept exploiting endogenous serum albumin as a drug carrier for cancer chemotherapyJ Med Chem20004371253125610753462
- KratzFWarneckeAScheuermannKProbing the cysteine-34 position of endogenous serum albumin with thiolbinding doxorubicin derivatives. Improved efficacy of an acid-sensitive doxorubicin derivative with specific albumin-binding properties compared to that of the parent compoundJ Med Chem200245255523553312459020
- BeyerURothern-RutishauserBUngerCWunderli-AllenspachHKratzFDifferences in the intracellular distribution of acid-sensitive doxorubicin-protein conjugates in comparison to free and liposomal formulated doxorubicin as shown by confocal microscopyPharm Res20011812938 Erratum appears in Pharm Res 2001 May; 18(5):71911336350
- MeredithA-MDassCRIncreasing role of the cancer chemotherapeutic doxorubicin in cellular metabolismJ Pharm Pharmacol201668672974110.1111/jphp.1253926989862
- KratzFRothTFichinerISchumacherPFiebigHHUngerCIn vitro and in vivo efficacy of acid-sensitive transferrin and albumin doxorubicin conjugates in a human xenograft panel and in the MDA-MB-435 mamma carcinoma modelJ Drug Target20008530531810.3109/1061186000899790811328658
- PrasadVVTSGopalanROGContinued use of MDA-MB-435, a melanoma cell line, as a model for human breast cancer, even in year, 2014Npj Breast Cancer201511500210.1038/npjbcancer.2015.228721362
- GraeserREsserNUngerHINNO-206, the (6-maleimidocaproyl hydrazone derivative of doxorubicin), shows superior antitumor efficacy compared to doxorubicin in different tumor xenograft models and in an orthotopic pancreas carcinoma modelInvest New Drugs2010281141910.1007/s10637-008-9208-219148580
- KratzFFichtnerIGraeserRCombination therapy with the albumin-binding prodrug of doxorubicin (INNO-206) and doxorubicin achieves complete remissions and improves tolerability in an ovarian A2780 xenograft modelInvest New Drugs20123041743174910.1007/s10637-011-9686-521590366
- SanchezELiMWangCAnti-myeloma effects of the novel anthracycline derivative INNO-206Clin Cancer Res201218143856386710.1158/1078-0432.CCR-11-313022619306
- LebrechtDGeistAKetelsenUPThe 6-maleimidocaproyl hydrazone derivative of doxorubicin (DOXO-EMCH) is superior to free doxorubicin with respect to cardiotoxicity and mitochondrial damageInt J Cancer2006120492793410.1002/ijc.22409
- KratzFEhlingGKauffmannHMUngerCAcute and repeat-dose toxicity studies of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin (DOXO-EMCH), an albumin-binding prodrug of the anticancer agent doxorubicinHum Exp Toxicol2007261193510.1177/096032710707382517334177
- UngerCHaringBMedingerMPhase I and pharmacokinetic study of the (6-maleimidocaproyl)hydrazone derivative of doxorubicinClin Cancer Res200713164858486610.1158/1078-0432.CCR-06-277617699865
- SpethPAvan HoeselQGHaanenCClinical pharmacokinetics of doxorubicinClin Pharmacokinet1988151153110.2165/00003088-198815010-000023042244
- KratzFBeyerUSerum proteins as drug carriers of anticancer agents: a reviewDrug Deliv19985428129910.3109/1071754980906575919569996
- MitaMMNataleRBWolinEMPharmacokinetic study of aldoxorubicin in patients with solid tumorsInvest New Drugs201533234134810.1007/s10637-014-0183-525388939
- MordenteASilvestriniAMartoranaGETavianDMeucciEInhibition of anthracycline alcohol metabolite formation in human heart cytosol: a potential role for several promising drugsDrug Metab Dispos201543111691170110.1124/dmd.115.06511026265744
- ChawlaSPChuaVSHendifarAFA phase 1B/2 study of aldoxorubicin in patients with soft tissue sarcomaCancer2015121457057910.1002/cncr.2908125312684
- SankhalaKKChawlaSChuaVSPhase 1b study of aldoxorubicin + gemcitabine in metastatic solid tumorsJ Clin Oncol201634supplabstr.#252310.1200/JCO.2016.34.15_suppl.2523
- ChawlaSPPapaiZMukhametshinaGFirst-line aldoxorubicin vs doxorubicin in metastatic or locally advanced unresectable soft-tissue sarcoma: a phase 2b randomized clinical trialJAMA Oncol2015191272128010.1001/jamaoncol.2015.310126378637
- ChawlaSPGanjooKNSchuetzeSPhase III study of aldoxorubicin vs investigators’ choice as treatment for relapsed/refractory soft tissue sarcomasJ Clin Oncol201735supplabstr.#1100010.1200/JCO.2017.35.15_suppl.11000
- ChawlaSPSankhalaKKChawlaSA phase 1/2 study of continuous infusion ifosfamide/mesna+aldoxorubicin in sarcoma patientsJ Clin Oncol201634supplabstr.#e2254710.1200/JCO.2016.34.15_suppl.e22547
- EilberFCSankhalaKKChawlaSPAdministration of aldoxorubicin and 14 days continuous infusion of ifosfamide/mesna in metastatic or locally advanced sarcomasJ Clin Oncol201735supplabstr.#1105110.1200/JCO.2017.35.15_suppl.11051
- GrovesMDPortnowJBoulmayBCPhase 2 study of aldoxorubicin in relapsed glioblastomaJ Clin Oncol201634supplabstr.#202710.1200/JCO.2016.34.15_suppl.2027
- MarreroLWyczechowskaDMustoAETherapeutic efficacy of aldoxorubicin in an intracranial xenograft mouse model of human glioblastomaNeoplasia (New York, NY)2014161087488210.1016/j.neo.2014.08.015
- ParsonsCChawlaSDinhHTreatment of HIV-associated Kaposi’s sarcoma with aldoxorubicinJ Clin Oncol201534supplabstr.#11038
- ChawlaSPCranmerLDVan TineBAPhase II study of the safety and antitumor activity of the hypoxia-activated prodrug TH-302 in combination with doxorubicin in patients with advanced soft tissue sarcomaJ Clin Oncol201432293299330610.1200/JCO.2013.54.366025185097
- LaraPNRedmanMWThe hazards of randomized phase II trialsAnn Oncol20122317910.1093/annonc/mdr56722190670
- WtaVDGBlayJ-YChawlaSPPazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trialLancet (London, England)201237998291879188610.1016/S0140-6736(12)60651-5
- DemetriGDvon MehrenMJonesRLEfficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trialJ Clin Oncol201634878679310.1200/JCO.2015.62.473426371143
- DemetriGDSchöffskiPGrignaniGActivity of eribulin in patients with advanced liposarcoma demonstrated in a subgroup analysis from a randomized phase III study of eribulin versus dacarbazineJ Clin Oncol201735303433343910.1200/JCO.2016.71.660528854066