
Abstract
Purpose
This study aimed to investigate the key long non-coding RNAs (lncRNAs) associated with colon cancer and elucidate their possible mechanisms of action.
Patients and methods
Eight early-stage (ES) colon tumor tissues, eight late-stage (LS) colon tumor tissues, and eight normal tissues were collected, and they were subjected to high-throughput RNA sequencing. Subsequently, comprehensive bioinformatics analyses, including the identification of differentially expressed mRNAs and lncRNAs, functional enrichment analysis, and construction of a protein–protein interaction network and an miRNA–lncRNA–mRNA regulatory network were performed. Additionally, the expression of key lncRNAs was verified using real-time quantitative PCR (qPCR).
Results
In total, 549 common differentially expressed mRNAs and 30 common differentially expressed lncRNAs were identified in both the ES and LS colon cancer samples upon comparison with the normal samples. Functional enrichment analysis showed that KIAA0125 was significantly enriched in the PI3K-Akt signaling pathway and that MSTRG.35002.1 was markedly enriched in BMP signaling-related functions. Moreover, key miRNA–lncRNA–mRNA relationships, such as hsa-miR-29b-3p-KIAA0125-BCL2 and hsa-miR-29b-3p-MSTRG.35002.1-MMP2, were identified. Notably, the qPCR assay confirmed that KIAA0125 and MSTRG.35002.1 were significantly downregulated in both ES and LS colon tumor tissues compared with normal colon tissues.
Conclusion
Our findings indicate that key lncRNAs, including KIAA0125 and MSTRG.35002.1, may be involved in colorectal cancer (CRC) development. Downregulation of KIAA0125 may contribute to CRC development via sponging of hsa-miR-29b-3p to regulate BCL2 expression or regulating the PI3K-Akt signaling pathway. Downregulation of MSTRG.35002.1 may promote CRC development via sponging of hsa-miR-29b-3p to regulate MMP2 expression or regulating the BMP signaling pathway.
Introduction
Colorectal cancer (CRC) is a common malignant tumor that ranks as the third leading cause of mortality worldwide.Citation1 The 5-year survival rate of patients with CRC decreases from >90% in stage I tô10% in stage IV.Citation2 Because of its high prevalence and poor prognosis, it remains imperative to explore promising molecular biomarkers or targets to improve prognostic prediction and therapeutic outcomes for CRC.
Non-coding RNAs (ncRNAs) have been revealed to be major components of the human transcriptome, and they are mainly divided into miRNAs (<200 nucleotides) and long ncRNAs (lncRNAs, >200 nucleotides) based on their length. In CRC pathogenesis, the crucial roles of several lncRNAs have been shown to be altered and to play critical roles in tumor biology, including oncogenic lncRNAs, 91H, CCAT1, DANCR, FEZF1-AS1, HOTAIR, HOTTIP, and MALAT1Citation3–Citation10 as well as tumor suppressive lncRNA, GAS5, MEG3, LINC01296, RP11-462C24.1, and TUSC7.Citation11–Citation15 For example, upregulation of the lncRNA PANDAR (promoter of CDKN1A antisense DNA damage-activated RNA) promotes the metastasis of CRC by epithelial–mesenchymal transition pathway,Citation16 and silencing of lncRNA PANDAR switches curcumin-induced senescence to apoptosis.Citation17
Furthermore, in 2011, lncRNAs were first revealed to regulate the encoding protein gene level by functioning as a competing endogenous RNA (ceRNA) to sponge miRNAs.Citation18 It has been reported that lncRNA SPRY4-IT1 can function as a ceRNA of miR-101-3p to regulate the proliferation and invasion of CRC cells,Citation19 and lncRNA UICLM can act as a ceRNA of miR-215 to regulate ZEB2 expression, thus promoting liver metastasis in CRC.Citation20 These findings imply that lncRNA, miRNA, and mRNA have interactive roles in CRC development. However, the key lncRNAs associated with CRC are largely unknown, as is their regulatory mechanism. Further identification of the key lncRNAs involved in CRC progression is of great importance.
In this study, high-throughput RNA sequencing of colon tumor tissues and normal colon tissues and subsequent comprehensive bioinformatics analyses were performed to identify key lncRNAs associated with colon cancer, followed by the validation of dysregulated lncRNAs through real-time quantitative PCR (qPCR). Our findings may provide novel insights into the molecular mechanisms underlying colorectal carcinogenesis.
Patients and methods
Patient samples
Eight early-stage (ES) colon tumor tissues were collected from patients who were diagnosed with stage I or II colon cancer (seven men and one woman, age 56.75±11.66 years), and eight late-stage (LS) colon tumor tissues were obtained from patients with stage III or IV colon cancer (four men and four women, age 58.25±11.07 years). The diagnosis of colon cancer was confirmed by surgery, and patients who did not receive radiotherapy or chemotherapy and volunteered to participate in the study and subsequent follow-up were enrolled. Patients with a medical history of diabetes mellitus, hypertension, hepatitis, and pulmonary tuberculosis were excluded. In addition, eight adjacent normal colon tissues, defined as a >5.0 cm distance from the tumor edge, were also obtained from four patients with ES and four patients with LS colon cancer (five men and three women, age 59.86±8.84 years) and used as normal controls (N). All collected clinical tissue samples were quickly frozen in liquid nitrogen and stored at −80°C. This study was conducted in accordance with the Declaration of Helsinki and approved by the ethics review board of China-Japan Union Hospital of Jilin University. Each patient provided signed informed written consent for inclusion in this study and for publication.
Construction of the cDNA library and high-throughput RNA sequencing
Total RNA was extracted from clinical tissue samples using an RNAiso Plus Kit (TaKaRa, Shiga, Japan), followed by the detection of its purity and concentration using agarose gel electrophoresis and a Nanodrop spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA). A poly (A) tail enrichment cDNA library was constructed following standard methods. RNA sequencing was carried out using the Illumina HiSeq 3000 platform. The generated sequencing data, with accession code SRP158734, were deposited in the NCBI database.
Quality control and read alignment
Quality control to obtain clean reads was performed using Trimmomatic (v3.6).Citation21 Subsequently, the obtained clean reads were aligned with the human GRCh38 reference using Tophat 2 version 2.1.1Citation22 using the following parameters: read-mismatches, 2; read-edit-dist, 2; and library-type, fr-firststrand.
Transcriptome assembly and identification of differentially expressed mRNAs and differentially expressed lncRNAs
The read alignment results of 24 samples were then subjected to transcriptome assembly using stringtie (v1.33b)Citation23 with the human genome annotation file (v28, GENCODE) as the reference. The gene expression levels in each sample were quantified using cuffquant and cuffnorm.Citation24 After transcriptome assembly, the transcripts with the class codes of types i, u, and x were considered unknown: type i indicated that the predicted transcripts were located in introns, type u indicated that the predicted transcripts were located in the gene area of the reference transcripts, and type x indicated that the exons of the predicted transcripts overlapped with reference transcripts but in the opposite direction. The predicted novel_lncRNAs, named using tracking_id, were then identified by prediction of the coding ability of the transcriptome sequence using Coding Potential Calculator (CPC), Coding-Non-Coding-Index (CNCI), Coding Potential Assessment Tool (CPAT), and PFAM.
The differentially expressed mRNAs and differentially expressed lncRNAs in the ES vs N, LS vs N, and LS vs ES groups were, respectively, identified using the cuffdiffCitation24 tool with a threshold value of q_value <0.05. Clustering heatmap analysis for differentially expressed mRNAs and differentially expressed lncRNAs was then conducted using the pheatmap version 1.0.8 in R3.4.1. Moreover, common differentially expressed mRNAs and common differentially expressed lncRNAs between the ES vs N and LS vs N groups were then identified using a Venn diagram.
Functional enrichment analysis of differentially expressed mRNAs
To understand the biological functions of the differentially expressed mRNAs, Gene Ontology (GO) functions, including biological process (BP), cellular component (CC), and molecular function (MF), as well as Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were completed using clusterProfiler v3.2.1.Citation25 A P value of <0.05 calculated from the hypergeometric test was set as the threshold value.
Construction of a protein–protein interaction (PPI) network
Based on the information in the Search Tool for the Retrieval of Interacting GenesCitation26 database that provides both predicted and experimental interactions between proteins, the PPI pairs between common differentially expressed mRNAs were obtained with the required confidence >0.4. Then, the PPI network was constructed using Cytoscape software.Citation27 The network topology property indicators, including degree centrality, betweenness centrality, and closeness centrality, were analyzed using CytoNCACitation28 in Cytoscape software. A node with a higher score of network topology property indicators indicated a more important role in that node in the PPI network, which was considered as a hub node.
Correlation analysis of lncRNAs and mRNAs
The correlation of gene expression levels between differentially expressed mRNAs and differentially expressed lncRNAs was analyzed by calculating the Pearson correlation coefficient. The Benjamini and HochbergCitation29 procedure was carried out for multiple-testing adjustment. The p.adjust <0.01 and |cor| >0.5 indicated a significant correlation between lncRNAs and mRNAs, suggesting that these mRNAs were potential targets of lncRNAs.
Prediction of lncRNA function
To thoroughly understand the functions of the lncRNAs, GO BP, and KEGG function enrichment analyses for the target mRNAs of common differentially expressed lncRNAs were also conducted using clusterProfiler v3.2.1, with a cut-off value of P<0.05.
Construction of an miRNA–lncRNA–mRNA regulatory network
The miRNAs targeting differentially expressed mRNAs were predicted using the miRTarBase_2017 database in Enrichr toolCitation30 with p.adjust <0.05, and the miRNA–mRNA regulatory relationships were obtained. Moreover, the binding sites between the above-mentioned miRNAs and lncRNAs were predicated using miRandaCitation31 with the parameters of -sc 140, -en −20, and –strict, and the miRNA–lncRNA regulatory relationships were obtained. By combining the miRNA–mRNA and miRNA–lncRNA regulatory relationships, the miRNA–lncRNA–mRNA regulatory network was then established using Cytoscape software.
Real-time qPCR analysis
To further verify the key differentially expressed lncRNAs identified earlier, qPCR was performed to detect the expression levels in eight ES colon tumor tissues, eight LS colon tumor tissues, and eight normal colon tissues. Real-time qPCR was conducted to detect the expression of lncRNAs using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific, Waltham, MA, USA). The primer sequences (forward and reverse) used for the amplification of targets were as follows: KIAA0125: 5′-TGGCAAAGGCAAGTGAC-3′ and 5′-GGCAGAAGGATGAACCC-3′; MSTRG.35002.1: 5′-TTCATTATCCGAACAAC-3′ and 5′-TGATGCCACTAACCAG-3′; and GAPDH: 5′-TGACAACTTTGGTATCGTGGAAGG-3′ and 5′-AGGCAGGGATGATGTTCTGGAGAG-3′. By normalization with the internal control GAPDH, the relative gene expression was calculated using the 2−ΔΔCT method.
Statistical analysis
The obtained data are presented as the mean ± SD. Using GraphPad Prism 5 (Graphpad Software, San Diego, CA, USA), significant differences between groups were evaluated with Student’s t-test or one-way ANOVA. A statistically significant result was obtained when P<0.05.
Results
Prediction of novel lncRNAs
In total, 2,292, 2,269, 2,042, and 2,368 transcripts were, respectively, identified from the CPC, CNCI, CPAT, and PFAM analyses. An integrated analysis using CPC, CNCI, CPAT, and PFAM analyses identified a total of 1,993 novel_lncRNAs, named using the tracking_id, from 2,368 unknown transcripts.
Identification of differentially expressed mRNAs and differentially expressed lncRNAs
With the cut-off value of q_value <0.05, a total of 805 differentially expressed mRNAs (428 up- and 377 downregulated) and 78 differentially expressed lncRNAs (55 up- and 23 downregulated, Table S1) were identified in the ES vs N group, and 810 differentially expressed mRNAs (480 up- and 330 downregulated) and 65 differentially expressed lncRNAs (41 up- and 24 downregulated, Table S1) were identified in the LS vs N group. However, differentially expressed mRNAs and differentially expressed lncRNAs were not screened out in the LS vs ES group with a q_value <0.05. The cut-off value was then changed to P<0.05, and 235 differentially expressed mRNAs (143 up- and 92 downregulated) and 82 differentially expressed lncRNAs (28 up- and 54 downregulated) were identified in the LS vs ES group. Moreover, heatmap analysis revealed that ES or LS samples could be significantly distinguished from normal (N) tissue samples according to the expression levels of differentially expressed mRNAs or differentially expressed lncRNAs (Figure S1). A subsequent Venn diagram analysis indicated that there were 549 common differentially expressed mRNAs (Figure S2A) and 30 common differentially expressed lncRNAs (Figure S2B) between the ES vs N and LS vs N groups.
Functional enrichment analysis of differentially expressed mRNAs
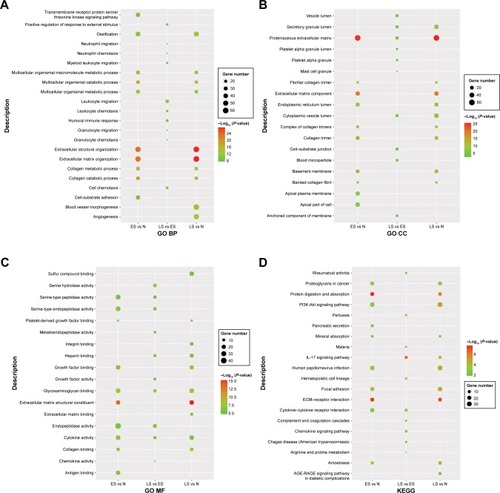
The results of GO and KEGG pathway enrichment analyses (top ten) are shown in . The differentially expressed mRNAs in the ES vs N and LS vs N groups were all markedly enriched in GO functions associated with extracellular matrix organization (BP function), proteinaceous extracellular matrix (CC function), and extracellular matrix structural constituent (MF function). The differentially expressed mRNAs in the LS vs ES group were enriched in the prominent GO functions related to leukocyte migration (BP function), proteinaceous extracellular matrix (CC function), and endopeptidase activity (MF function). In addition, the differentially expressed mRNAs in the ES vs N and LS vs N groups were all significantly enriched in several KEGG pathways, such as the PI3K-Akt signaling pathway, focal adhesion, and extracellular matrix–receptor interaction; the differentially expressed mRNAs in the LS vs ES group were markedly enriched in several KEGG pathways, including the IL-17 signaling pathway, the chemokine signaling pathway, and cytokine–cytokine receptor interaction.
Figure 1 Functional enrichment analyses of differentially expressed mRNAs identified from the ES vs N, LS vs N, and LS vs ES groups. (A) Gene Ontology (GO) biological process (BP); (B) GO cellular component (CC); (C) GO molecular function (MF); (D) Kyoto Encyclopedia of Genes and Genomes (KEGG). Node size: gene ratio; node color: P-value.
Abbreviations: ES, early stage; LS, late stage; N, normal samples.
PPI network analyses
The PPI network constructed using the common differentially expressed mRNAs between the ES vs N and LS vs N groups consisted of 377 nodes and 1,324 interaction pairs (Figure S3). After analysis of network topology properties, the top 15 nodes with the highest scores of degree centrality, betweenness centrality, and closeness centrality were, respectively, identified (), among which MMP2, MMP9, BCL2, SPP1, BMP2, and CCND1 belonged to the top 15 calculated from the three network topological properties.
Table 1 The hub nodes in the protein–protein interaction network according to three network topology properties
Correlation analysis of lncRNAs and mRNAs
Using Pearson’s correlation analysis, the top ten lncRNAs were MSTRG.9732.1 (number = 402), CTB-28J9.3 (number = 390), MSTRG.25642.1 (number = 372), MAFG-AS1 (number = 312), MSTRG.225.1 (number = 306), MSTRG.35002.1 (number = 302), MSTRG.1647.1 (number = 284), MSTRG.29201.1 (number = 278), MSTRG.25557.1 (number = 275), and RP11-400N13.3 (number = 233) according to the number of their target mRNAs.
Functional analysis of lncRNAs
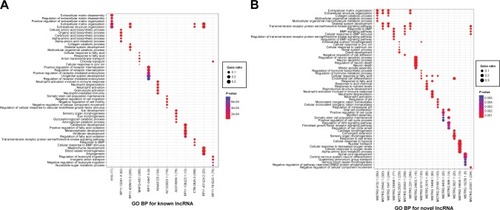
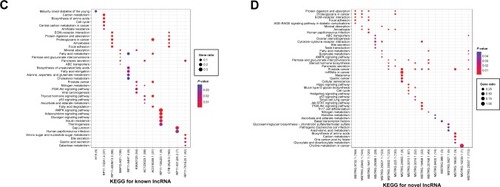
To better understand the functions of the lncRNAs, GO BP and KEGG pathway enrichment analyses for the target mRNAs of the differentially expressed lncRNAs were conducted (). Notably, functional enrichment analysis showed that KIAA0125 was significantly enriched in neutrophil activation (GO BP) and the PI3K-Akt signaling pathway (KEGG). MSTRG.35002.1 (RP1-290B4.1) was markedly enriched in BMP signaling-related functions (GO BP) and steroid hormone biosynthesis (KEGG).
Figure 2 Functional enrichment analyses of common differentially expressed lncRNAs identified from the ES vs N and LS vs N groups. (A) GO BP for common differentially expressed lncRNAs; (B) GO BP for common novel differentially expressed lncRNAs; (C) KEGG for common differentially expressed lncRNAs; (D) KEGG for common novel differentially expressed lncRNAs. Node size: gene ratio; node color: P-value.
Abbreviations: ES, early stage; LS, late stage; N, normal samples; lncRNAs, long non-coding RNAs; GO, Gene Ontology; BP, biological process; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Construction of an miRNA–lncRNA–mRNA ceRNA regulatory network
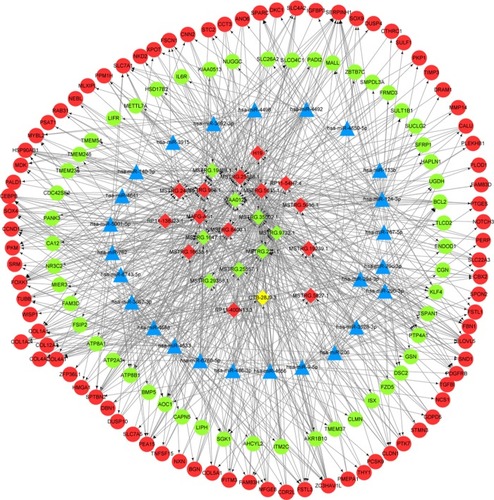
The miRNAs targeting common differentially expressed mRNAs were predicted using the miRTarBase_2017 database in Enrichr tool, and the top five miRNAs with the highest number of target mRNAs were identified, including hsa-miR-124-3p, hsa-miR-29a-3p, hsa-miR-29b-3p, hsa-miR-29c-3p, and hsa-miR-4533 (). By integrating the miRNA–mRNA and miRNA–lncRNA regulatory relationships, the miRNA–lncRNA–mRNA network was then constructed. The miRNA–lncRNA–mRNA regulatory network is shown in , in which several ceRNA relationships were revealed. For example, the ceRNA relationships hsa-miR-29b-3p-H19-FSCN1, hsa-miR-29b-3p-KIAA0125-BCL2, and hsa-miR-29b-3p-MSTRG.35002.1-MMP2 were identified. The downregulated lncRNAs MSTRG.35002.1, MSTRG.9732.1, MSTRG.225.1, and KIAA0125 and the upregulated lncRNAs MSTRG.5015.1, MSTRG.25168.1, and MAFG-AS1 were the central nodes in this network, indicating their important roles in this network.
Table 2 The target mRNAs for the top5 miRNAs
Figure 3 The miRNA–lncRNA–mRNA ceRNA network. The blue triangle node represents miRNA, the diamond node represents lncRNA, and the circular node represents mRNA. Red indicates upregulation, and green indicates downregulation.
Abbreviations: lncRNAs, long non-coding RNAs; ceRNA, competing endogenous RNA.
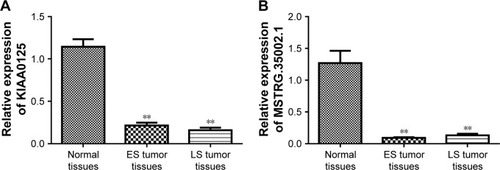
Validation of the differential expression of KIAA0125 and MSTRG.35002.1 by qPCR
To further verify the differential expression of the key differentially expressed lncRNAs identified earlier, qPCR was conducted to determine their expression levels in ES and LS colon tumor tissues. As shown in , KIAA0125 and MSTRG.35002.1 were significantly downregulated in both ES and LS colon tumor tissues compared with normal colon tissues, which was in line with the results of the aforementioned differential expression analysis.
Figure 4 qPCR confirmed the differential expression of (A) KIAA0125 and (B) MSTRG.35002.1 in ES and LS colon tumor tissues compared with that in normal tissues. Data are presented as the mean ± SD. **P<0.01 compared with normal tissues.
Abbreviations: qPCR, quantitative PCR; ES, early stage; LS, late stage.
Discussion
In this study, we investigated the mechanism underlying CRC by high-throughput RNA sequencing of colon cancer samples and normal samples and subsequent bioinformatics analysis. A total of 549 common differentially expressed mRNAs and 30 common differentially expressed lncRNAs were identified in both ES and LS colon cancer samples after comparison with normal samples. MMP2 and BCL2 were identified as hub nodes in the PPI network. In addition, functional analysis of lncRNAs revealed that KIAA0125 was significantly enriched in the PI3K-Akt signaling pathway and that MSTRG.35002.1 was markedly enriched in BMP signaling-related functions. Moreover, key miRNA–lncRNA–mRNA relationships, such as hsa-miR-29b-3p-H19-FSCN1, hsa-miR-29b-3p-KIAA0125-BCL2, and hsa-miR-29b-3p-MSTRG.35002.1-MMP2, were identified in the ceRNA network. Notably, the qPCR assay confirmed the differential expression of KIAA0125 and MSTRG.35002.1.
LncRNA H19, located at chromosome 11p15.5 locus, is expressed in the cell nucleus and cytoplasm.Citation32 It has been shown to act as an oncogene in various cancers, including CRC.Citation33–Citation35 In addition, several lncRNA H19 and miR-29b associated ceRNA relationships have been reported to play important roles in carcinogenesis. For example, LncRNA H19/miR-29b-3p/PGRN axis promoted epithelial–mesenchymal transition of CRC cells by acting on Wnt Signaling.Citation36 LncRNA H19 regulates E2F1 expression by competitively sponging endogenous miR-29a-3p in clear-cell renal cell carcinoma.Citation37 In this study, we found that lncRNA H19 was significantly upregulated in CRC tissues compared with adjacent normal control. Upregulation of H19 might regulate FSCN1 expression by competitively sponging miR-29b-3p. FSCN1 is an actin-binding protein required for the formation of cytoplasmic bundles of microfilaments.Citation38 FSCN1 was reported to be an oncogene in several cancers.Citation39–Citation42 These results indicated our results are reliable.
Increasing evidence has revealed that the aberrant expression of BCL2 is involved in several malignancies, including CRC.Citation43 Given the role of BCL2 in CRC, we believe that the aberrant expression of BCL2 contributes to the development of CRC. In addition, several studies have confirmed that hsa-miR-29b-3p is involved in several human cancers.Citation44 Basal levels of circulating hsa-miR-29b-3p have been shown to be associated with overall survival in patients with metastatic CRC.Citation45 Moreover, hsa-miR-29b-3p is considered to be a promising miRNA-based therapy against CRC, and an miR-29b byproduct sequence has been shown to affect tumor suppressive activities in KRAS mutant colon cancer cells.Citation46 These findings imply that hsa-miR-29b-3p plays a key role in CRC development. Moreover, lncRNA KIAA0125 was first identified to play a role in neurogenesis.Citation47 However, the role of KIAA0125 in CRC has not been determined. In this study, hsa-miR-29b-3p-KIAA0125-BCL2 was a key miRNA–lncRNA–mRNA relationship detected in the ceRNA regulation network, and KIAA0125 was experimentally verified to be downregulated in colon cancer samples. We hypothesize that the downregulation of KIAA0125 may contribute to CRC development via the regulation of BCL2 expression by sponging hsa-miR-29b-3p.
Furthermore, KIAA0125 was found to be significantly enriched in the PI3K-Akt signaling pathway in this study. The PI3K-Akt signaling pathway is involved in regulating cell growth and survival under pathological conditions, including cancer. Accumulating evidence has highlighted the critical role of PI3K-Akt signaling in CRC development and progression.Citation48 Moreover, targeting the PI3K/AKT/mTOR pathway has been suggested to be a promising therapeutic strategy for CRC therapy.Citation49 Given the key role of the PI3K-Akt signaling pathway in CRC, we speculate that KIAA0125 might have significant roles in CRC development.
On the other hand, the hsa-miR-29b-3p-MSTRG.35002.1-MMP2 regulatory relationship was identified in the ceRNA network. Plasma MMP-2 expression was found to be increased in patients with lymph node-positive CRC, and it may be used as a potential predictor in this malignancy.Citation50 Considering the crucial role of hsa-miR-29b-3p and MMP2, we suggest that MSTRG.35002.1, a novel lncRNA, may contribute to CRC development by regulating hsa-miR-29b-3p and MMP2. Furthermore, MSTRG.35002.1 was markedly enriched in BMP signaling-related functions. BMPs and their receptors are widely involved in the pathogenesis of some solid tumors. BMP signaling has been shown to promote cell invasion and bone metastasis in breast cancer via the SMAD pathway.Citation51 Moreover, loss of SMAD4 can regulate BMP signaling to enhance the metastasis of CRC.Citation52 Based on our results, we speculate that MSTRG.35002.1 may regulate CRC development via the BMP signaling pathway.
There are some limitations of this study that should be noted. First, the sample size is relatively small in our study. Only 24 samples were sequenced in this study, and the average age of the included patients was relatively young. Second, because of difficulty in collecting qualified samples, sex distribution was unbalanced in samples of ES group. Third, we only performed qPCR to detect the expression of KIAA0125 and MSTRG.35002.1. The ceRNA relationships, including hsa-miR-29b-3p-KIAA0125-BCL2 and hsa-miR-29b-3p-MSTRG.35002.1-MMP2, and their role in CRC development should be further verified through in vitro and in vivo experiments.
Conclusion
In summary, our findings indicate that key lncRNAs, including KIAA0125 and MSTRG.35002.1, may be involved in CRC development. Downregulation of KIAA0125 may contribute to CRC development by sponging hsa-miR-29b-3p to regulate BCL2 expression or by regulating the PI3K-Akt signaling pathway. Downregulation of MSTRG.35002.1 may promote CRC development by sponging hsa-miR-29b-3p to regulate MMP2 expression or by affecting the BMP signaling pathway. Further functional experiments are needed to verify these observations.
Acknowledgments
This study was supported by the Norman Bethune Program of Jilin University (No 2012201), Science and Technology Development Project of Jilin Province (No 20180101124JC), Special Project for Health Research of Jilin Province (No 2018SCZ031), and Health Technology Innovation Project of Jilin Province (No 3D517ED43430).
Disclosure
The authors report no conflicts of interest in this work.
References
- SiegelRLMillerKDFedewaSAColorectal cancer statistics, 2017CA Cancer J Clin2017673104117
- TanoueYTanakaNNomuraYPrimary site resection is superior for incurable metastatic colorectal cancerWorld J Gastroenterol201016283561356620653065
- DengQHeBGaoTUp-regulation of 91h promotes tumor metastasis and predicts poor prognosis for patients with colorectal cancerPLoS One201497e10302210.1371/journal.pone.010302225058480
- AlaiyanBIlyayevNStojadinovicADifferential expression of colon cancer associated transcript1 (CCAT1) along the colonic adenoma-carcinoma sequenceBMC Cancer20131319610.1186/1471-2407-13-19623594791
- WangYLuZWangNLong noncoding RNA DANCR promotes colorectal cancer proliferation and metastasis via mir-577 spongingExp Mol Med20185055710.1038/s12276-018-0082-529717105
- BianZZhangJLiMLncRNA-FEZF1-AS1 promotes tumor proliferation and metastasis in colorectal cancer by regulating PKM2 signalingClin Cancer Res201824194808481910.1158/1078-0432.CCR-17-296729914894
- LuoZFZhaoDLiXQClinical significance of hotair expression in colon cancerWorld J Gastroenterol201622225254525910.3748/wjg.v22.i22.525427298568
- TatangeloFDi MauroAScognamiglioGPosterior hox genes and hotair expression in the proximal and distal colon cancer pathogenesisJ Transl Med201816135010.1186/s12967-018-1725-y30541551
- RuiYHuMWangPLncRNA HOTTIP mediated DKK1 downregulation confers metastasis and invasion in colorectal cancer cellsHistol Histopathol20181804310.14670/hh-18-043
- XuYZhangXHuXThe effects of lncRNA MALAT1 on proliferation, invasion and migration in colorectal cancer through regulating sox9Mol Med20182415210.1186/s10020-018-0050-530285605
- ChengKZhaoZWangGWangJZhuWLncRNA GAS5 inhibits colorectal cancer cell proliferation via the miR1825p/FOXO3a axisOncol Rep20184042371238010.3892/or.2018.658430066886
- ZhuYChenPGaoYMEG3 activated by vitamin D inhibits colorectal cancer cells proliferation and migration via regulating clusterinEBioMedicine20183014815710.1016/j.ebiom.2018.03.03229628342
- YuanZYuXNiBOverexpression of long non-coding RNA-CTD903 inhibits colorectal cancer invasion and migration by repressing Wnt/beta-catenin signaling and predicts favorable prognosisInt J Oncol20164862675268510.3892/ijo.2016.344727035092
- ShiDZhengHZhuoCLow expression of novel lncRNA RP11-462c24.1 suggests a biomarker of poor prognosis in colorectal cancerMed Oncol20143173110.1007/s12032-014-0374-024908062
- XuJZhangRZhaoJThe novel long noncoding RNA TUSC7 inhibits proliferation by sponging MiR-211 in colorectal cancerCell Physiol Biochem201741263564410.1159/00045793828214867
- LuMLiuZLiBWangGLiDZhuYThe high expression of long non-coding RNA PANDAR indicates a poor prognosis for colorectal cancer and promotes metastasis by EMT pathwayJ Cancer Res Clin Oncol20171431718110.1007/s00432-016-2252-y27629879
- ChenTYangPWangHHeZYSilence of long noncoding RNA PANDAR switches low-dose curcumin-induced senescence to apoptosis in colorectal cancer cellsOnco Targets Ther20171048349110.2147/OTT.S12754728176943
- SalmenaLPolisenoLTayYKatsLPandolfiPPA ceRNA hypothesis: the Rosetta stone of a hidden RNA language?Cell2011146335335810.1016/j.cell.2011.07.01421802130
- JinJChuZMaPMengYYangYLong non-coding RNA SPRY4-IT1 promotes proliferation and invasion by acting as a ceRNA of miR-101-3p in colorectal cancer cellsTumour Biol2017397101042831771625010.1177/101042831771625028720069
- ChenDLLuYXZhangJXLong non-coding RNA UICLM promotes colorectal cancer liver metastasis by acting as a ceRNA for microRNA-215 to regulate ZEB2 expressionTheranostics20177194836484910.7150/thno.2094229187907
- BolgerAMLohseMUsadelBTrimmomatic: a flexible trimmer for illumina sequence dataBioinformatics201430152114212010.1093/bioinformatics/btu17024695404
- KimDPerteaGTrapnellCPimentelHKelleyRSalzbergSLTopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusionsGenome Biol2013144R3610.1186/gb-2013-14-4-r3623618408
- PerteaMPerteaGMAntonescuCMChangTCMendellJTSalzbergSLStringTie enables improved reconstruction of a transcriptome from RNA-seq readsNat Biotechnol201533329029510.1038/nbt.312225690850
- TrapnellCWilliamsBPerteaGTranscript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiationNat Biotechnol201028551151510.1038/nbt.162120436464
- YuGWangLGHanYHeQYClusterprofiler: an R package for comparing biological themes among gene clustersOMICS201216528428710.1089/omi.2011.011822455463
- SzklarczykDFranceschiniAWyderSSTRING v10: protein–protein interaction networks, integrated over the tree of lifeNucleic Acids Res201543Database issueD447D45210.1093/nar/gku100325352553
- KohlMWieseSWarscheidBCytoscape: software for visualization and analysis of biological networksMethods Mol Biol201169629130310.1007/978-1-60761-987-1_1821063955
- TangYLiMWangJPanYWuFXCytoNCA: a cytoscape plugin for centrality analysis and evaluation of protein interaction networksBiosystems2015127677210.1016/j.biosystems.2014.11.00525451770
- BenjaminiYHochbergYControlling the false discovery rate: a practical and powerful approach to multiple testingJ R Stat Soc1995571289300
- ChenEYTanCMKouYEnrichr: interactive and collaborative HTML5 gene list enrichment analysis toolBMC Bioinformatics201314112810.1186/1471-2105-14-12823586463
- JohnBEnrightAJAravinATuschlTSanderCMarksDSHuman microRNA targetsPLoS Biol2004211e36310.1371/journal.pbio.002036315502875
- GaboryAJammesHDandoloLThe H19 locus: role of an imprinted non-coding RNA in growth and developmentBioessays201032647348010.1002/bies.20090017020486133
- LiJHuangYDengXLong noncoding RNA H19 promotes transforming growth factor-beta-induced epithelial-mesenchymal transition by acting as a competing endogenous RNA of miR-370-3p in ovarian cancer cellsOnco Targets Ther20181142744010.2147/OTT.S14990829403287
- WangMHanDYuanZLong non-coding RNA H19 confers 5-Fu resistance in colorectal cancer by promoting SIRT1-mediated autophagyCell Death Dis2018912114910.1038/s41419-018-1187-430451820
- HuangZLeiWHuHBZhangHZhuYH19 promotes non-small-cell lung cancer (NSCLC) development through STAT3 signaling via sponging miR-17J Cell Physiol2018233106768677610.1002/jcp.2653029693721
- DingDLiCZhaoTLiDYangLZhangBLncRNA H19/miR-29b-3p/PGRN axis promoted epithelial-mesenchymal transition of colorectal cancer cells by acting on Wnt signalingMol Cells201841542343510.14348/molcells.2018.225829754471
- HeHWangNYiXTangCWangDLong non-coding RNA H19 regulates E2F1 expression by competitively sponging endogenous miR-29a-3p in clear cell renal cell carcinomaCell Biosci201776510.1186/s13578-017-0193-z29214011
- HashimotoYSkacelMAdamsJCRoles of fascin in human carcinoma motility and signaling: prospects for a novel biomarker?Int J Biochem Cell Biol20053791787180410.1016/j.biocel.2005.05.00416002322
- HankerLCKarnTHoltrichUPrognostic impact of fascin-1 (fscn1) in epithelial ovarian cancerAnticancer Res201333237137723393326
- DarnelADBehmoaramEVollmerRTFascin regulates prostate cancer cell invasion and is associated with metastasis and biochemical failure in prostate cancerClin Cancer Res20091541376138310.1158/1078-0432.CCR-08-178919228738
- LiangZWangYShenZFascin 1 promoted the growth and migration of non-small cell lung cancer cells by activating YAP/TEAD signalingTumour Biol2016378109091091510.1007/s13277-016-4934-026886283
- ZhaoWGaoJWuJExpression of fascin-1 on human lung cancer and paracarcinoma tissue and its relation to clinicopathological characteristics in patients with lung cancerOnco Targets Ther201582571257610.2147/OTT.S8191526451116
- LiaoWTYeYPZhangNJMicroRNA-30b functions as a tumour suppressor in human colorectal cancer by targeting KRAS, PIK3CD and BCL2J Pathol2014232441542710.1002/path.430924293274
- YanWYangWLiuZWuGCharacterization of microRNA expression in primary human colon adenocarcinoma cells (SW480) and their lymph node metastatic derivatives (SW620)Onco Targets Ther2018114701470910.2147/OTT.S16923330127618
- UliviPCanaleMPassardiACirculating plasma levels of miR-20b, miR-29b and miR-155 as predictors of bevacizumab efficacy in patients with metastatic colorectal cancerInt J Mol Sci201819110.3390/ijms19010307
- InoueAMizushimaTWuXA miR-29b byproduct sequence exhibits potent tumor-suppressive activities via inhibition of NF-κB signaling in KRAS-mutant colon cancer cellsMol Cancer Ther201817597798710.1158/1535-7163.MCT-17-085029545333
- UhrigMIttrichCWiedmannVNew Alzheimer amyloid beta responsive genes identified in human neuroblastoma cells by hierarchical clusteringPLoS One200948e677910.1371/journal.pone.000677919707560
- SlatteryMLMullanyLESakodaLCThe PI3k/AKT signaling pathway: associations of miRNAs with dysregulated gene expression in colorectal cancerMol Carcinog201857224326110.1002/mc.2275229068474
- WangXWZhangYJTargeting mTOR network in colorectal cancer therapyWorld J Gastroenterol201420154178418810.3748/wjg.v20.i15.417824764656
- LangenskioldMHolmdahlLFalkPIvarssonMLIncreased plasma MMP-2 protein expression in lymph node-positive patients with colorectal cancerInt J Colorectal Dis200520324525210.1007/s00384-004-0667-415592677
- KatsunoYHanyuAKandaHBone morphogenetic protein signaling enhances invasion and bone metastasis of breast cancer cells through SMAD pathwayOncogene200827496322633310.1038/onc.2008.23218663362
- VoorneveldPWKodachLLJacobsRJLoss of SMAD4 alters BMP signaling to promote colorectal cancer cell metastasis via activation of Rho and ROCKGastroenterology20141471196208.e11310.1053/j.gastro.2014.03.05224704720