
Abstract
Acute myeloid leukemia (AML) is a clonal disorder of myeloid progenitors characterized by the acquisition of chromosomal abnormalities, somatic mutations, and epigenetic changes that determine a consistent degree of biological and clinical heterogeneity. Advances in genomic technologies have increasingly shown the complexity and heterogeneity of genetic and epigenetic alterations in AML. Among the genetic alterations occurring in AML, frequent are the genetic alterations at the level of various genes involved in the epigenetic control of the DNA methylome and histone methylome. In fact, genes involved in DNA demethylation (such as DNMT3A, TET2, IDH1, and IDH2) or histone methylation and demethylation (EZH2, MLL, DOT1L) are frequently mutated in primary and secondary AML. Furthermore, some histone demethylases, such as LSD1, are frequently overexpressed in AML. These observations have strongly supported a major role of dysregulated epigenetic regulatory processes in leukemia onset and development. This conclusion was further supported by the observation that mutations in genes encoding epigenetic modifiers, such as DMT3A, ASXL1, TET2, IDH1, and IDH2, are usually acquired early and are present in the founding leukemic clone. These observations have contributed to development of the idea that targeting epigenetic abnormalities could represent a potentially promising strategy for the development of innovative treatments of AML. In this review, we analyze those proteins and their inhibitors that have already reached the first stages of clinical trials in AML, namely the histone methyltransferase DOT1L, the demethylase LSD1, and the MLL-interacting protein menin.
Introduction
Epigenetic modification of DNA
In the nucleus of eukaryotic cells, DNA is packaged together with structural proteins (histones) to form a supramolecular complex known as chromatin. Chromatin is arranged in repeating units, known as nucleosomes: each nucleosome is formed by a sequence of DNA wound around eight histone cores. Chromatin may have a condensed conformation, known as heterochromatin, is essentially transcriptionally repressed, or a decondensed conformation known as euchromatin, and is transcriptionally active. The regulation of chromatin conformation is a fundamental biologic process and regulates, at specific sites, accessibility to DNA, a step required to allow gene transcription, replication, recombination, and DNA repair. Epigenetic mechanisms determine heritable changes in gene expression that are due to alterations in the DNA sequence. Various epigenetic mechanisms play an essential role in the control of chromatin conformation, and are mainly represented by posttranslational modifications of histone, adenosine triphosphate (ATP)-dependent nucleosome remodeling, DNA modifications, and replacement of histone with histone variants. Posttranslational modifications of histones represent one of the main epigenetic mechanisms to control gene expression.
Histones are highly basic nuclear proteins. They represent spools around which DNA winds, and play an essential role in the control of gene expression.Citation1–Citation3 There are five main families of histones: H1/H5, H2A, H2B, H3, and H4. H1/H5 are known as linker histones, while H2A, H2B, H3, and H4 are known as core histones. The linker histones bind the nucleosome at the entry and exit sites of the DNA, thus maintaining DNA in place. The core histones form dimers, and four dimers form one octameric nucleosome core. Each canonical histone protein – H3, H4, H2A, and H2B – shares a common structural domain consisting of 3α helices (α1, α2, and α3), separated by two loops (L1 and L2) called the histone fold, facilitating the heterodimerization of H2A with H2B and H3 with H4. These heterodimeric interactions form the dimeric structural unit of nucleosomes. There are variants for the core histones H3, H2AB, and H2B: the histone variant H3.3 differs from the canonical histone H3 in five amino acids required for the genomic localization of H3.3 (usually H3.3 occupies gene promoters), and H2A. Z has considerable amino-acid variation compared to H2A.
Histone proteins undergo many modifications, mainly rep-resented by lysine methylation, arginine methylation, arginine citrullination, lysine acetylation, and serine/threonine/ tyrosine phosphorylation. Each nucleosome is composed of a short segment of DNA of about 145–147 base pairs and is engulfed by a histone octamer core, consisting of four dimers of each core histone (H2A, H2B, H3, and H4). The core histone is essentially a globular structure, except for histone tails, composed of about 30 amino acids. These tails are modified by numerous posttranslational modifications, including methylation, phosphorylation, acetylation, ubiquitination, SUMOylation, ADP ribosylation, deamination, and crotonylation. These epigenetic, posttranslation modifications of histone are highly controlled, governed by three categories of enzymatic proteins: “writers” are involved in the addition of chemical groups to histone tails, “erasers” involved in the removal of these chemical groups, and “readers” are proteins that specifically recognize these histone modifications. Notable examples of writers are represented by histone acetyltransferases (HATs) and histone methyltransferases, while erasers are represented by histone deacetylases and lysine demethylases; examples of readers are given by bromodomain-containing proteins, methyl-lysine and methyl-arginine-binding domain-containing proteins and PDH-containing proteins.
Histone acetylation is a dynamic process regulated by two families of enzymes: HATs and histone deacetylases (HDACs).Citation4 HATs catalyze the transfer of an acetyl group to the ε-amino group of lysine side chains of the histone protein; these enzymes use acetyl-CoA as a cofactor. This reaction determines the shift of lysine from positively charged to a more neutral condition, reducing the affinity of the histone tail protruding from the nucleosome. As a consequence of these changes, chromatin adopts a more relaxed structure, more suitable for transcriptional activity through the binding of transcription machinery. Particularly, the BET proteins BRD2, BRD3, and BRD4 recognize and bind to the acetylated histone-lysine residues. HDACs act in an opposite way, removing the acetyl groups from histone lysines. HATs comprise many enzymes, including CREBBP (CBP), EP300 (p300), KAT2B (PCAF), KAT5 (Tip60), and KAT6A (Mo2); HDACs include HDAC1–11 and SIRT1–7.
Histone methylation is a very important biologic process through which the expression of many genes is controlled. Histones can be methylated at the level of either lysine or arginine residue.Citation5 Lysine residue can be monomethylated, demethylated, and trimethylated. Arginine residues can be monomethylated or demethylated. Methylation is a dynamic process. Methyl units to histone molecules are added by S-adenosyl-l-methionine (SAM)-dependent methyltransferase and erased by specific demethylases represented by flavin-dependent LSD1 (also known as KDM1A) and LSD2 (also known as KDM2A) or by the Jmj family of 2-oxoglutarate-dependent demethylase.Citation5
Histone-lysine methylation can lead either to transcriptional activation or transcriptional repression. Methylation of histone H3K4, H3K36, and H3K79 is associated with transcriptional activation, while demethylation and trimethylation of H3K9 and H3K27 are associated with transcriptional repression. Methylation at specific lysine residues is catalyzed by specific methyltransferases: H3K4 methylation is catalyzed by the SET domain-containing methyltransferases MLL1 (KMT2A) and MLL2 (KMT2D), H3K79 is methylated by DOT1L, H3K27 methylation is catalyzed by EZH1 and EZH2, H3K36 methylation is performed by SETD2 and WHSC1, H3K9 methylation is induced by MECOM (EVI1) and PRDM16, and H3K9 dimethylation and trimethylation is induced by SUV39H1 and EHMT2. It is important to note that methylation occurring at the level of H3K9 and H3K27 represents the two main mechanisms of gene silencing in mammalian cells. EZH1 and EZH2 make up part of PRC2 in association with two other subunits: EED and SUL12.
It is important to point out that in addition to epigenetic modifications of histone, the epigenetic modification of DNA through methylation represents another key mechanism of gene regulation.Citation6 DNA methylation is associated with transcriptional repression and occurs at the level of the C5 carbon of cytosine in DNA to form 5-methylcytosine (5-mC), through a reaction catalyzed by three types of DNA methyltransferase: DNMT1, DNMT3A, and DNMT3B. TET enzymes (TET1, TET2, and TET3) promote the oxidation of 5-mC to 5-hydroxymethylcytosine (5-hmC), promoting the demethylation process. IDH1 and IDH2 enzymes catalyze the conversion from isocitrate to α-ketoglutarate (αKG), which is required for the catalytic function of TET enzymes.
Alterations in epigenetic modifiers in acute myeloid leukemia
A considerable number of genetic studies have been performed in acute myeloid leukemia (AML), allowing identification of the most frequent genetic alterations observed in these leukemia types. Comprehensive whole-genome sequencing, exome sequencing, and targeted sequencing studies have shown that mutations at the level of the complex epigenetic gene machinery are frequent in AML. DNMT3A is mutated in 20%–25% of adult AML, and frequently involves the substitution of the amino acid arginine at position 882. DNMT3A mutation is frequently found in association with FLT3ITD, NPM1, IDH1, and IDH2 mutations.Citation7 DNMT3AR882H has dominant negative activity that markedly reduces DNA methylation. DNA hypomethylation is an initiating event in the development of DNMT3A-mutated AML and thus represents the cause and not the consequence of leukemia development. Furthermore, CpG-island hypermethylation, a phenomenon frequently observed in DNMT3AWT AML, is dramatically reduced in DNMT3A-mutated AML.Citation8 In line with these observations, conditional knock-in mice develop AML with enlarged hematopoietic stem-cell compartments and involvement of the mTOR pathway. Consequently, DNMT3A-mutated leukemic cells are sensitive to the mTOR inhibitor rapamycin.Citation9 DNMT3A mutations occur during the preleukemic phase of AML pathogenesis, supporting the key role for aberrant DNA methylation and consequent epigenetic reprogramming in malignant transformation of hematopoietic cells. The presence of DNMT3A mutations in AML is associated with poor outcome and reduced survival.Citation10,Citation11 DNMT1 is not mutated in AML, but is frequently overexpressed.Citation12 DNMT1, which methylates hemimethylated DNA, is involved in the differentiation of normal hemopoietic stem cells (HSCs) and maintenance of leukemic SCs through epigenetic silencing of genes that inhibit self-renewal and leukemogenesis.Citation13 Recent research has suggested that DNMT1A could represent a therapeutic target for some AML. In fact, DNMT1A expression can be targeted in leukemic cells by inhibitors of FABP4 (upregulated in AML and stimulates DNMT1A expression in these cells)Citation14 or by inhibitors of receptor tyrosine kinases.Citation15 These treatments result in inhibition of tumor growth, induction of cell differentiation, and impairment of leukemic progress in leukemia animal models.Citation14,Citation15 Very interestingly, a recent study provided evidence that MUC1-C, a transmembrane oncoprotein aberrantly expressed in leukemic SCs (where it is coexpressed with DNMT1), drives DNMT1 transcription.Citation16 Targeting MUC1-C with a specific monoclonal antibody, together with the DNMT1 inhibitor decitabine, markedly reduces DNMT1 expression and impairs the survival of AML cells.Citation16
The ASXL1 gene encodes a chromatin-binding protein and is mutated in about 3%–5% of AML. The incidence of these mutations is higher in patients with intermediate risk and particularly with high-risk and secondary AML, where it is mutated in about 16% of patients. ASXL1 mutations, as a single prognostic factor, are associated with a negative outcome.Citation17 ASXL1 gene mutations are particularly frequent (20%) in RUNX1-mutated AMLCitation18 and are often associated with myelodysplasia-related changes.Citation19
EZH2, a catalytic component of PRC2, which mediates transcriptional silencing through di- and trimethylation of lysine 27 of histone H3, playing an important role as a histone–lysine N-methyltransferase enzyme, is mutated in about 2% of adult AML, where inactivating mutations are usually observed.Citation20 Some AML displays a decrease in EZH2 protein levels due to a posttranslational mechanism triggered by EZH2 phosphorylation induced by CDK1.Citation21 EZH2 inactivation is frequently observed at disease relapse, and is associated with HOX gene derepression and resistance to multiple drugs.Citation21 EZH2 loss is frequently associated with a decrease in H3K27me3 levels.Citation21 EZH2 mutations are more frequent in RUNX1-mutatedCitation18 and secondary AML, and are often associated with increased HOXA9 expression.Citation22
About 3%–5% of de novo AML patients display partial tandem duplication of the MLL gene, which is characterized by internal tandem duplication of exons 3–9 or 3–11.Citation23 MLLPTD acts as an oncogene by upregulating the expression of HOX genes. These AML types display frequent mutations of other epigenetic regulators, such as TET2 (16%), EZH2 (10%), IDH1/2 (31%), and ASXL1 (6%). Furthermore, a typical feature of MLLPTD consists of the absence of NPM1 mutations and frequent RUNX1 (23%) and STAG2 mutations (16%).Citation23 BCOR is a component of the variant-group polycomb-repressive complex, mutated in about 4% of karyotype-normal AML. BCOR plays an important role in the control of hematopoiesis by inhibiting myeloid-cell proliferation and differentiation and regulates HOX gene expression.Citation24
Interestingly, in a recent molecular classification of AML based on the analysis of a large set of samples, one of the largest groups was represented by AML with mutated chromatin, RNA-splicing genes or both, characterized by mutations of genes regulating chromatin (ASXL1, STAG2, BCOR, MLLPTD, and PHF6), RNA splicing (SRSF2, SF3B1, U2AF1, and ZRSR2), or transcription (RUNX1).Citation25 As such, no single molecular lesion defines this group. Interestingly, overlap-ping patterns of comutations are observed among the defining genes of this AML group.Citation25 It is important to note that as stated earlier, this AML subgroup also contains AML types with mutations of the splicing-factor machinery. Interestingly, mutations at the level of these genes interfere with pre-mRNA splicing of genes that are involved in the epigenetic machinery, such as BCOR, MLL2, and EZH2, and through this mechanism affect hematopoiesis.Citation26,Citation27 It is important to underline that most patients in the chromatin-spliceosome group were older, with lower white-cell and blast-cell counts and response rates and negative long-term clinical outcome.Citation25 Therefore, it is evident that most of these patients must be classified as high-risk AML.Citation28
About 3% of de novo adult AML harbors a translocation of the MLL1 gene fused to various partners, including AF4, AF9, ENL, AF10, ELL and AF6; secondary MLL-rearranged AML is observed in patients treated with topoisomerase inhibitors.Citation29 These AML types have a negative prognosis, and are thus classified as high-risk AML. The main pathogenic mechanism of these AML types is related to the capacity of the MLL-fusion proteins to aberrantly regulate MLL-target genes, such as HOXA and MEIS1, and to modify the genetic program of proliferation and differentiation of HSC/HPC.Citation30
TET2 is a protein that acts as an epigenetic modifier to convert methylcytosine to hydroxymethylcytosine. This protein is mutated in about 10%–20% adult AML,Citation31 and its frequency is significantly higher in older AML patients.Citation32 TET2 mutations in AML were not associated with distinct clinical or genetic features, except for IDH mutations, which were almost mutually exclusive with TET2 mutations.Citation33 At the clinical level, it is unclear whether the presence of TET2 mutations represents a factor affecting patient out-come. The mutation of such genes as TET2, IDH1, and IDH2 causes defective conversion of 5-methylcytosine to 5-hmC, impairing demethylation of DNA. Recent immunocytochemical and biochemical studies have shown that AML with TET2 mutations shows reduced 5-hmC levels; however, 5-hmC levels were not predictive of survival in AML patients with normal-karyotype AML.Citation34,Citation35 Importantly, TET2 mutations are found also in the white blood cells of otherwise-normal adults with clonal hematopoiesis, a condition related to aging and associated with myeloid-lineage bias and increased risk of development of myelodysplastic syndrome (MDS) or AML.Citation36 These observations have led to a hypothesis that TET2 mutations represent a preleukemic abnormality required for the initial steps of leukemic trans-formation, enabling disease progression. In line with this hypothesis, a recent study provided evidence that TET2 mutations are essential to induce the survival and aberrant self-renewal of leukemic SCs.Citation37 Interestingly, vitamin C, able to enhance 5-hmC in TET2-deficient cells, drives DNA hypomethylation, induces the expression of a TET2-dependent gene signature, inhibits colony formation of TET2-mutated human leukemic cells, and blocks leukemia progression in primary leukemia patient-derived xenografts.Citation37 Finally, vitamin C strongly synergizes with PARP inhibitors to induce the killing of TET2-mutated leukemic cells.Citation37
The NADP+-dependent isocitrate dehydrogenases (IDH1 and IDH2) are critical metabolic enzymes involved in the interconversion of isocitrate to αKG. Recurrent somatic mutations of IDH1 occur in 6%–10% of adult AML cases: these mutations affect the arginine residue at position 132 or 170. IDH2 mutations occur in 8%–12% of adult AML, affecting the arginine residue at position 140 or 172. Leukemia-associated IDH1/IDH2 mutations confer the neomorphic activity of reducing αKG to the oncometabolite 2-hydroxyglutarate. The accumulation of this oncometabolite inhibits αKG-dependent dioxygenases, including histone demethylases and methylcytosine dioxygenases of the TET family.Citation38 The consequent epigenetic deregulation results in DNA and histone hypermethylation, altered gene expression, and blocked cell differentiation.Citation28 The presence of IDH mutations does not confer specific properties to leukemic cells, apart from IDH2Citation172 mutations. In fact, IDH2Citation172-mutant AML represents a subgroup of AML corresponding to about 1% of all AML typesCitation25 and displaying gene-expression and DNA-methylation profiles that differ from the profiles for other IDH mutations, and display peculiar aberrations in metabolic activity.Citation39,Citation40 Specific inhibitors of mutant IDH1 and IDH2 enzymes have been developed and introduced into clinical trials. These inhibitors have demonstrated a remarkable single-agent activity in relapsed/refractory AML patients. Patients with relapsing/refractory IDH2-mutant AML displayed in 40% of cases a clinical response to treatment with enasidenib (AG221), an IDH2-mutant-specific inhibitor. Importantly, 19% of these patients achieved a complete response, with median survival of about 20 months.Citation41 Interestingly, the responding patients displayed blast-cell leukemic-cell differentiation.Citation41 Mutations of some genes involved in the epigenetic DNA machinery are frequent in AML, and among these mutations, TET2, ASXL1, and DNMT3A are associated with reduced overall survival.Citation42
In parallel with these observations, other studies have analyzed the occurrence of gene mutations in AML at the clonal level and during the history of disease (ie, at diagnosis and relapse). The results of these analyses have defined the clonal evolution of the AML process. The most relevant results of these studies showed that mutations in genes encoding epigenetic modifiers, such as DNMT3A, TET2, ASXL1, IDH1, and IDH2, were acquired during the early steps of leukemia development and were present in the founding clone, while other mutations, such as those involving NPM1, FLT3, and RAS, were secondary events occurring at late steps of leukemic development. Importantly, these mutations of epigenetic modifiers are observed also in aging individuals in the context of clonal expansion of hematopoiesis, a preleukemic condition associated with an increased risk of developing leukemia. Furthermore, these mutations usually persist after therapy, lead to clonal expansion during remission, and contribute together with new mutations to disease relapse.Citation43,Citation44 The analysis of large sets of older people of different geographical regions without evidence of hematologic malignancies has confirmed a high incidence of clonal hematopoiesis (5%–10% at 70 years; about 20% at 90 years), and has shown that the most commonly mutated genes are DNMT3A, TET2, and ASXL1.Citation45,Citation46
Several recent studies have explored the pattern of epigenetic changes occurring in the various AML types and attempted to classify these leukemias according to this pattern and define possible associations between these epigenetic changes and patient outcome. Large-scale genome-wide DNA-methylation profiling has revealed the existence of distinct DNA-methylation patterns in AML, thus indicating that these leukemias are composed of epigenetically distinct diseases.Citation47 Different cytogenetic and molecular AML subtypes were found to display different DNA-methylation profiles.Citation47–Citation50 AML characterized by specific translocation events, such as t(8;21)-AML1/ETO, inv(16)-t(16;16)-CBFB-MYH11, t(15;17)-PML-RARα, and t(v;11q23)-MLL, was characterized by unique DNA-methylation signatures.Citation47–Citation50 The group of NPM1-mutated AML was heterogeneous for the methylation pattern: four methylation clusters were identified, one hypermethylated and three both hyper- and hypomethylated,Citation47 this methylation heterogeneity seemingly reflecting the molecular heterogeneity of this karyotype-normal AML subgroup and dictated by the co-occurrence of DNMT3A, FLT3, and IDH1/IDH2 mutations.Citation47,Citation48 DNMT3A mutations and particularly DNMT3AR882 mutations were associated with hypermethylation.Citation51 TET2 and IDH1/IDH2 mutations were associated with a genome-wide hypermethylation signature, particularly pronounced for IDH1/IDH2-mutated AMLs.Citation51,Citation52
Various studies have supported a possible role of DNA methylation pattern as a prognostic index for predicting clinical outcome in AML patients. In this context, particularly interesting was a recent study by Luskin et al. These authors performed a multilocus DNA assessment using an xMELP assay and calculated a methylation statistic (M score), showing that: the M score was lower in patients surviving after 2 years compared to that observed in dead patients, and the same applies for complete remission; low-M-score AML patients had better overall survival than high-score patients; and the M score was not associated with established molecular markers, such as NPM1 and FLT3ITD mutations, but was clearly associated with mutations in DNMT3A and IDH1.Citation53
In this context, Li et al explored epigenetic heterogeneity and possible links among genetic heterogeneity, genetics, and epigenetics in AML.Citation54 They observed that the degree of methylation variation, defined as epiallele burden, at the same loci between samples represented a predictor of relapse risk among AML patients. Patients were subdivided according to the level of allelic burden, and those with high allelic burden relapsed more rapidly, compared with those with low epiallele burden. Importantly, the prognostic significance of the level of allelic burden was independent of cytogenetics and white-blood-cell counts at diagnosis. Comparison between AML blasts and normal bone-marrow cells showed that 100% leukemic samples displayed epigenetic allele shifting com-pared to normal bone marrow. Furthermore, 92% of patients showed epigenetic allele shifting between diagnosis and relapse. Importantly, there was no link between epigenetic burden and mutation burden. Finally, there was no increase in genes that regulate methylation (DNMT1, DNMT3A, TET1, TET2, IDH1, IDH2) in those samples with higher level of epiallele burden.Citation54
Constant advancements in the identification of molecular abnormalities of AML has allowed the proposal of new classifications of AML neoplasia, encapsulating information on genetic abnormalities, morphology, immunophenotype, and clinical presentation. The first French–American–British classification defined eight AML subtypes (M0–M7) according to morphological and cytochemical features: M0 (undifferentiated acute myeloblastic leukemia), M1 (acute myeloblastic leukemia with minimal maturation), M2 (acute myeloblastic leukemia with maturation), M3 (acute promyelocytic leukemia [APL]), M4 (acute myelomonocytic leukemia), M4E (acute myelomonocytic leukemia with eosinophilia), M5 (acute monocytic leukemia), M6 (acute erythroid leukemia), and M7 (acute megakaryoblastic leukemia). In 2008, the World Health Organization (WHO) introduced a new classification based on the integration of molecular and cellular criteria.Citation55 Finally, in 2016 a new revised updated version of the WHO classification of myeloid neoplasia was released.Citation56
The integration of cytogenetic and molecular criteria allows the stratification of AML into three prognostic subgroups: 1) favorable prognosis for various AML subtypes identified according to the cytogenetic and molecular features t(8;21) with no c-Kit mutations, inv(16), t(15;17), mutated NPM1 without FLT3ITD (karyotype cytogenetically normal [CN]-AML), and mutated biallelic CEBPA (CN-AML); 2) intermediate prognosis, including t(8;21) with c-Kit mutation, t(9,11), CN-AML other than those included in the favorable or adverse prognostic group, and cytogenetic abnormalities not included in the favorable or adverse prognostic group; and 3) adverse prognosis, including TP53 mutation, regardless of cytogenetic profile, CN-AML with FLT3ITD; CN-AML with DNMT3A, CN-AML with KTM2APTD, inv(3), t(6,9), −5, or del(5q), −7, complex karyotype, and 11q abnormalities other than t(9;11).
A recent studyCitation25 based on a very large set of primary adult AML (1,540) patients proposed a detailed molecular classification of these neoplasia, with the identification of 13 subgroups:
AML with NPM1 mutations (about 27%), frequently displaying also DNMT3A, FLT3ITD, NRAS, and TET2 mutations
AML with mutations in genes encoding chromatin, RNA-splicing genes, or both (about 18%), including RUNX1, MLL, SRSF2, DNMT3A, ASXL1, and STAG2 mutations
AML with TP53 mutations, chromosome aneuploidy, or both (about 13%), including TP53 mutations, complex karyotype −5/5q, −7/7q, −12/12p, and +8/8q
AML with inv(16) or t(16;16), CBFB–MYH11 (about 9%)
AML with biallelic CEBPA mutations (about 4%)
AML with t(15;17), PML-RARα (about 4%)
AML with t(8;21)(q22;q22), AML1–ETO (about 4%)
AML with MLL fusion genes, t(x;11)(x;q23) (about 3%)
AML with iv(3) or t(3;3), GATA2, MECOM (about 1%)
AML with IDH2R172 mutations (about 1%)
AML with driver mutations, but no detected class-defining lesions (about 11%), frequently displaying FLT3ITD or DNMT3A mutations
AML with no detected driver mutations (4%)
AML meeting criteria for multiple genomic subgroups (4%).
Adult AML patients with AML usually receive a standard treatment based on 7 days of cytarabine and an anthracycline for 7 days. Using this standard treatment, some groups of AML patients have an approximate “cure” probability (favorable risk), whereas other groups have a survival comprised in a range of 6–18 months. First-generation epigenetic drugs, such as azacitidine and decitabine, currently used for the treatment of MDS, are also used for treating AML patients not eligible for treatment with intensive chemotherapy and with stem-cell transplantation. Under this epigenetic treatment, only 15%–20% of patients display a complete response. More recently, guadecitabine was introduced, a next-generation hypomethylating agent that is not metabolized by cytidine deaminase and the enzyme that degrades decitabine. A recent study showed that >50% of treatment-naïve AML patients >65 years old (77 years mean age) displayed a complete response to treatment with guadecitabine.Citation57 Responding patients displayed a median survival >500 days, with an acceptable drug-related toxicity profile. Analysis of the various types of patients enrolled in this study provided evidence about the existence of genetic determinants underlying the response of AML patients to guadecitabine. Patients with RAS and IDH2 mutations had much less chance of getting a complete response to guadecitabine treatment than those without these mutations. In contrast, patients with mutations of other epigenetic regulators, such as DNMT3A, ASXL1, EZH2, TET2, U2AF1, or WT1, had a comparable chance of developing a complete response, as well as those with AML without these mutations.Citation58 Furthermore, the frequency of complete responders was higher among patients with naïve AML (56%) than those with relapsed–refractory AML (22%).Citation58 Although patients with relapsed–refractory AML displayed a lower rate of complete responses, responder patients displayed long-term survival, and their overall survival was significantly better than that observed for nonresponder patients.Citation59 In the whole AML-treated population, 19% of patients survived after 2 years; median overall survival was 6.5 months among nonresponder patients and >29 months in responder patients.Citation59 These results are encouraging, and strongly support the use of hypomethylating agents in older AML patients.
AML with TP53 mutation has a very negative prognosis; these leukemias are usually associated with adverse karyotypes and are more frequent among older AML patients. Welch et al treated 88 AML patients with a 10-day regimen of decitabine and reported high rates of morphological remission (46%).Citation60 Interestingly, they noted higher response rates among AML patients with an unfavorable cytogenetic profile than among those with intermediate or favorable cytogenetic profiles (67% versus 37%) and among patients with TP53 mutations compared to those without TP53 mutations (100% versus 41%).
Two recent studies have explored the clinical activity of guadecitabine in high-risk myelodysplasia patients refractory or relapsing after azacitidine and observed only modest response rates.Citation61 The second study explored a population of untreated high-risk myelodysplasia, and provided evidence that guadecitabine was active in this set of patients (with 28% complete responses), even in patients with adverse biologic features, such as high frequency of complex karyotype, therapy-related disease, and TP53 mutations.Citation62 Targeting epigenetic abnormalities could represent a potentially promising strategy for the development to innovative AML treatments. Here, we analyze some epigenetic modifiers and their inhibitors, focusing on those that have reached the first stages of clinical trials in AML.
LSD1
Structure and function
Histone methylation is a dynamic process. Histone lysine and arginine residues are N-methylated at the level of H4; some nonhistone proteins, such as p53, can be also methylated. The effect of methylation on gene transcription is variable in that it can result either in induction or repression of gene transcription, depending on the extent of methylation and the position of the methylated residue. Therefore, examples of methylation typically associated with active gene expression are given by methylation of lysine 4 or 36 of histone H3, while examples of repressive methylation are represented by methylation of lysine 9 or 27 of histone H3. The combination of all the histone-modification events, including methylation, acetylation, phosphorylation, and ubiquitination, determine the final chromatin conformation and the transcriptional activation of a given gene. More than 20 lysine demethylases have been discovered, pertaining to two main gene families: the KDM1 subfamily containing the LSD enzymes and the KDM2–KDM7 subfamilies, consisting of JmjC-containing enzymes. The KDM1 LSDs are mainly represented by LSD1 and LSD2, which are flavin-dependent amine oxidases related to monoamine and polyamine oxidases: these enzymes are dependent on a single electron pair within the lysine for catalysis and consequently can demethylate only mono- and dimethylated lysines. In contrast, JmjC KDMs are Fe2+- and 2-oxoglutarate-dependent deoxygenases capable of demethylating mono-, di-, and trimethylated lysines. LSD1 is specifically involved in the demethylation of monomethylated and dimethylated lysine 4 residues on histone 3. LSD1 can demethylate the lysine residues also of some nonhistone proteins, such as p53 and DNMT1. LSD1 can also methylate H3K9 when it is complexed with the androgen receptor.Citation63 LSD2 specifically demethylates histone H3K4 me1–2 inside areas of its target genes.
The molecular structure of LSD1 and LSD2 proteins has been determined. LSD2 is a homologue of LSD1 based on its sequence, and shares about 30% sequence similarity with LSD1. Several structural/functional domains have been characterized. Some of these domains are present in both LSD1 and LSD2 proteins: an AOL domain, essential for enzymatic activity (the AOL domain has two lobes, one forming a noncovalent FAD-binding site and the other a substrate-binding and -recognition site), and a SWIRM domain, typically observed in chromatin-associated proteins (the SWIRM domain is involved in protein–protein interactions; it is also involved in the association with the androgen receptor). Other domains are specific to LSD1, such as the TOWER domain, while others are specific to LSD2, such as the amino zinc-finger domain, whose function is unclear ().Citation64 As mentioned, the catalytic activity of both LSD1 and LSD2 enzymes is in the AOL domain and requires the cofactor FAD.
Figure 1 Schematic representation of enzymatic activity and linear structure of human LSD1 and LSD2.
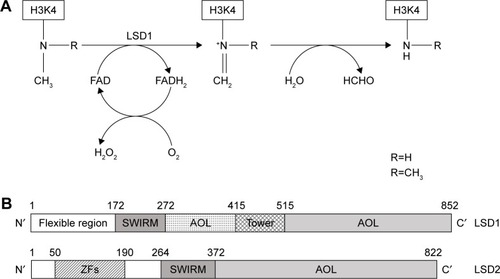
The determination of the three-dimensional structure of LSD1 considerably helped in the understanding of the function of this protein at the molecular level. LSD1 forms a highly symmetric, packed domain structure from which a long helical tower domain protrudes. The SWIRM domain, which contributes to the stability of the protein, packs together with the AOL domain through numerous interactions. The active site cavity present at the level of the AOL domain is spacious and capable of accommodating several residues of the histone-tail substrate.Citation65,Citation66
The chemical demethylation reaction catalyzed by LSD1 is complex. During all the reactions catalyzed by LSD1, at each demethylation cycle a molecule of formaldheyde and H2O2 is produced and O2 is consumed. The chemical reaction involves the initial conversion of methylated lysine to an iminium cation by loss of a hydride anion captured by the oxidized FAD prosthetic groups (). The imine cation is then hydrolyzed to produce a carbinol amine, decomposing to formaldehyde, and the demethylated residue. The reduced FAD produced during the initial step of the reaction is reoxidized by O2 to generate a molecule of H2O2 and regenerated oxidized FAD ().Citation67,Citation68
The capacity of LSD1 to form molecular complexes with other nuclear proteins and transcriptional factors is an essential property of its activity and the regulation thereof. In this context, it is important to point out that LSD1 was initially discovered as a molecular partner of HDAC2 in HeLa cells.Citation69 Subsequently, many other studies have shown that LSD1 participates in the formation of multisubunit complexes involving LSD1, CoREST (also known as RCOR1), HDAC1, HDAC2, ZNF217, PHF21A and HMG20B. This complex is commonly known as the CoREST transcription-repressor complex.Citation70,Citation71 The functional implications of all these interactions are not completely defined. However, the LSD1–CoREST interactions are required to protect LSD1 from proteosomal degradation, while the association with HDAC1/HDAC2 is required for both demethylase and HDAC activity.Citation72 The molecular protein complex formed by LSD1 was covered in two recent reviews on LSD1.Citation73,Citation74
The LSD1 gene contains 19 exons, highly conserved among vertebrates. Through a process of alternative RNA splicing, two additional exons, E2a and E8a, can be included into mature mRNA and generate four possible LSD1 isoforms, called LSD1, LSD1-2a, LSD1-8a, and LSD1-2a+8a.Citation75 The inclusion of exon E2a can occur in all tissues, while LSD1 transcripts containing E8a are found only in brain and testis.Citation73 While insertion of the 60-nucleotide-long E2a exon does not modify the enzymatic activity of LSD1, insertion of the 12-nucleotide-long exon E8a could modify LSD1 enzymatic activity (in fact, the LSD1-8a isoform plays a role in mediating H3K9 and H4K20 demethylation).
Role of LSD1 in normal hematopoietic differentiation
Gene-knockout studies have contributed greatly to our understanding of the role of LSD1 in the control of normal hematopoiesis. Conditional knockout studies have shown that a deficit of LSD1 expression in the hematopoietic system determines an expansion of HSCs and hematopoietic progenitor cells (HPCs) not associated with a significant increase in marrow cellularity and only a slight increase in white blood cells.Citation76 Analysis of bone marrow of LSD1-deficient animals showed an inhibition of granulopoiesis and a stimulation of monocytopoiesis, thus suggesting that LSD1 plays an important role in lineage choice during granulo/monocytic differentiation.Citation76 Analysis of erythroid-cell lineage has shown that LSD1 knockdown results in anemia associated with perturbed terminal erythropoiesis and expansion of early erythroid progenitors.Citation70 Other studies have supported the role of LSD1 as an indispensable epigenetic governor of hematopoietic differentiation.Citation77 This function is mainly exerted through the capacity of LSD1 to repress hematopoietic stem and progenitor gene-expression programs during hematopoietic cell differentiation.Citation77 Deficient LSD1 expression causes a failure to silence HSC and HPC genes fully, compromising the different steps of the hematopoietic differentiation-cell program.Citation77
The activity of LSD1 during hematopoietic differentiation and its regulatory effects on hematopoiesis are regulated by its interaction with CoREST/REST (Rcor) corepressors. Upadhyay et al showed that the three Rcor proteins (Rcor1, Rcor2, and Rcor3) regulate LSD1 activity and cellular differentiation in hematopoietic cells. All the three Rcor proteins interact with LSD1 and the transcription factor Gfi1b: while Rcor1 and Rcor2 facilitate LSD1-mediated nucleosome demethylation, Rcor3 inhibits demethylation.Citation78 In line with these observations, Rcor1 and Rcor2 favor differentiation, while Rcor3 inhibits differentiation.Citation78 Rcor1 and Rcor3 levels are differentially regulated in erythroid and megakaryocytic cells during terminal stages of maturation.Citation78
The capacity of LSD1 to interact with some transcription factors acting as key master regulators of hematopoiesis is essential to explain its effects on hematopoietic differentiation. In hematopoietic cells, LSD1 and Rcor1 associate with Gfi1 and Gfi1b and repress most Gfi1b targets in erythroid cells.Citation79 Gfi-transcription factors are master regulators of hematopoietic cell differentiation: Gfi1 is essential for neutrophil differentiation, while Gfi1b is required for the development of erythroid and megakaryocytic lineages.Citation80 Furthermore, LSD1–Rcor1 also associates with other transcription factors, such as Scl1/Tal1,Citation81,Citation82 Bcl11A,Citation83 and GATA2,Citation84 in erythroid cells, inducing repression of their target genes. The excessive and uncoordinated function of all these transcription factors induced by LSD1 deficiency in HPCs determines a derangement of erythroid and megakaryocytic cell differentiation. The interaction between Gfi1 and LSD1 involves the SNAG domain of Gfi1: this domain needs to be methylated at the level of lysine 8 for optimal binding capacity to LSD1.Citation85
Finally, recent studies provided evidence that LSD1 plays a relevant functional role at the level of events that determine the initial specification of hematopoietic cell lineage during embryonic development. In vertebrates, the initial appearance of HSCs with long-term repopulation and multilineage-differentiation capacities occurs at the level of the aorta–gonad–mesonephros region. This primitive population of HSCs is generated from a rare subpopulation of endothelial cells, known as hemogenic endothelia and is capable of transdifferentiation through a process of endothelial to hematopoietic transition. This unique transdifferentiation process requires a molecular orchestration requiring from one side the repression of the endothelial differentiation program and from the other side the induction of the hematopoietic differentiation program. Gfi1 and Gfi1b proteins mark the hemogenic endothelia, and are strictly required for the hemogenic activity of the hemangioblasts: the Gfi proteins, through the recruitment of LSD1 protein, exert their repressive effects on the endothelial differentiation program.Citation86 Furthermore, LSD1 activity in the hemangioblast is essential for the inhibition of the endothelial differentiation program through downregulation of the transcription factor Etv2, an essential regulator of vasculogenesis.Citation87
Gfi1 and Gfi1B act as transcriptional repressors by recruiting histone-modifying enzymes to promoters and enhancers of target genes, and thus can be considered epigenetic regulators that modify chromatin structure. Several rare hematological diseases are associated with acquired or inheritable mutations in the GFI1 and GFI1B genes; particularly, some patients with severe congenital neutropenia carry mutations in GFI1 that determine the disruption of the C-terminal zinc-finger domains.Citation88 Furthermore, recent studies have suggested a possible role of GFi1 in human leukemias. Hones et al analyzed a large number of AML samples for GFi1 expression, showing that about 10% of these expressed low GFi1 levels.Citation89 These leukemia types have a poor outcome and frequently display an adverse cytogenetic FABM0 phenotype, NRAS mutations, elevated EVI1 expression, and a leukemic stem-cell signature at the level of the gene-expression profile.Citation83 In experimental mouse models, low GFi1 expression accelerates leukemia development driven by oncofusion proteins, such as MLLAF9.Citation89 Low GFi1 confers sensitivity of leukemic cells to histone methyltransferase inhibitors, associated with resistance to HDAC inhibitors.Citation89 Recently, Volpe et al analyzed AML with normal karyotype for GFi1 expression and observed that those displaying high expression of this transcription factor have frequent FLT3ITD and NPM1 mutations, display an FLT3ITD signature, and exhibit high expression of some leukemia-related genes, such as HOXA9, MEIS1, and PBX3.Citation90 At variance with the findings of Hönes et alCitation89 obtained in the whole AML population, in karyotype-normal AML, high GFi1 expression was associated with a worse outcome.Citation90
Very interestingly, mice deleted for Rcor1 were shown to be markedly anemic, showing a block of erythroid pre-cursors at the transition from proerythroblasts to basophilic erythroblasts.Citation91 Erythroid progenitors purified from Rcor1-null bone marrow cultured in vitro form myeloid colonies, but fail to form erythroid colonies;Citation91 mutant proerythroblasts aberrantly express genes typically expressed in myeloid cells and in SCs/PCs.Citation91 Evaluation of the myelomonocytic lineage of Rcor1−/− animals showed absence of mature neutrophils associated with an increase in monocytes, a feature observed also in LSD1−/− mice.Citation92
LSD1 in leukemia
LSD1 is not mutated in acute leukemias or in other blood neoplastic disease. However, LSD1 is overexpressed in many hematologic diseases, including AML, acute lymphoblastic leukemia (ALL), myeloproliferative neoplasms, chronic myelomonocytic leukemia, and MDS.Citation93 In AMLs, LSD1 was overexpressed in about 60% of cases.Citation93
Using a model of human MLLAF9 leukemia, Harris et al identified LSD1 as a key regulator of leukemic stem-cell potential, in that this demethylase acts at the level of the genomic loci bound by MLLAF9, favoring the effect of the MLL-fusion protein and thus preventing cell differentiation and apoptosis.Citation94 In line with this finding, primary AML cells bearing various MLL rearrangements were shown to be markedly inhibited by small-molecule LSD1 inhibitors.Citation94 Interestingly, LSD1 was expressed in MLL-rearranged AML, as well as in other molecular subtypes of AML. In addition to MLL-rearranged AML, AML associated with PML-RARA and RUNX1–RUNX1T1 was shown to be sensitive to the LSD1 inhibitors.Citation94 Consistent with these findings, LSD1 was found among the 5% most highly expressed genes in prospectively purified immunophenotypic leukemic SCs from a variety of distinct AML subtypes.Citation95
According to these observations, elevated LSD1 expression in AML and in other hematological neoplasia may contribute to leukemia development. To test this hypothesis, Wada et al evaluated the effect of LSD1 overexpression in HSCs and HPCs by generating transgenic mice that over-express LSD1 in HSCs/HPCs under control of the Sca1 promoter.Citation96 First, these authors showed that among acute leukemias LSD1 is expressed at the highest levels in T-cell ALL.Citation88 The overexpression of the short LSD1 isoform, which lacks E2a and E8a, induced a marked increase in the self-renewal activity of HSCs via upregulation of HOXA genes, but retained multidifferentiation capacities. Transgenic mice overexpressing LSD1 did not develop any hematological neoplasia; however, these mice developed high frequency T-cell ALL after γ-irradiation.Citation96
Schenk et al explored the capacity of LSD1 inhibitors to confer to non-APL cells the capacity to differentiate in response to all-trans retinoic acid (ATRA). In fact, while APL cells have the remarkable property to differentiate under treatment with ATRA, non-APL cells fail to undergo differentiation. Schenk et al showed that non-APL cell lines, as well as primary AML cells, undergo granulocytic differentiation when incubated with ATRA and an LSD1 inhibitor.Citation97 Analysis of leukemic cells treated with LSD1 inhibitors showed that these drugs did not lead to a large-scale increase of H3K4me across the genome, but increased H3K4me and gene expression at the level of selected genes, particularly some genes involved in myeloid differentiation.Citation97
LSD1 inhibitors
The development of specific LSD1 inhibitors has been the objective of many recent studies. These inhibitors represent a fundamental tool to improve our understanding of the role of LSD1 in normal and pathological conditions, and offer a unique strategy to develop preclinical studies to evaluate the therapeutic impact of LSD1 inhibition in animal models of various pathologic conditions and to translate these studies in the clinic.
Some recent papers have analyzed in detail the structure and mechanism of action of the main LSD1 inhibitors that have been developed in the last 10 years.Citation95 Here, we analyze the main LSD1 inhibitors and particularly those under clinical development, with emphasis on studies focused on AML treatment (). According to Niwa and Umehara, LSD1 inhibitors can be subdivided into two main groups: irreversible covalent inhibitors and reversible noncovalent inhibitors.Citation98 The irreversible covalent inhibitors can be subdivided into three subgroups: (±)-trans-2-phenylcyclopropylamine (2-PCPA), N-alkylated 2-PCPA derivatives, and inhibitors other than 2-PCPA derivatives.
Table 1 Main LSD1 inhibitors introduced into a plan of clinical development
Studies on the MAO inhibitor tranylcypromine have led to the discovery that this compound exhibited better LSD1-inhibitory activity than the other MAO inhibitors known at the time.Citation99 It is important to briefly discuss the properties of Tranylcypromine (TCP), the compound initially investigated for its capacity to inhibit LSD1. TCP was approved as a drug for the treatment of depression. In combination with ATRA, TCP is under exploration in three clinical trials in AML patients: TCP in combination with ATRA in AML patients who cannot undergo chemotherapy (NCT2261779), TCP at four doses (10, 20, 40, and 60 mg) in combination with ATRA in AML and MDS patients (NCT2273102), and TCP in combination with ATRA and the chemotherapy agent cytarabine (DRKS00006055) ().
The 2-PCPA scaffold represents the basic structure used for the development of the large majority of irreversible LSD1 inhibitors.Citation100 The introduction of various chemical modifications to this basic structure has led to the identification of many new compounds, some exhibiting higher LSD1-inhibitory activity and a reduced MAO-inhibitory activity.Citation98 Particularly, chemical substitutions on the phenyl ring of tranylcypromine have considerably improved the potency and selectivity of these LSD1 inhibitors.Citation99
Most LSD1 inhibitors under clinical development pertain to the family of N-alkylated 2-PCPA derivatives. This category of compounds was discovered by Oryzon Genomics (Barcelona, Spain), showing that LSD1 inhibition is markedly improved by the development of N-alkylated 2-PCPA derivatives, exhibiting LSD1-inhibitory potency in the nanomolar range. One of these compounds, ORY1001, displayed potent LSD1-inhibitory activity (IC50 of about 18 nM) and specificity (>1,000-fold selectivity over MAOs and LSD2).Citation97 A cellular assay on the THP1 cell line (MLL – AF9 cells) displayed pharmacologic activity of ORY1001 (as evaluated by methylation and cell differentiation assay) at subnanomolar concentrations.Citation100 Leukemic cells with MLL translocations were particularly sensitive to the inhibitory effects of this compound. Daily oral doses <20 μg/kg have shown a potent antileukemic effect in mice transplanted with MV (4;11) cells.Citation100 Finally, preclinical pharmacologic and toxicologic studies showed good oral bioavailability and a good safety profile.Citation101 Given this favorable pharmacologic profile, ORY1001 entered a plan of clinical development screening to investigate its safety profile and possible antitumor effects in patients with non-small-cell lung cancer and AML ().
A preliminary report on the ongoing Phase I/II clinical study on AML patients (EUDRACT 2013-002447-29) was presented at the last meeting of the American Society of Hematology in December 2016. Patients with refractory/ relapsed AML were enrolled in this study, and the dose-escalation section allowed definition of a recommended dose of ORY1001 for the expanded phase, of 140 μg/m2/day.Citation102 At this dose, the drug was relatively well tolerated, and the most frequent adverse events were asthenia, febrile neutropenia, constipation, and peripheral edema.Citation102 Importantly, ORY1001 was evaluated in an extension cohort of 14 AML patients with relapsing AML subtypes predicted to be more sensitive according to preclinical studies (ie, ten AML MLL/ translocated and four acute erythroleukemia/M6). Objective responses were seen in 36% and blast-cell differentiation in blood observed in 64% of these patients.Citation102 These observations further support additional studies exploring the anti-leukemic effects of ORY1001.
After the end of this Phase I study in AML, further development of ORY1001 will be carried out by Roche (Basel, Switzerland) and the compound name will be RG6016. RG6016 will be additionally evaluated in AML patients and in small-cell lung cancer patients in the context of a Phase I study (NCT2913443). Another LSD1 inhibitor, ORY2001, generated by Oryzon is under evaluation in a Phase I clinical study in neurodegenerative disease (Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease). This study was preceded by one initiated in early 2016 to determine the safety, tolerability, and kinetics of ORY2001 in healthy volunteers.
Other N-alkylated compounds, RN1 and RN7, are currently being evaluated in preclinical models. Both those compounds displayed a selective and potent LSD1 inhibitory capacity (IC50 10–30 nM); an important property of these compounds is in their brain penetration capacity, eliciting inhibitory effects on long-term, but not short-term memory formation.Citation103 RN1 was evaluated in preclinical models for its capacity to act as an agent stimulating fetal hemoglobin synthesis in adult erythroid cells and for its antileukemic properties. Concerning the first point, RN1 was evaluated in a mouse model of sickle-cell disease, showing that drug administration induced fetal hemoglobin synthesis in erythroid cells, reduced disease pathology,Citation104 and reduced oxidative-induced damage to erythroid cells.Citation105 Preclinical studies in baboons support the efficacy (evaluated by fetal hemoglobin increase) and safety of long-term administration of RN1 in baboons.Citation106,Citation107 These observations support further development of the LSD1 inhibitor RN1 in the treatment of sickle-cell disease.Citation106,Citation107
Other studies on RN1 were focused on evaluating the antileukemic effects of this inhibitor. Studies performed on leukemic cell lines showed that leukemic cells with MLL translocations or bearing RUNX1–RUNX1T1 (AML1–ETO) translocations are particularly sensitive to cell death, cell differentiation, and inhibition of cell proliferation in vitro and in vivo induced by RN1.Citation106 The in vitro assay indicated that RN1 is a potent LSD1 inhibitor with an IC50 assayed on inhibition of cell proliferation corresponding to 1–5 nM.Citation108 These observations suggest that both MLL-rearranged and RUNX1-rearranged AML may converge on similar downstream oncogenic pathways that are impacted by LSD1 inhibition.Citation108
In 2015, the Cancer Epigenetic Department of Glaxo-SmithKline (Philadelphia, PA, USA) performed a screening on a library of 2.5 million compounds, and discovered a series of small molecules with LSD1-inhibitory activity that led to the development of three potent LSD1 inhibitors: GSK2879552, GSKLSD1 and GSK2699537. All three molecules are N-alkylated cyclopropylamine derivatives and act as potent irreversible LSD1 inhibitors. The pharmacokinetic and pharmacodynamic properties of GSK2879552 indicated that this compound was suitable for clinical development. GSK2879552 treatment of a large panel of tumor cell lines indicated that AML and small-cell lung cancer cells were uniquely sensitive.Citation109 The antitumor effect against small-cell lung cancer was characterized in detail, providing evidence that the sensitivity of these tumor cells to GSK2879552 correlated with DNA hypomethylation of a signature set of probes.Citation109 GSK2879552 is under investigation in the context of a Phase I study in a subset of cancer patients with relapsed/ refractory small-cell lung cancer (NCT2034123) ().
Incyte (Palo Alto, CA, USA) recently developed a new LSD1 inhibitor, INCB059872, displaying properties suitable for clinical development. This inhibitor was potent and selective for LSD1 and was orally bioavailable. Initial pharmacodynamic studies confirmed the capacity of this inhibitor, like other LSD1 inhibitors, to induce apoptosis and differentiation of leukemic cells displaying MLL rearrangements.Citation108 Studies in xenograft mice transplanted with human MLLAF9 leukemic cells showed prolonged in vivo effects of INCB059872, allowing its administration with an alternate-day regimen.Citation110 Preclinical studies with INCB059872 showed a synergistic interaction with ATRACitation111 and with a BET inhibitorCitation112 in inducing cell differentiation and inhibiting the growth of non-APL AML cells. In another set of studies, preliminary evidence was provided on possible synergy between INCB059872 and various signal-transduction inhibitors (such as PIM-kinase inhibitors, JAK1/2 inhibitors, or PI3Kδ-selective inhibitor) in some AML models.Citation113 Other studies were focused on exploring the possible antitumor effects of INCB059872 in some solid tumors. Interestingly, preclinical studies showed significant antitumor effects of this LSD1 inhibitor alone or in combination with chemotherapy in Ewing sarcoma cell lines.Citation113 In vivo studies in mice xenografted with human Ewing sarcoma cell lines confirmed their sensitivity to the antitumor effects of INCB059872.Citation114 Interestingly, using patient-derived xenograft models, evidence was provided that tumors with EWS–FLI translocations were particularly inhibited in their growth by the LSD1 inhibitor.Citation115 Finally, other preclinical studies confirmed the observations made with ORY1001 and showed clear sensitivity of small-cell lung cancer to LSD1 inhibitors.Citation116 On the basis of these observations, Incyte has launched two clinical trials based on the administration of INCB059872: a Phase I/II study of dose escalation/dose expansion in patients with advanced cancer (NCT2712905), and a Phase I dose-escalation study in sickle-cell disease (NCT3132324) ().
Recently, new LSD1 inhibitors pertaining to this chemical subtype have been reported. These compounds are promising for their potency, specificity, and pharmacologic properties. Takeda (Osaka, Japan) reported a new LSD1 inhibitor, T3775440, a tranylcypromine derivate characterized by high LSD1-inhibitory activity (EC50 2 nM). A screening of the effects of this compound against a large panel of leukemic cell lines showed a sensitivity of erythroid and megakaryocytic leukemic cell lines. The growth-inhibitory effect observed on these cells seems to be related to a trans-differentiating effect consisting of the induction of monocyte differentiation.Citation117 In vivo experiments in normal mice showed that T3775440, as well as other LSD1 inhibitors, exerted an inhibitory effect on erythroid and megakaryocytic progenitors, resulting in thrombocytopenia.Citation117 This inhibitor effect on erythromegakaryopoiesis could be related to the capacity of T3775440 to disrupt Gfi1b-containing transcriptional complexes.Citation111 These observations support the hypothesis that LSD1 inhibitors could be evaluated for the treatment of M6 and M7 AML. In line with this observation, as mentioned, M6 AML patients were shown to be frequent responders to treatment with the ORY1001 LSD1 inhibitor.Citation102
Ogasawara et al developed novel LSD1 inhibitors, NCD25 and NCD38, which consist of two moieties: a tranylcypromine-based moiety with LSD1-inhibiting activity and a lysine that is designed to recognize, with high affinity, an LSD1 enzymatic pocket, thus allowing selective inhibitory activity restricted to LSD1 and not extended to other MAO enzy-mes.Citation118 These two LSD1 inhibitors were recently characterized for their anticancer properties. Sugino et al explored the antileukemic activity of NCD25 and NCD38.Citation119 NCD25 and NCD38 inhibited LSD1 with EC50 of about 500 nM. Through the study of a panel of sensitive leukemic cell lines and gene-expression studies in LSD1 inhibitor-treated cells, the conclusion was reached that these inhibitors derepress superenhancers of hematopoietic regulators (such as Gfi1, ERG, and CEBPA) that are abnormally silenced by LSD1 and via this mechanism inhibit leukemic programs and promote cell differentiation.Citation119 Furthermore, NCD38 showed a marked inhibitory effect on colony formation by primary leukemic cells derived from MLL-rearranged AML, erythroleukemia, and MDS.Citation119 These findings provide a rationale for clinical trials of LSD1 inhibitors in MDSs.Citation119 Interestingly, NCD38 and NCL1, another lysine-specific LSD1 inhibitor,Citation120 were shown to inhibit glioma SCs.Citation121
The use of noncovalent reversible LSD1 inhibitors could provide some advantages compared to irreversible inhibitors, particularly as regards a safer metabolic profile. Only a limited number of noncovalent LSD1 inhibitors have been developed, and some are promising in preclinical assay in terms of development of antileukemic drugs.Citation122 Several compounds with these properties and submicromolar potency have been developed. Among these compounds, particularly interesting is the inhibitor SP2509, containing a benzohydrazide scaffold, and with a Ki on LSD1 activity of about 30 nM.Citation123
The effect of SP2509 on leukemic cell lines was tested, providing evidence about effects comparable to those induced by irreversible LSD1 inhibitors (ie, inhibition of proliferation, induction of apoptosis and cell differentiation).Citation124 This compound was tested also on primary AML cells, providing evidence that NPM1 and MLL-rearranged leukemic cells are particularly sensitive to the effects of SP2509.Citation124 Interestingly, cotreatment with an HDAC inhibitor (panobinostat) and SP2509 was synergistically inhibitory for AML blasts.Citation124
Another reversible LSD1 inhibitor, JL1037, was recently identified through computer-aided drug-design technology.Citation125 This compound displayed a reversible and selective LSD1-inhibitory activity with IC50 of 100 nM and antileukemic effects like those observed using other LSD1 inhibitors.Citation125 Exploration of the inhibitory activity of JL1037 on leukemic cells revealed induction of apoptotic and autophagic responses.Citation125
From high-throughput screening, a new series of com-pounds was identified, named 4H-thieno[3,2-b]pyrrole-5-carboxamide, with inhibitory activity against LSD1.Citation126 Structure-guided optimization of this chemical series led to the isolation of some compounds (46, 49, and 50) displaying selective inhibitory activity for LSD1 and pharmacological effects comparable to those observed for other LSD1 inhibitors.Citation126
A high throughput scree and subsequent in silico screening led to the development of (4-cyanophenyl) glycine derivatives as potent reversible LSD1 inhibitors.Citation127 The scaffold-hopping method was used to develop structurally new LSD1 inhibitors: a series of 4-(pyrrolidin-3-yl) benzonitrile derivatives that act as compounds mimicking the GSK690 LSD1 inhibitor, with improved selectivity over the HERG ion channel and no activity against related MOA enzymes.Citation128 Among these inhibitors, particularly promising is the compound 21G.Citation128
Recently, the development of tranylcypromine derivatives targeting both LSD1 and HDAC was reported.Citation129 Particularly, compound 7 displayed potent inhibitory activity against HDAC1 and HDAC2, with IC50 of 15 nM and 13 nM, respectively, as well as potent inhibition against LSD1, with IC50 of 1.20 μM.Citation123 At the biochemical level, this compound increased cellular H3K4 and H3K9 methylation, as well as acetylation.Citation129
DOT1L
Structure and function
One of the most important and studied histone-modification pathways is represented by lysine methylation, occurring as mono-, di-, or tri methylation. The biologic consequences of histone–lysine methylation are variable in that they can result in both repression or derepression of gene expression. There are two distinct classes of histone–lysine KMTs: a first class of proteins is characterized by the presence in its sequence of an evolutionary conserved SET catalytic methyltransferase domain; a second class lacks an SET domain and is represented by a single member – DOT1/DOT1L.
DOT1 was originally identified in yeast as a gene that disrupts telomeric silencing when overexpressed. In mammalians, this protein is called DOT1L. DOT1L is the only known KMT able to methylate H3K79: this methylation is associated with active transcription and occurs on the nucleosome core, not on the histone tail.Citation130 No demethylase has been identified to remove H3K79 methylation, thus indicating that DOT1L is the key determinant for H3K79 methylation. In addition to its role in DNA methylation, DOT1L is also involved in DNA-repair mechanisms and cell-cycle regulation.Citation124 DOT1L is a unique enzyme in the KMT superfamily for its structure. Although DOT1L is a lysine methyltransferase, sequence characteristics indicate similarity to the arginine methyltransferase family.Citation131
The three-dimensional structure of DOT1L reveals an elongated molecule with two domains different from those observed in SET-containing proteins and like those observed in non-SET domain-containing methyltransferases;Citation132 the SAM-binding site of DOT1L presented at the level of the C-terminal α/β domain ().Citation132 The DOT1L protein is a dynamic enzyme able to have different conformational states: particularly, following SAM binding, the protein displays a more closed conformation.Citation132 Study of the protein conformation, when DOT1L binds various types of competitive SAM inhibitors, indicates that loops in SAM binding and near this binding site adopt different conformations. Though not mutated, DOT1L plays and important role in leukemogenesis. In fact, MLL fusion proteins interact directly or indirectly with DOT1L and result in inappropriate recruitment of this KMT at the level of the gene targets of MLL fusion proteins, such as HOXA and MEIS1, resulting in their transcriptional activation.Citation133,Citation134
Figure 2 Schematic representation of the DOT1L protein.
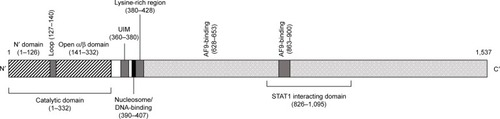
Recent studies have in part clarified the mechanism through which DOT1L modulates transcriptional activity and contributes to the deregulated gene expression observed in MLL-rearranged leukemias. The capacity of DOT1L to interact with other proteins to form multiprotein complexes is fundamental for its physiological function of transcriptional regulator and for its relevant contribution to the pathogenesis of MLL-rearranged leukemias. DOT1L has been found to be involved in the formation of complexes with an ENL family protein and an AF10 family protein, such as AF10 or AF9. The association of DOT1L with these proteins increases the methyltransferase activity of DOT1L to produce highly methylated H3K79 me2/3 markers.Citation135
Many of these proteins complexing with DOT1L are also partners of MLL in the formation of MLL-fusion proteins. In MLL-rearranged leukemias, these partners interact with DOT1L, and through this interaction drive the DOT1L complex at the level of MLL-fusion target genes, such as HOXA and MEIS1, thus inducing their high H3K79 methylation and consequently their high expression. This is the basic mechanism through which DOT1L contributes in a relevant way to MLL-fusion leukemia, enabling a high expression of MLL target genes. The AF9 and AF10 proteins act as readers of histone modifications.Citation136,Citation137
AF9 interacts with DOT1L, forming a stable complex with its C-terminus (). AF9 interacts with H3K9ac, which is enriched in transcriptionally active genes.Citation126 Most AF9-bound genes are enriched for both H3K9 ac and H3K79 me3.Citation137 Importantly, AF9 knockdown leads to decreased H3K79 methylation on target genes.Citation137 AF10 determines higher H3K79 methylation states, and thus enhances the methyltransferase activity of DOT1L.Citation135 AF10 interacts with unmodified H3K27, but not methylated H3K27, thus explaining why H3K27 methylation does not often co-occur with H3K79 methylation on the genetic region.Citation136
Another important element in the understanding of the mechanism through which DOT1L exerts its leukemogenic function derives from a recent study showing that DOT1L prevents SIRT1-mediated gene silencing at the level of gene regions recognized by MLL-fusion proteins. Particularly, DOT1L recruited at the level of MLL-fusion target loci prevents SIRT1 from deacetylating H3K9.Citation138
When DOT1L is inactivated by chemical inhibitors, SIRT1 reacquires the capacity to bind to these genetic loci and promotes deacetylation of H3K9, which leads to H3K9 methylation and consequent downregulation of MLL-fusion target-gene expression.Citation138 Finally, SIRT1 activators synergize with DOT1L inhibitors and accelerate downregulation of MLL-fusion target genes.Citation138 The mechanisms through which DOT1L contributes to MLL-driven leukemogenesis are very complex, and this is in large part related to the capacity of MLL-fusion proteins to form large complexes interacting with numerous other nuclear proteins. More than 70 genes have been shown to fuse with MLL. In most cases, MLL is fused with a component of the AF4–ENL–P-TEFb (AEP) complex or the DOT1L–AF10–ENL complex. These complexes involve a member of the ENL family, such as ENL and AF9. The ENL proteins contain a YEATS domain, recognizing H3K9/18/27 Ac. The AEP complex contains AF4 protein families, which provide various interaction platforms for cofactors involved in initiation and elongation of transcription. The mechanism through which the MLL–AF10–ENL complex transforms HSCs operates through the DOT1L-interaction domain,Citation133 while the MLL–ENL–AF9 involves the ANCI-homology domain, which interacts with both DOT1L and AF4.Citation139
A recent study provided evidence that MLL–F10–DOT1L acts through a more complex mechanism that does not need the simple recruitment of DOT1L to the target promoters, but involves also the capacity of DOT1L through its ENL-binding motifs to recruit AF4.Citation132 According to these observations, it was suggested that the simultaneous inhibition of the MLL fusion-AF4 complex and DOT1L may provide more effective treatment of MLL-rearranged leukemia.Citation140
Interestingly, recent studies have shown that DOT1L may represent a therapeutic target for the treatment of DNMT3A-mutant AML. DNMT3A is mutated in about 20% of AML and usually associated with a negative prognosis. In 60% of cases, DNMT3A mutations involve the arginine at amino-acid position 882 and determine a marked loss of cellular DNA methyltransferase activity. Ablation of DNMT3A in HSCs determines a pronounced expansion of HSCs and a progressive block in cell differentiation. HSCs deficient in DNMT3A overexpress DOT1L and have increased H3K79 methylation, particularly at the level of DNA regions corresponding to genes highly dysregulated in leukemia.Citation135 These observations have suggested that DOT1L could represent a target in DNMT3A-mutated AML. In line with this hypothesis, DOT1L inhibitors blocked the growth of DNMT3A-mutated cell lines and primary AML cells and induced cell differentiation.Citation141
DOT1L and hematopoiesis
Gene-knockout studies in mice have supported a role for DOT1L in the control of normal hematopoiesis. DOT1L−/− mice developed more slowly and died on embryonic days 10.5–13.5, exhibiting pronounced anemia.Citation140 Analysis of the hematopoietic tissue of DOT1L-deficient mice displayed a selective defect at the level of erythroid but not myeloid progenitors: erythroid progenitors failed to develop normally, displayed delayed progression through the cell cycle, accumulated in G0/G1, and exhibited a great tendency toward apoptosis, even in the presence of erythroid growth factors.Citation142 GATA2 expression was greatly decreased in erythroid cells of DOT1L-deficient mice, while PU1 levels were increased.Citation142
Subsequently, two additional studies demonstrated that DOT1L plays an important role in maintaining normal adult hematopoiesis, using conditional DOT1L-knockout mice, coupled with serial bone-transplant assays.Citation140,Citation141 Knockout of DOT1L in bone marrow causes pancytopenia and depletes HSCs and various progenitor cell (PC) populations, thus indicating an essential role for DOT1L in maintaining HSCs and HPCs.Citation143,Citation144
Daigle et al explored the effect of a potent DOT1L inhibitor (EPZ004777) on in vivo murine hematopoiesis, showing that the administration of this inhibitor at tolerable pharmacodynamically active doses elicited only slight effects consisting of a moderate and unexplained increase of neutrophils, monocytes, and lymphocytes not accompanied by any significant change at the level of frequency of bone-marrow colony-forming cells. HSCs and granulomonocytic progenitors were not affected by the drug.Citation145 Differences observed in knockout vs drug-inhibition studies are seemingly related to differences in DOT1L silencing achieved by these different procedures.
DOT1L inhibitors
Given the important role of DOT1L in MLL-rearranged leukemia, many recent studies have investigated the development and characterization of DOT1L inhibitors and explored their potential role in leukemia therapy (). Three compounds were initially reported – EPZ004777, EPZ5676, and SGC0946 – and selected for their specific inhibitory activity against DOT1L.Citation145–Citation147
Table 2 Main properties of DOT1L inhibitors
All these three compounds act as competitive inhibitors of SAM, the cofactor required for the methyltransferase activity of DOT1L. More recently, new DOT1L inhibitors have been identified. Wang et al reported the identification of a new class of DOT1L inhibitors, characterized by a scaffold of [1,2,4]-triazolo-[3,4-b] [1,3,4]-thiadiazole. The most active of these compounds, compound 6 is a selective DOT1L inhibitor with on IC50 of 8.3 μM.Citation148
The DOT1L inhibitor EPZ004777 is characterized by a considerable inhibitory potency (IC50 400 pM). This inhibitor was designed to mimic the SAM enzyme cofactor. EPZ was competitive with SAM and not with the peptide substrate. EPZ represents the standard DOT1L inhibitors with several typical pharmacological effects, consisting of global reduction of H3K79 me2 levels, inhibition of the expression of HOXA9 and MEIS1, and inhibition of the growth in vitro and in vivo of MLL-rearranged leukemic cells.Citation145 Taking advantage of the crystal structure of the DOT1L–EPZ004777 complex, SG0946 inhibitors with increased in vitro and in vivo cellular potencies were developed.Citation145 SG0946 contains a bromo-substitution at position 7 compared to EPZ004777 and was more potent in chemical and cellular assays.Citation147 A major advancement in the field was represented by the discovery of the EPZ5676 DOT1L inhibitor by Daigle et al in 2013.Citation146 This inhibitor binds the DOT1L enzyme at the level of the cofactor-binding site with an affinity of 0.03 nM and reduces H3K79 me2 levels in MV411 cells with an IC50 of 3 nM.Citation147 Preclinical studies have shown that the continuous infusion of EPZ5676 was able to induce complete tumor regression in immunocompromised mice xenografted with MLL-rearranged MV411 cells.Citation146 The efficacy of EPZ5676 in preclinical models has triggered the clinical development of this inhibitor as an anticancer drug. In clinical studies, the inhibitor is administered by continuous intravenous infusion. Alternatively, EPZ5676 can be administered by subcutaneous bolus formulation, but this mode of administration has been used only in preclinical studies.Citation149 Interestingly, in MLL-rearranged AML cells, EPZ5676 synergizes with standard antileukemic drugs and the hypomethylating agent azacitidine, eg, AraC and daunorubicin, to inhibit the growth of leukemic cells.Citation150
The metabolism and disposition of EPZ5676 has been explored in preclinical animals (rats and dogs) and humans, showing that fecal excretion of the intact drug and its metabolites is responsible for the majority of drug-related elimination, with low renal excretion.Citation151 Despite the pharmacokinetic improvements in EPZ5676 compared to other LSD1 inhibitors, this compound still shows low oral bioavailability.
A first Phase I study (completed) was designed to evaluate safety profile, pharmacokinetics, and pharmacodynamics and obtain preliminary indications on clinical activity in adult patients with relapsed/refractory acute leukemia (either myeloid or lymphoid) ().Citation152 A total of 49 patients were treated (either in the dose-escalation or expansion phase), and six patients displayed objective response, including one morphologic complete remission, one cytogenetic complete response, one partial response, and three resolutions of leukemia cutis.Citation152 Drug administration is usually well tolerated, and about 15% of treated patients display adverse events.Citation152 In another Phase I study, pinometostat was evaluated in a cohort of pediatric patients with relapsed or refractory MLL-rearranged acute leukemia.Citation147 No objective responses were observed in these patients, and only transient reductions in blast-cell counts were observed in 40% of treated patients.Citation153
Menin
Menin structure and function
Menin is a 67 kDa protein encoded by the MEN1 gene localized on chromosome 11q13.Citation154 Menin is a nuclear protein, ubiquitously expressed.Citation155 Menin interacts directly with the N-terminal region of MLL;Citation156–Citation158 this N-terminal MLL region is retained in all MLL-fusion proteins and plays a key role in the binding of MLL and MLL-fusion proteins to target genes, such as HOXA9.Citation156,Citation157,Citation159,Citation160 The capacity of MLL-fusion proteins to bind menin is strictly required for their leukemogenic activity: in fact, mutations at the level of the N-terminal region of MLL-fusion proteins block their capacity to induce leukemia and to upregulate HOXA gene expression.Citation154 These observations have indicated that the menin–MLL interaction represents a potentially important target in the treatment of MLL-rearranged AML. Analysis of the structure of the menin protein is essential for a better understanding of its function. Human menin is composed of 615 amino acids and it is a globular protein. The linear structure of the protein shows four structured regions that form the structured core and represent regions conserved during evolution, interspaced by three loop regions. In contrast to the structured regions, the loop regions and C-terminal region are variable among species and contain sites of posttranslational modification of the protein, such as phosphorylation and SUMOylation ().Citation161 Structural studies have shown that menin is an α-helical protein with three folds based on three tetratricopeptide repeats:Citation162 these repeats form a large central cavity that represents the binding site for the protein– protein interactions with MLL and JunD8127. This central cavity is a relatively rigid structure that does not change conformation upon binding of protein ligand.
Figure 3 Linear structure of human menin.
As stated, an essential function of menin is dependent upon its capacity to interact with the N-terminal region of MLL: at the level of this region of the MLL molecule, two menin-binding motifs have been discovered: high-affinity MBM1, encompassing residues 6–13, and low-affinity MBM2, encompassing residues 24–40 ().Citation158 The MBM1 pocket on menin represents a major site for targeting by small-molecule inhibitors.
Menin acts as an oncogenic cofactor of MLL-fusion proteins in leukemia. This function is related to the capacity of menin to interact with the N-terminus of MLL, an MLL region retained in all MLL-fusion proteins. Through this mechanism, menin drives MLL and MLL-fusion proteins to target genes.Citation156,Citation157,Citation159,Citation160 The essential role of menin in supporting the oncogenic activity of MLL-fusion proteins is directly shown by the observation that loss of menin binding by MLL-fusion proteins abolishes their capacity to upregulate HOX gene expression and to induce leukemia in vivo in mice.Citation161 Furthermore, expression of a dominant-negative inhibitor composed of an amino-terminal MLL sequence acts as a blocking agent of the oncogenic potential of MLL-fusion proteins.Citation162 Finally, small-molecule inhibitors able to bind to menin and disrupt the binding of these molecules to MLL inhibit the proliferation of MLL-fusion-bearing leukemic cells and induce their differentiation.Citation163
The requirement of menin for leukemogenic activity of MLL-fusion proteins is strongly supported by various experi-mental studies, and represents a strong rationale to develop small-molecule inhibitors to target the menin–MLL interactions. The analysis of a large set of different MLL-fusion proteins clearly supported the capacity of a potent menin inhibitor, MI2-2, to induce growth arrest, differentiation, and downregulation of MLL-fusion target genes in leukemia cells transformed with various MLL fusions.Citation164
Importantly, this menin inhibitor elicited its inhibitory activity on AML cells displaying different types of MLL-fusion genes. As mentioned, MLL-rearranged AML is sensitive to DOT1L inhibitors. However, DOT1L inhibitors act slowly on these leukemia cells, and their effect on many MLL-rearranged cell lines is limited. To bypass this important limitation, it was discovered that a combined inhibition of both DOT1L and menin resulted in strong, complementary inhibition of MLL-rearranged leukemic cells, as supported by induction of cell killing and differentiation.Citation165 At the gene-expression level, the combination of the two drugs elicited a more marked inhibitory effect on MLL-fusion and MYC target genes.Citation165
Analysis of HOXA and HOXB cluster-gene expression in AML allows identification of four AML subgroups:
a subgroup not expressing HOXA or HOXB, represented by AML, characterized by PML–RARα and AML1–ETO fusion transcripts
a subgroup expressing only HOXA, represented by AML, characterized by MLL-fusion genes and by frequent complex karyotypes
a subgroup expressing only HOXB, represented by AML, frequently characterized by CBFB–MYH11 rearrangements
a subgroup expressing both HOXA and HOXB, repre-sented by AML, frequently characterized by NPM1 mutations in most cases and more rarely by MLL-PTD.Citation166
NPM1-mutated AML represents the largest group of HOX-expressing AML. A recent study provided evidence that pharmacologic inhibition of menin and DOT1L inhibited HOX and FLT3 expression and promoted differentiation of NPM1-mutated AML.Citation167 Menin is involved also in the genesis of other tumors in addition to leukemias. Therefore, mutations of the menin gene are responsible for the development of a peculiar syndrome called multiple endocrine neoplasia type 1 syndrome, an autosomal-dominant disorder in which variable combinations of endocrine and nonendocrine tumors may occur, in the context of varied phenotypic and clinical features.Citation168 The large majority of menin gene mutations observed in these patients are inactivating and private mutations, without any preferential distribution among selected regions of the menin gene.Citation168
Enhanced expression of the menin–MLL complex plays an important tumorigenic role in other neoplasia such as Ewing’s sarcomaCitation169 and prostate cancer.Citation170 In Ewing’s sarcoma, expression of both menin and MLL is increased and determines enhanced HOXD expression: inhibition of MLL–menin interaction in these tumor cells decreases their tumorigenicity and inhibits HOXD expression.Citation167 In prostate cancer cells, the overexpressed MLL–menin complex acts as a coactivator of androgen-receptor signaling and contributes to tumorigenicity. Importantly a menin inhibitor blocks androgen-receptor signaling and reduces growth of castration-resistant tumors in vivo.Citation170
Role of MLL and menin in hematopoiesis
The study of specific knockout mice has contributed to elucidation of the role of MLL and menin 1 in normal hematopoiesis. A basic observation derived from these studies was that while normal hematopoiesis was markedly impaired in the absence of MLL, less pronounced and superimposable defects were observed in menin-deficient mice. During embryonic life, MLL expression is strictly required for the development of both primitive and definitive hematopoiesis.Citation171–Citation173 Furthermore, MLL is required also for adult hematopoiesis, particularly for HSC self-renewal and HPC proliferation.Citation174,Citation175
Menin 1 knockout results in mouse lethality during mid-gestation for multiple developmental defects. The possible consequence of a loss of menin 1 expression for hematopoiesis was investigated using a conditional knockout strategy. These studies showed no overt defects of hematopoiesis in menin 1-deficient mice: these mice displayed only a modest decrease in white blood-cell counts, associated with decreased clonogenic capacity of HPCs in standard methylcellulose assaysCitation176 and reduced repopulating capacity of HSCs in competitive transplantation assays.Citation177
The analysis of regulation of hematopoiesis in mice deficient in the interaction between MLL1 and menin 1 came to the conclusion that MLL1 and its cofactor menin 1 act independently in regulating hematopoiesis at the level of HSCs.Citation178 Both MLL1 and menin 1 play an important role in the control of B-lymphocytic cell differentiation and act largely through independent mechanisms (interestingly, some genes, such as HOXA9 and MEIS1, are regulated by both proteins, but other sets of genes are specifically regulated by either MLL1 or menin 1).Citation178 These findings support the view that these proteins are largely independent for their effects on the control of normal hematopoiesis and the disruption of menin 1–MLL interactions may represent a safe strategy to try to target selectively the aberrant gene expression deriving from the interaction between MLL-fusion proteins and menin 1.Citation178
Developmentally induced MLL1 loss showed that deficient mice exhibit phenotypically normal fetal hematopoiesis, but rarely survive past 3 weeks of age. Surviving animals were anemic, thrombocytopenic, and exhibited a reduction in HSC number.Citation179 The role of histone methyltransferase activity in MLL1−/− phenotypes was investigated using the MLL1 gene deleted from the SET domain, corresponding to the catalytic site, and provided evidence that histone methyltransferase activity is dispensable for the effects of MLL on hematopoiesis.Citation180
The role of MLL1 in HSC biology was better delineated in additional studies. Artinger et al identified the transcriptional network of MLL in the hematopoietic system and showed that it extends beyond HOX genes and involves also a number of other transcriptional regulators, such as MECOM, Prdm16, Pbx1, Eya1, and others involved in HSC maintenance and proliferation.Citation181 Furthermore, the MLL-transcription-dependent network contains genes that are both dependent and independent of menin.Citation172 The maintenance of MLL activity on gene transcription and HSC function requires the capacity of this protein to recruit an H4k16 acetyltransferase; in fact, H4k16 deacetylase inhibitors are sufficient to restore MLL1-dependent gene expression in MLL1−/− cells.Citation181
Menin 1 inhibitors
The study and understanding of the molecular basis of MLL1–menin 1 interactions have been of fundamental importance in the development of specific small molecule inhibitors. The interaction between these two proteins is dictated by the binding of MLL to a large hydrophobic cavity present in the menin 1 molecule through two binding motifs: a high-affinity motif encompassing MLL residues 6–13 (MBM1), and a low-affinity motif encompassing MLL residues 24–40 (MBM2) ().Citation182 The MBM1 pocket is of fundamental importance for the interaction between MLL and menin 1, and represents a major site for targeting by small-molecule inhibitors. The mutation of F9 to alanine in MBM1 has a very marked impact on MLL–menin interaction.Citation182 The role of MBM2 in MLL–menin interaction is less defined. The MBM2 motif carries a positive charge due to the presence of four arginine residues, and is seemingly involved in the mediation of electrostatic interaction with the central cavity of menin 1, which is negatively charged.Citation183
In line with these observations, inhibitors able to target MBM1 are capable of blocking MLL–menin interaction and suitable for preclinical and clinical studies. The first small-molecule inhibitors of the MLL–menin interaction were identified used a technology based on fluorescence polarization-based high-throughput screening.Citation177 Among the various inhibitors identified using this approach, the most potent was the MI2 inhibitor, with an IC50 of 446 nM, able to block the interaction between MLL and menin and to inhibit the leukemogenic potential of MLL-fusion proteins.Citation184
Analysis of the crystal structure of human menin complexed with MI2 allowed the design and the production of a more powerful inhibitor, Mi2-2, with a clear improvement in IC50 of about tenfold.Citation179 The introduction of a cyanoindole ring connected to a thienopyrimidine core allowed the definition of a new chemical core structure suitable for the development of new specific and more potent menin inhibitors.Citation185 This strategy led to the synthesis and characterization of two new menin inhibitors: MI463, with an IC50 of about 15 nM, and MI503, with an IC50 of about 15 nM.Citation179 Both these com-pounds exhibit a suitable pharmacological profile based on standard preclinical studies.Citation177 He et al identified another class of hydroxy- and aminomethyl piperidine compounds capable of mimicking the three hydrophobic interactions between MLL and menin: the most notable of these compounds, MIV6, displayed an IC50 of about 56 nM.Citation186
Other studies were based on completely different approaches. Zhou et al designed some macrocyclic peptidomimetic inhibitors, such as MCP1, able to bind the menin pocket as like the native MBM1 MLL motif: two of these compounds exhibited high IC50 in the 5–20 nM range but their high molecular weight hampered their further preclinical development.Citation187 Finally, the screening of existing drugs allowed the identification of two drugs, the aminoglycosidic antibiotic neomycin and tobramycin, two low-affinity inhibitors with IC50 in the 20–60 μM range.Citation188
More recently, Xu et al, using pharmacophore-based and structure-based approaches, developed a new screening strategy to discover new inhibitors targeting the MLL–menin interface. M123, one of these compounds, displayed potent inhibitor activity with an IC50 of about 5 nM and the capacity to inhibit the growth of MLL-rearranged leukemia cells.Citation189 Borkin et al recently reported the synthesis and structure-based optimization of a new thienopyrimidine class of compounds able to block the interaction between MLL and menin.Citation190 This approach led to the identification of a compound (MI136) that showed marked inhibitory activity in vitro and in vivo and seems suitable for preclinical studies. This compound displays a favorable pharmacokinetic profile.Citation190
Conclusion
Studies carried out in the last few years have provided strong and clear evidence that multiple dysregulations of epigenetic regulatory mechanisms have a central role in leukemia onset and progression. Many epigenetic modifications are suitable for pharmacological interventions. Histone methylation is a very important process in the context of epigenetic processes of control of gene expression, and the study of these processes in leukemia has led to the identification of lysine methyltransferases and demethylases as promising targets for therapeutic interventions. In this review, we have analyzed those enzymes (LSD1 and DOT1L) or molecules (menin) and their inhibitors that have already reached the stage of initial clinical development in leukemia therapy. Although the first indications with these inhibitors are promising, future studies will clarify the real impact of these inhibitors in the treatment of leukemia and other cancers.
Disclosure
The authors report no conflicts of interest in this work.
References
- LawrenceMDaujatSSchneiderRLateral thinking: how histone modifications regulate gene expressionTrends Genet2016321425626704082
- VankateshSWorkmanJLHistone exchange, chromatin structure and the regulation of transcriptionNature Rev Mol Cell Biol201516317818925650798
- HarrJCGonzalez-SandovalAGasserSMHistone and histone modifications in perinuclear chromatin anchoring: from yeast to manEMBO Rep201617213915526792937
- VerdinEOttM50 Years of protein acetylation: from gene regulation to epigenetics, metabolism and beyondNat Rev Mol Cell Biol201516425826425549891
- DuJJohnsonLMJacobsenSEPatelDJDNA methylation pathways and their crosstalk with histone methylationNat Rev Mol Cell Biol201516951953226296162
- GreerELShiYHistone methylation: a dynamic mark in health, disease and inheritanceNat Rev Genet201213534335722473383
- Russler-GermainDASpencerDHYoungMAThe R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramersCancer Cell201425444245424656771
- SpencerDHRussler-GermainDAKetkarSCpG island hypermethylation mediated by DNMT3A is a consequence of AML progressionCell2017168580181628215704
- DayYJWangYYHuangJYConditional knockin of DNMT3A R878H initiates acute myeloid leukemia with mTOR pathway involvementProc Natl Acad Sci U S A2017114205237524228461508
- ShenYZhuYMFanXGene mutation patterns and their prognostic impact in a cohort of 1185 patients with acute myeloid leukemiaBlood2011118205593560321881046
- LinPHLiHYFanSCA targeted next-generation sequencing in the molecular risk stratification of adult acute myeloid leukemia: implications for clinical practiceCancer Med20176234936028070990
- MizunoSChijwaTOkamuraTExpression of DNA methyltransferases DNMT1A, 3A and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemiaBlood20019751172117911222358
- TrowbridgeJJSinhaAUZhuNLiMArmstrongSAOrkinSHHaploinsufficiency of Dnmt1 impairs leukemia stem cell function through derepression of bivalent chromatin domainsGenes Dev201226234434922345515
- YanFShenNPangJXA vicious loop of fatty acid-binding protein 4 and DNA methyltransferase 1 promotes acute myeloid leukemia and acts as a therapeutic targetLeukemia Epub20171010
- ShenNYanFPangJXInactivation of receptor tyrosine kinases reverts aberrant DNA methylation in acute myeloid leukemiaClin Cancer Res201723206254626628720666
- TagdeARajabiHStroopinskyDMUC1-C induces DNA methyltransferase 1 and represses tumor suppressor genes in acute myeloid leukemiaOncotarget2016726389743898727259275
- ArgoteJADasanuCAASXL1 mutations in myeloid neoplasms: pathogenetic considerations, impact on clinical outcome and survivalCurr Med Res Opin Epub2017124
- GaidzikVITeleanuVPapaemmanuilERUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic featuresLeukemia201630112160216827137476
- OhgamiRSMaLMerkerJDNext-generation sequencing of acute myeloid leukemia identifies the significance of TP53, U2AF1, ASXL1, and TET2 mutationsMod Pathol201528570671425412851
- LundKAdamsPDCoplandMEZH2 in normal and malignant hematopoiesisLeukemia2014281444924097338
- GöllnerSOellerichTAgrawal-SinghSLoss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemiaNat Med2017231697827941792
- GaoLSunJLiuFZhangHMaYHigher expression levels of the HOXA9 gene, closely associated with MLL-PTD and EZH2 mutations, predict inferior outcome in acute myeloid leukemiaOnco Targets Ther2016971172226929642
- SunQYDingLWTanKTOrdering of mutations in acute myeloid leukemia with partial tandem duplication of MLL (MLL-PTD)Leukemia201731111027389053
- CaoQGearhartMGerySBCOR regulates myeloid cell proliferation and differentiationLeukemia20163051155116526847029
- PapaemmanuilEGerstungMBullingerLGenomic classification and prognosis in acute myeloid leukemiaN Eng J Med20163742322092221
- ShiraiCILeyJNWhiteBSMutant U2AF1 expression alters hematopoiesis and pre-mRNA splicing in vivoCancer Cell201527563164325965570
- KimEIlaganJOLiangYSRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognitionCancer Cell201527561763025965569
- DöhnerHEsteyEGrimwadeDDiagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panelBlood2017129442444727895058
- WintersABerntKMLL-rearranged leukemias: an update on science and clinical approachesFront Pediatr20175428232907
- BasilicoSGöttgensBDysregulation of haematopoietic stem cell regulatory program in acute myeloid leukemiaJ Mol Med (Berl)201795771972728429049
- RoseDHaferlachTSchnittgerSPerglerováKKernWHaferlachCSubtype-specific patterns of molecular mutations in acute myeloid leukemiaLeukemia2017311111727285584
- TsaiCHHouHATangJLGenetic alterations and their clinical implications in older patients with acute myeloid leukemiaLeukemia20163071485149227055875
- GaidzikVIPaschkaPSpäthDTET2 mutations in acute myeloid leukemia (AML): results from a comprehensive genetic and clinical analysis of the AML study groupJ Clin Oncol201230121350135722430270
- MagotraMSakhdariALeePJImmunohistochemical loss of 5-hydroxymethylcytosine expression in acute myeloid leukemia: relationship to somatic gene mutations affecting epigenetic pathwaysHistopathology201696610551065
- AhnJSKimHJKimYK5-Hydroxymethylcytosine correlates with epigenetic regulatory mutations, but may not have prognostic value in predicting survival in normal karyotype acute myeloid leukemiaOncotarget2017858305831428039446
- SperlingASGibsonCJEbertBLThe genetics of myelodysplastic syndrome: from clonal hematopoiesis to secondary leukaemiaNat Rev Cancer201717151927834397
- CimminoLDolgalevIWangYRestoration of TET2 function blocks aberrant self-renewal and leukemia progressionCell2017170710781095.e20
- MedeirosBCFathiATDiNardoCDPollyeaDAChanSMSwordsRIsocitrate dehydrogenase mutations in myeloid malignanciesLeukemia201731227228127721426
- ChenCLiuYLuCCancer-associated IDH2 mutants drive an acute myeloid leukemia that is susceptible to Bdr4 inhibitionGenes Dev201327181974198524065765
- MarcucciGMaharryKWuYZIDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B studyJ Clin Oncol201028142348235520368543
- SteinEMDiNardoCDPollyeaDAEnasidenib in mutant-IDH2 relapsed or refractory acute myeloid leukemiaBlood2017130672273128588020
- LinPHLiHYFanSCA targeted next-generation sequencing in the molecular risk stratification of adult acute myeloid leukemia: implications for clinical practiceCancer Med20176234936028070990
- BullingerLDöhnerKDöhnerHGenomics of acute myeloid leukemia diagnosis and pathwaysJ Clin Oncol201735993494628297624
- HacklHAstaninaKWieserRMolecular and genetic alterations associated with therapy resistance and relapse of acute myeloid leukemiaJ Hematol Oncol20171015128219393
- KumimotoHNakajimaHEpigenetic dysregulation of hematopoietic stem cells and preleukemic stateInt J Hematol20171061344428555413
- ZinkFStaceySNNorddahlGLClonal hematopoiesis, with and without candidate driver mutations, is common in the elderlyBlood2017130674275228483762
- FigueroaMELugthartSLiYDNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemiaCancer Cell2010171132720060365
- Cancer Genome Atlas Research NetworkGenomic and epigenomic landscape of adult de novo acute myeloid leukemiaN Engl J Med2013368222059207423634996
- AkalinAGarrett-BakelmanFEKormakssonMBase-pair resolution DNA methylation sequencing reveals profoundly diver-gent epigenetic landscapes in acute myeloid leukemiaPLoS Genet201286e100278122737091
- SchoofsTBerdelWEMüller-TidowCOrigins of aberrant DNA methylation in acute myeloid leukemiaLeukemia201428111423958917
- MarcucciGYanPMaharryKEpigenetics meets genetics in acute myeloid leukemia: clinical impact of a novel seven-gene scoreJ Clin Oncol201432654855624378410
- FigueroaMEAbdel-WahabOLuCLeukemic IDH1 and IDH2 mutations result in hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiationCancer Cell201018655356721130701
- LuskinMRGimottyPASmithCA clinical measure of DNA methylation predicts outcome in de novo acute myeloid leukemiaJCI Insight201619e8732327446991
- LiSGarrett-BakelmanFChungSDistinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemiaNat Med2012227792799
- VardimanJWThieleJArberDAThe 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changesBlood2009114593795119357394
- ArberDAOraziAHasserjianRThe 2016 revision to the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemiaBlood2016127202391240527069254
- KantajianHRobozGKropfPGuadecitabine (SG-110) in treatment-naïve patients with acute myeloid leukemia: phase 2 results from a multicenter, randomized, phase 1–2 trialLancet Oncol2017101813171326
- KropfPLChungWBKellyADGenetic determinants of response to guadecitabine (SG-110) in AMLBlood2016128221680
- DaverNKantarjianHMRobozGJLong term survival and clinical complete responses of various prognostic subgroups in 103 relapsed/refractory acute myeloid leukemia (r/r AML) patients treated with guadecitabine (SG-110) in phase 2 studiesBlood201612822904
- WelchJSPettiAAMillerCATP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromesN Engl J Med21163752120232036
- SebertMBallyCPeterlinPResults of a phase II study of guadecitabine (SGI-110) in higher risk MDS, CMML or low blast count AML patients refractory to or relapsing after azacitidine (AZA) treatmentBlood201612822347
- Montaban-BravoGBosePAlvaradoYInitial results of a phase 2 study of guadecitabine (SGI-110), a novel subcutaneous (SC) hypomethylating agent, for patients with previously untreated intermediate-2 or high risk myelodysplastic syndromes (MDS) or chronic myelomonocytic leukemia (CMML)Blood201612822346
- MetregerLWissmannMYinNLSD1 demethylates repressive histone marks to promote androgen receptor-dependent transcriptionNature2005437705743643916079795
- HøjfeldtJWAggerKHelinKHistone lysine demethylases as targets for anticancer therapyNat Rev Drug Discov2013121291793024232376
- StavropoulosPBlobelGHoelzACrystal structure and mechanism of human lysine-specific demethylase-1Nat Struct Mol Biol200613762663216799558
- ChenYYangYWangFCrystal structure of human histone lysine-specific demethylase 1 (LSD1)Proc Natl Acad Sci U S A200610338139561396116956976
- FornerisFBindaCVanoniMAHuman histone demethylase LSD1 reads the histone codeJ Biol Chem200528050413604136516223729
- FornerisFBindaCVanoniMAMatteviABattaglioliEHistone demethylation catalyzed by LSD1 is a flavin-dependent oxidative processFEBS Lett2005579102203220715811342
- TongJKHassigCASchnitzlerGRKingstonRESchreiberSLChromatin deacetylation by an ATP-dependent nucleosome remodeling complexNature199839567059179219804427
- HumphreyGWWangYRussanovaVRStable histone deacetylase complexes distinguished by the presence of SANT domain protein CoREST/kiaa0071 and Mta-L1J Biol Chem200127696817682411102443
- YouATongJKGrozingerCMSchreiberSLCoREST is an integral component of the CoREST-human histone deacetylase complexProc Natl Acad Sci U S A20019841454145811171972
- HakimiMADongYLaneWSSpeicherDWShiekhattarRA candidate X-linked mental retardation gene is a component of a new family of histone deacetylase-containing complexesJ Biol Chem200327897234723912493763
- DiazAMSomervailleTLSD1: biologic roles and therapeutic targetingEpigenomics2016881103111627479862
- ZhengYCMaJWangZA systematic review of histone lysine-specific demethylase 1 and its inhibitorsMed Res Rev20153551032107125990136
- ZibettiCAdamoABindaCAlternative splicing of the histone demethylase LSD1/KDM1 contributes to the modulation of neurite morphogenesis in the mammalian nervous systemJ Neurosci20103072521253220164337
- SprüsselASchulteJHWeberSLysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiationLeukemia20122692039205122699452
- KerenyiMAShaoZHsuYJHistone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturationElife20132e0063323795291
- UpadhyayGChowdhuzyAVaidyanathanBKimDSalequeSAntagonistic actions of Rcor proteins regulate LSD1 activity and cellular differentiationProc Natl Acad Sci U S A2014111228071807624843136
- SalequeSKimJRookeHMOrkinSHEpigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1Mol Cell200727456257217707228
- AnguitaECandelFJChaparroARoldán-EtcheverryJJTranscription factor GFl1B in health and diseaseFront Oncol201775428401061
- HuXLiXValverdeKLSD1-mediated epigenetic modification is required for TAL-1 function and hematopoiesisProc Natl Acad Sci U S A200910625101411014619497860
- LiYDengCHuXDynamic interaction between TAL1 oncoprotein and LSD1 regulates TAL1 junction in hematopoiesis and leukemogenesisOncogene201231485007501822310283
- XuJBauerDEKerenyiMACorepressor-dependent silencing of fetal hemoglobin expression by BCL11AProc Natl Acad Sci U S A2013110166518652323576758
- GuoYFuXJinYHistone demethylase LSD1-mediated repression of GATA-2 is critical for erythroid differentiationDrug Des Devel Ther2015931533162
- VelinderMSingerJBareyanDGFl1 functions in transcriptional control and cell fate determination require SNAG domain methylation to recruit LSD1Biochem J2016473193355336927480105
- ThambyrajahRMazanMPatelRGFl1 proteins orchestrate the emergence of haematopoietic stem cells through recruitment of LSD1Nat Cell Biol2016181213226619147
- TakeuchiMFuseYWatanabeMLSD1/KDM1A promotes hematopoietic commitment of hemangioblasts through downregulation of Etv2Proc Natl Acad Sci U S A201511245139221392726512114
- MöröyTVanenLWilkesBKhandanpourCFrom cytopenia to leukemia: the role of GFi1 and GFi1b in blood formationBlood201512642561256926447191
- HönesJMBotezatuLHelnessAGFI1 as a novel prognostic and therapeutic factor for AML/MDSLeukemia20163021237124526847026
- VolpeGWaltonDSGraingerDEPrognostic significance of high GFI1 expression in AML of normal karyotype and its association with a FLT3-ITD signatureSci Rep2017711114828894287
- YaoHGoldmanDCNechiporukTCorepressor Rcor1 is essential for murine erythropoiesisBlood2014123203175318424652990
- YaoHGoldmanDCFanGThe corepressor Rcor1 is essential for normal myeloerythroid lineage differentiationStem Cells201533113304331426119982
- NiebelDKirfelJJanzenVHöllerTMajoresMGütgemannILysine-specific demethylase 1 (LSD1) in hematopoietic and lymphoid neoplasmsBlood2014124115115224993879
- HarrisWJHuangXLynchJTThe histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cellsCancer Cell201221447348722464800
- GoardonNMarchiEAtzbergerACoexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemiaCancer Cell201119113815221251617
- WadaTKoyamaDKikuchiJHondaHFurukawaYOverexpression of the shortest isoform of histone demethylase LSD1 primes hemopoietic stem cells for malignant transformationBlood2015125243731374625904247
- SchenkTChenWCGöllnerSInhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemiaNat Med201218460561122406747
- NiwaHUmeharaTStructural insight into inhibitors of flavin adenine dinucleotide-dependent lysine demethylasesEpigenetics201712534035228277979
- LeeMGWynderCSchmidtDMMcCaffertyDGShiekhattarRHistone H3 lysine 4 demethylation is a target of nonselective antidepressive medicationChem Biol200613656356716793513
- ZhengYCYuBChenZSLiuYLiuHMTCPs: privileged scaffolds for identifying potent LSD1 inhibitors for cancer therapyEpigenomics20168565166627102879
- MaesTTirapuIMascaroAPreclinical characterization of a potent and selective inhibitor of the histone demethylase KDM1A for MLL leukemiaJ Clin Oncol20133115 Supple13543
- SomervailleTSalameroOMontesimosPSafety, pharmacokinetics (PK), pharmacodynamics (PD) and preliminary activity in acute leukemia of Ory-1001, a first-in-class inhibitor of lysine-specific histone demethylase 1A (LSD1/KDM1A): initial results from a first-in-human phase 1 studyPoster presented at: 58th American Society of Hematology Annual MeetingDecember 3–6, 2016San Diego, CA
- NeelamegamRRicqELMalvaezMBrain-penetrant LSD1 inhibitors can block memory consolidationACS Chem Neurosci20123212012822754608
- CuiSLimKCShiLThe LSD1 inhibitor RN-1 induces fetal hemoglobin synthesis and reduced disease pathology in sickle cell miceBlood2015126338639626031919
- JagadeeswaranRVazquezBAThiruppathiMPharmacological inhibition of LSD1 and mTOR reduces mitochondrial retention and associated ROS levels in the red blood cell of sickle cell diseaseExp Hematol201750465228238805
- RiversAVaitkusKIbanezVThe LSD1 inhibitor RN-1 recapitulates the fetal pattern of hemoglobin synthesis in baboons (P. anubis)Haematologica2016101668869726858356
- IbanezIVaitkusKRiversAEfficacy and safety of long-term RN-1 treatment to increase HbF in baboonsBlood2017129226026327908882
- McGrathJWilliamsonKEBalasubramanianSPharmacological inhibition of the histone lysine demethylase KDM1A suppresses the growth of multiple acute myeloid leukemia subtypesCancer Res20167671975198826837761
- MohammadHPSmithemanKNKamatCDA DNA hypom-ethylation signature predicts antitumor activity of LSD1 inhibitors in SCLCCancer Cell2015281576926175415
- LeeSHStubbsMLiuXMDiscovery of INCB059872, a novel FAD-directed LSD1 inhibitor that is effective in preclinical models of human and murine AMLCancer Res20167614 Suppl4712
- YeMLiuMLuYThe LSD1 inhibitor INCB059872 is synergistic with ATRA in models of non-APL acute myelogenous leukemiaCancer Res20167614 Suppl469626980761
- LiuXStubbsMYeMCombination of BET inhibitor INCB054329 and LSD1 inhibitor INCB059872 is synergistic for the treatment of AML in vitro and in vivoCancer Res20167614 Suppl4702
- LeeSHStubbsMJuvekarACombination of epigenetic regulation via LSD1 inhibition with signal transduction inhibitors significantly enhances anti-tumor activity in models of hematologic malignanciesCancer Res20177713 Suppl2032
- WelchDKahenECubittCLReedDRLSD1 inhibition alone and in combination with chemotherapy in Ewing sarcoma cell linesCancer Res20177713 Suppl2034
- RomanVDYeMLiuHThe evaluation of INCB059872, an FAD-directed covalent inhibitor of LSD1, in preclinical models of Ewing sarcomaCancer Res20177713 Suppl1162
- LeeSHLiuXMDiamondMThe evaluation of INCB059872, on FAD-directed inhibitor of LSD1, in preclinical models of human small cell lung cancerCancer Res20177614 Suppl4704
- IshikawaYGamoKYabukiMA novel LSD1 inhibitor T-3775440 disrupts GFI1B-containing complex leading to transdifferentiation and impaired growth of AML cellsMol Cancer Ther201616227328427903753
- OgasawaraDItohYTsumotoHLysine-specific demethylase 1-selective inactivators: protein targeted drug delivery mechanismAngew Chem Int Ed Engl201352338620862423824985
- SuginoMKawaharaMTatsumiGA novel LSD1 inhibitor NCD38 ameliorates MDS-related leukemia with complex karyotype by attenuating leukemia programs via activating super-enhancersLeukemia201731112303231428210006
- OgasawaraDSuzukiTMinoKSynthesis and biological activity of optically active NCL-1, a lysine-specific demethylase 1 selective inhibitorBioorg Med Chem201119123702370821227703
- SareddyGRViswanadhapalliSSurapaneniPSuzukiTBrennerAVadlamudiRKNovel KDm1A inhibitors induce differentiation and apoptosis of glioma stem cells via unfolded protein response pathwayOncogene201736172423243427893719
- MouldDPMcGouagleAEWisemanDHWilliamsELJordanAMReversible inhibitors of LSD1 as therapeutic agents in acute myeloid leukemia: clinical significance and progress to dateMed Res Rev201535358661825418875
- SornaVTheisenERStephensBHigh-throughput virtual screening identifies novel N′-(1-phenylethylidene)-benzohydrazides as potent, specific, and reversible LSD1 inhibitorsJ Med Chem201356239496950824237195
- FiskusWSharmaSShahBHighly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cellsLeukemia201428112155216424699304
- LiuSALuWTLiSYIdentification of JL1037 as a novel, specific, reversible lysine-specific demethylase 1 inhibitor that induce [sic] apoptosis and autophagy of AML cellsOncotarget2017819319013191428404874
- VianelloPSartoriLAmigoniFThieno [3,2-b]pyrrole-5- carboxiamides as new reversible inhibition of histone lysine demethylase KDM1A/LSD1 – part 2: structure-based drug design and structure-activity relationshipJ Med Chem20176051693171528186757
- MouldDPAlliCBrembergUDevelopment of (4-cyanophenyl) glycine derivatives as reversible inhibitors of lysine specific demethylase 1J Med Chem201760197984799928892629
- MouldDPBrembergUJordanAMDevelopment and evaluation of 4-(pyrrolidin-3-yl) benzonitrile derivatives as inhibitors of LSD1Bioorg Med Chem Lett201727204755475928927796
- DuanYCMaYCQinWPDesign and synthesis of tranylcypromine derivatives as novel LSD1/HDAC dual inhibitors for cancer treatmentEur J Med Chem201714039240228987602
- McLeanCMKaremakerIDvan LeeuwenFThe emerging roles of DOT1L in leukemia and normal developmentLeukemia201428112131213824854991
- FengQWangHNgHHMethylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domainCurr Biol200212121052105812123582
- MinJFengQLiZZhangYXuRMStructure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferaseCell2003112571172312628190
- OkadaYFengQLinYhDOT1L links histone methylation of leukemogenesisCell2005121216717815851025
- NguyenATTaranovaOHeJZhangYDOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesisBlood2011117256912692221521783
- DeshpandeAJDeshpandeASinhaAUAF10 regulates progressive H3K79 methylation and HOX gene expression in diverse AML subtypesCancer Cell201426689690825464900
- ChenSYangZWilkinsonAWThe PZP domain of AF10 senses unmodified H3K27 to regulate DOT1L-mediated methylation of H3K79Mol Cell201560231932726439302
- LiYYWenHXiYXAF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylationCell2014159355857125417107
- ChenCWKocheRPSinhaAUDOT1L inhibits SIRT1-mdiated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemiaNat Med201521433534325822366
- KuntimaddiAAchilleNJThorpeJDegree of recruitment of DOT1L to MLL-AF9 defines level of H3K79 di- and tri-methylation on target genes and transformation potentialCell Rep201511580882025921540
- OkudaHStanogevicBKanaiACooperative gene activation by AF4 and DOT1L drive MLL-rearranged leukemiaJ Clin Invest201712751918193128394257
- RanRRodriguezBLuoMDOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemiaBlood2016128797198127335278
- FengYYangYOrtegaMMEarly mammalian erythropoiesis requires the Dot1L methyltransferaseBlood2010116224483449120798234
- NguyenATHeJTaranovaOZhangYEssential role of DOT1L in maintaining normal adult hematopoiesisCell Res20112191370137321769133
- JoSYGranowiczEMMaillardIThomasDHessJLRequirement for Dot1l in murine postnatal hematopoiesis and leukemogenesis by MLL translocationBlood2011117184759476821398221
- DaigleSROlhavaEJTherkelsenCASelective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitorCancer Cell2011201536521741596
- DaigleSROlhavaEJTherkelsenCAPotent inhibition of DOT1L as treatment for MLL-fusion leukemiaBlood201312261017102523801631
- YuWChoryEJWernimontAKCatalytic site remodeling of the DOT1L methyltransferase by selective inhibitorsNat Commun20123128823250418
- WangYLiLZhangBDiscovery of novel disruptor of silencing telomeric 1-like (DOT1L) inhibitors using a target-specific function for the (s)-adenosyl-l-methionine (SAM)-dependent methyltransferase familyJ Med Chem20176052026203628165739
- WatersNJDaigleSRRehlaenderBNExploring drug delivery for the DOT1L inhibitor pinometostat (EPZ-5676): subcutaneous administration as an alternative to continuous IV infusion, in the pursuit of an epigenetic targetJ Control Release2015220Pt B75876526385168
- KlausCRIwanowiczDJohnstonDDOT1L inhibitor EPZ-5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged cellsJ Pharmacol Exp Ther2014350964665624993360
- WatersNJSmithSAOlhavaEJMetabolism and disposition of the DOT1L inhibitor, pinometostat (EPZ-5676), in rat, dog and humanCancer Chemother Pharmacol2016771436226645404
- SteinEMGarcia-ManeroGRizzieriDAA phase 1 study of the DOT1L inhibitor, pinometostat (EPZ-5676), in adults with relapse or refractory leukemia: safety, clinical activity, exposure and target inhibitionBlood2015126232547
- ShuklaNWetmoreCO’BrienMMFinal report of phase 1 study of the DOT1L inhibitor, pinometostat (EPZ-5676), in children with relapsed or refractory MLL-2 acute leukemiaBlood2016128222780
- ChandrasekharappaSCGuruSCManickamPPositional cloning of the gene for multiple endocrine neoplasia-type 1Science199727653114044079103196
- GuruSCGoldsmithPKBurnsALMenin, the product of the MEN1 gene, is a nuclear proteinProc Natl Acad Sci U S A1998954163016349465067
- YokoyamaASomervailleTCSmithKSRozenblatt-RosenOMeyersonMClearyMLThe menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesisCell2005123220721816239140
- CasliniCYangZEl-OstaMMilneTASlanyRKHessJLInter-action of MLL amino terminal sequences with menin is required for transformationCancer Res200767157275728317671196
- GrembeckaJBelcherAMHartleyTCierpickiTMolecular basis of the mixed lineage leukemia-menin interaction: implications for targeting mixed lineage leukemiasJ Biol Chem201028552406904069820961854
- ChenYXYanJKeeshanKThe tumor suppressor menin regulates hematopoiesis and myeloid transformation by influencing Hox gene expressionProc Natl Acad Sci U S A200610341018102316415155
- HughesCMRozenblatt-RosenOMilneTAMenin associated with a trithorax family histone methyltransferase complex and with the Hoxc8 locusMol Cell200413458759714992727
- CierpickiTGrembeckaJChanges and opportunities in targeting the menin-MLL interactionFuture Med Chem20146444746224635524
- MuraiMJChruszczMReddyGGrembeckaJCierpickiTCrystal structure of menin binding site for mixed lineage leukemia (MLL) proteinJ Biol Chem201128636317423174821757704
- GrembeckaJHeSShiAMenin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemiaNat Chem Biol20128327728422286128
- HeSMalikBBorkinDMenin-MLL inhibitors block oncogenic transformation by MLL-fusion proteins in a fusion partner-independent mannerLeukemia201630250851326084867
- DafflonCCraigVJMéreauHComplementary activities of DOT1L and menin inhibitors in MLL-rearranged leukemiaLeukemia20173161269127727840424
- SpencerDHYoungMALamprechtTLEpigenomic analysis of the HOX gene loci reveals mechanisms that may control canonical expression patterns in AML and normal hematopoietic cellsLeukemia20152961279128925600023
- KühnMWSongEFengZHTargeting chromatin regulators inhibits leukemogenic gene expression in NPM1 mutant leukemiaCancer Discov20166101166118127535106
- FalchettiAGenetics of multiple endocrine neoplasia type 1 syndrome: what’s new and what’s oldF1000Res2017673
- SvobodaLKBaileyNVan NoordRATumorigenicity of Ewing sarcoma is critically dependent on the trithorax proteins MLL1 and meninOncotarget20178145847127888797
- MalikRKhanAPAsanganiIATargeting the MLL complex in castration-resistant prostate cancerNat Med201521434435225822367
- HessJLYuBDLiBHansonRKorsmeyerSJDefects in yolk sac hematopoiesis in MLL-null embryosBlood1997905179918069292512
- YagiHDeguchiKAonoAKishimotoTKomoriTGrowth disturbance in fetal liver hematopoiesis of MLL-mutant miceBlood19989211081179639506
- ErnstPFisherJKAveryWWadeSFoyDKorsmeyerSJDefinitive hematopoiesis requires the mixed-lineage leukemia geneDev Cell20046343744315030765
- JudeCDClimerLXuDArtingerEFisherJKErnstPUnique and independent roles for MLL in adult hematopoietic stem cells and progenitorsCell Stem Cell20071332433718371366
- McMahonKAHiewSYHadjurSMLL has a critical role in fetal and adult hematopoietic stem cell self-renewalCell Stem Cell20071333834518371367
- MaillardIChenYXFriedmanAMenin regulates the function of hematopoietic stem cells and lymphoid progenitorsBlood200911381661166919228930
- MaillardIHessJLThe role of menin in hematopoiesisAdv Exp Med Biol2009668515720175452
- LiBGanTMeyersonMRabbittsTHErnstPDistinct pathways regulated by menin and by MLL1 in hematopoietic stem cells and developing B cellsBlood2013122122039204623908472
- GantTJudeCDZaffutoKErnstPDevelopmentally induced MLL1 loss reveals defects in postnatal haematopoiesisLeukemia201024101732174120724987
- MishraBPZaffutoKMArtingerELThe histone methyltransferase activity of MLL1 is dispensable for hematopoiesis and leukemogenesisCell Rep2014741239124724813891
- ArtingerELMishraBPZaffutoKMAn MLL-dependent network sustains hematopoiesisProc Natl Acad Sci U S A201311029120001200523744037
- GrembeckaJBelcherAMHartleyTCierpickiTMolecular basis of the mixed lineage leukemia-menin interaction: implication for targeting mixed lineage leukemiasJ Biol Chem201028552406904069820961854
- DhiAMuraiMJHeSStructural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemiaBlood2012120234461446922936661
- GrembeckaJHeSShiAMenin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemiaNat Chem Biol20128327728422286128
- BorkinDHeSMiaoHPharmacologic inhibition of the menin-MLL interaction blocks progression of MLL leukemia in vivoCancer Cell201527458960225817203
- HeSSenterTJPollockJHigh-affinity small-molecule inhibitors of the menin-mixed lineage leukemia (MLL) interaction closely mimic a natural protein-protein interactionJ Med Chem20145741543155624472025
- ZhouHLiuLHuangJStructure-based design of high-affinity macrocyclic peptidomimetics to block the menin-mixed lineage leukemia 1 (MLL 1) protein-protein interactionJ Med Chem20135631113112323244744
- LiLZhouRGengHDiscovery of two aminoglycoside antibiotics as inhibitors targeting the menin-mixed lineage leukemia interfaceBioorg Med Chem Lett20142492090209324709560
- XuYYueLWangYDiscovery of novel inhibitors targeting the menin-mixed lineage leukemia interface using pharmacophore-and docking-based virtual screeningJ Chem Inf Model20165691847185527513308
- BorkinDPollockJKempinskaKProperty focused structure-based optimization of small molecule inhibitors of the protein-protein interaction between menin and mixed lineage leukemiaJ Med Chem201659389291326744767
- SpurrSSBayleEDYuWNew small molecule inhibitors of histone methyl transferase DOT1L with a nitrile as a non-traditional replacement for heavy halogen atomsBioorg Med Chem Lett201626184518452227485386
- ChenCZhuHStaufferFDiscovery of novel Dot1L inhibitors through a structure-based fragmentation approachACS Med Chem Lett20167873574027563395
- MöbitzHMachauerRHolzerPDiscovery of potent, selective, and structurally novel Dot1L inhibitors by a fragment linking approachACS Med Chem Lett20178333834328337327