Abstract
In the last year, the armamentarium of melanoma therapeutics has radically changed. Recent discoveries in melanoma biology and immunology have led to novel therapeutics targeting known oncogenes and immunotherapeutic antibodies. Phase III clinical trials of these agents have reported measurable and meaningful benefits to patients with metastatic disease. In this article, we review recent findings and discuss their significance in melanoma therapy. As our understanding of melanoma biology grows, this initial therapeutic success may be enhanced through the use of molecular markers to select patients, and new targeted immunotherapies in sequential or combination drug regimens.
Epidemiology and treatment of metastatic melanoma
Malignant melanoma is an important health care issue. The incidence of melanoma has increased dramatically over the last four decades and melanoma is now one of the most common forms of cancer. In 2011, over 75,000 North Americans will be diagnosed with melanoma and over 9000 will die from it.Citation1,Citation2 Although less common than cancers of the breast, prostate, or colon, it is potentially more lethal. It has a high propensity for hematogenous and lymphatic dissemination to regional and distant sites and is poorly responsive to most systemic therapies. The 5-year survival rate for metastatic melanoma is dismal, ranging from 5% to 10% with a median survival of less than 8 months with treatment.Citation3
Until recently, the therapeutic options for patients with metastatic melanoma were limited. The only approved treatment options were dacarbazine and interleukin 2 (IL-2). Dacarbazine, an alkylating agent, has a response rate of <10%, with median response durations of 4–8 months.Citation4 Single-agent therapy with IL-2, an immune-modulatory agent, has similar overall response rates of 16%, with ~5% of patients achieving durable complete responses (CRs) that may result in long-term survival in selected patients.Citation5 Use of IL-2 is limited by its low overall response rate and its potential severe multiorgan toxicities requiring management in specialized cancer centers. With these agents, remissions are infrequent, usually of short duration, and treatment is primarily palliative, as neither agent has been shown definitively to improve survival. Improved treatments with high-risk resected and advanced metastatic disease are urgently needed.
In the last year, there have been major treatment advances for metastatic melanoma patients. Two agents, ipilimumab (Yervoy™, Bristol-Myers Squibb, Princeton, NJ) and vemurafenib (Zelboraf, Plexxikon/Roche, Auckland, NZ), have demonstrated improved survival in patients with advanced melanoma when compared with standard treatments. The clinical benefits of these targeted drugs have been realized after decades of research in the molecular pathogenesis of melanoma. This research has identified tumor and immune cellular signaling pathway abnormalities that promote melanoma development and progression. Once regarded as a cancer with a dismal record of negative Phase III treatment trials, melanoma is now a tumor type for the clinical evaluation of paradigm-shifting therapeutic strategies. In this review, we outline key molecular pathways and the agents targeting them.
Melanoma, the immune system, and immune targeting monoclonal antibodies
There is overwhelming evidence that melanoma is an immune-responsive cancer. Spontaneous regression of melanoma is observed and is probably due to immune processes. The identification of tumor-infiltrating lymphocytes and melanoma antigen-specific T-cells in peripheral blood from cancer patients are evidence that melanoma-specific immune recognition and activation occur.Citation6–Citation8 Moreover, melanoma exhibits cellular properties that can be explained by immune selection, such as downregulation of major histocompatibility complex class I expression or release of cytokines such as transforming growth factor-beta.Citation9,Citation10 Lastly, dramatic clinical responses have been demonstrated with immunemodulatory treatments, such as IL-2 and adoptive T-cell transfer in selected patients with metastatic melanoma, although neither of these treatments have demonstrated superiority over standard of care in randomized clinical trials.Citation4,Citation11 However, ipilimumab, a novel monoclonal antibody modulating the immune system, provides the first evidence that an immunotherapy strategy can change the clinical course of metastatic melanoma and result in improvement in patient survival.Citation12,Citation13
Ipilimumab is a modulator of immune system activation. T-cell activation occurs when an antigen is presented by a major histocompatibility complex molecule and a co-stimulatory molecule, B7.1 or B7.2, binds to CD28.Citation14 Simultaneously, downregulation of this process is initiated by B7-cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) binding. Inhibitory co-receptors and pathways restrict T-cell functions to prevent autoimmunity. In cancer patients, this restraint impedes antitumor immunity. Monoclonal antibodies that bind to CTLA-4 and block the interaction between B7 and CTLA-4 inhibit this negative signal, and may break peripheral tolerance to self-tissues and induce antitumor responses. Ipilimumab is a fully human, IgG1 monoclonal antibody that blocks CTLA-4. By inhibiting CTLA-4, ipilimumab potentiates T-cell activation and proliferation, promoting antitumor immunity. Proof of benefit from the approach is found in two recent randomized controlled Phase III trials that demonstrated improved survival in patients with metastatic melanoma treated with ipilimumab.Citation12,Citation13 However, the drug’s effect on the immune response is not tumor specific: ipilimumab treatment has been associated with severe and potentially fatal immunological adverse effects due to T-cell activation and proliferation. A risk evaluation and mitigation strategy has been set up to inform prescribers of the potential risks.
The first ipilimumab Phase III study randomized patients with advanced stage melanoma who progressed on standard treatments, to receive ipilimumab plus gp100 (a melanomaspecific antigen), ipilimumab alone, or gp100 alone.Citation12 The median overall survival (OS) was statistically superior among patients randomized to an arm containing ipilimumab, as compared with those patients receiving gp100 alone (approximately 10 months versus 6.4 months). No difference in OS was detected between the ipilimumab groups. This was the first study to demonstrate a survival benefit in the setting of advanced melanoma in over three decades, but the enthusiasm was tempered by the lack of a standard treatment in the control arm.
The second Phase III study, conducted in previously untreated metastatic melanoma patients and with an acceptable control treatment arm, corroborated the benefits observed with ipilimumab treatment.Citation13 This study randomly assigned patients with previously untreated metastatic melanoma to ipilimumab plus dacarbazine or placebo plus dacarbazine. OS was significantly longer in the group receiving ipilimumab plus dacarbazine than in the group receiving dacarbazine alone: 11.2 months versus 9.1 months, respectively. Interestingly, benefit from ipilimumab was seen across patient subgroups based on human leukocyte antigen types, tumor mutations, or prognostic factors such as serum lactate dehydrogenase. This confirmed the clinical benefit of ipilimumab and added to the growing number of immunotherapeutic strategies for patients with advanced melanoma.
These ipilimumab trials demonstrated proof of clinical benefit of immune modulation in metastatic melanoma. Key areas for future research focus on improving the risk–benefit of immunotherapy in metastatic disease and evaluating the agent in high-risk early stage disease. For the former, efforts focused on the following areas are required: (1) reduction in the frequency and severity and improved management of adverse effects, (2) identification of markers of tumor sensitivity and patient risk of severe side effects, and (3) the evaluation of combination strategies that either increase the tumor-specific immune response and/or target tumor proliferation and survival pathways. For the latter, trials are underway to evaluate the benefit of ipilimumab in resected melanoma patients. Agents and trial results of therapies targeting the immune system are summarized in . Tumor-targeting drugs are discussed further in the next section.
Table 1 Clinical trials of selected immune modulators in melanoma
The molecular pathways altered in melanoma cells
Crucial genetic alterations that enhance the oncogenic potential in melanoma have been identified. Key mutations that lead to constitutive activation of tumor growth and survival pathways occur in the receptor tyrosine kinase CKIT (CD117), and the RAS/RAF/MEK/ERK and phosphoinositide- 3-OH kinase (PI3K)/Protein Kinase B (AKT)/ phosphatase and tensin homolog deleted on chromosome 10 (PTEN) signal transduction systems (). Although convenient to conceptualize these pathways as independent, significant interactions occur and simultaneous activation of the pathways plays a role in melanoma pathogenesis. The demonstration that the mutant BRAF inhibitor vemurafenib improves survival in patients with metastatic melanoma proves that targeting aberrant proteins of signaling pathways with kinase inhibitors can lead to clinical benefit.Citation15 Below, we describe some of these key pathways and the agents designed to target the components of these pathways (see ).
Figure 1 Molecular pathogenesis of melanoma.
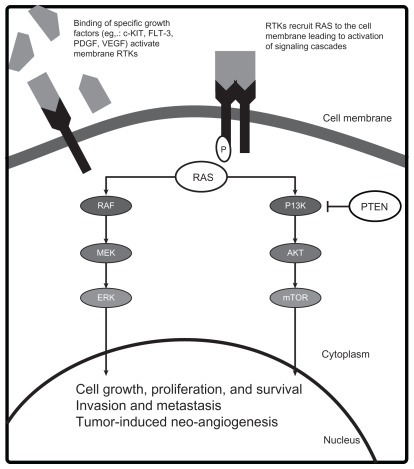
The RAS/RAF/MEK/ERK pathway
The RAS/RAF/MEK/ERK pathway plays a role in normal organogenesis; however, when aberrantly activated it can lead to malignant cellular proliferation, inhibition of apoptosis, and invasion.Citation16 This mitogen-activated protein kinase (MAPK) pathway relays extracellular signals from the plasma membrane of the cell to the nucleus via an ordered series of phosphorylation events.Citation17 Various extracellular stimuli, including growth factor-mediated activation of receptor tyrosine kinases (RTKs), lead to the sequential recruitment, phosphorylation, and activation of one of three RAS isoforms (designated KRAS, NRAS, HRAS), three RAF family members (ARAF, BRAF, CRAF) via a SRC-family tyrosine kinase,Citation18–Citation20 MEK (mitogen-activated ERK kinase),Citation21–Citation23 and ERK (extracellular signal-regulated kinase).Citation21–Citation23 Activated ERK translocates to the nucleus and phosphorylates several nuclear transcription factors necessary for cellular proliferation, differentiation, and survival.
Constitutive activation of the MAPK pathway occurs in 90% of melanomas.Citation24 The two most common mechanisms for MAPK pathway activation in melanoma are mutations in the BRAF (40%–60%) and NRAS genes (15%–30%).Citation25,Citation26 The BRAF V600E mutation accounts for approximately 90% of all activating BRAF mutations.Citation25 This protein product of the V600E mutation has demonstrated a 10.7-fold increase in kinase activity as compared with the wild-type protein.Citation25 Constitutive activation of BRAF and the MAPK pathway impart a proliferative and survival advantage to the cancer cell.Citation27 Mutations in ARAF and CRAF have not been found in melanoma.Citation28,Citation29 Vemurafenib is a potent inhibitor of the activated V600E BRAF mutant protein and wild-type BRAF, but is a weak inhibitor of the A and CRAF isoforms.Citation30
The second most common means for MAPK pathway activation in melanoma is through mutations in the NRAS gene.Citation26 Somatic RAS mutations usually occur in codons 12, 13, or 61 and maintain RAS protein in a constitutively active state.Citation31 Mutations in the HRAS and KRAS isoforms are rare in melanoma.Citation32–Citation34 Interestingly, BRAF and RAS activating mutations are often mutually exclusive events, suggesting only one mutation within the same pathway is sufficient for pathway activation and denoting the redundant mechanisms of activating this pathway in melanoma pathogenesis. Of note, oncogenic RAS can also bind and activate PI3K, resulting in increased AKT activity.Citation35 Thus, RAS activation leads to the upregulation of two major signaling cascades involved in melanoma: the MAPK and PI3K/AKT/mammalian target of rapamycin (mTOR) pathways. The mechanism of pathway activation and molecular response to specific targeted inhibition are likely to be determinants of sensitivity and clinical benefit to individual agents and combinations.
PI3K/AKT/mTOR pathway
The PI3K/AKT/mTOR pathway is another signaling transduction pathway that is aberrantly activated in several cancers, including melanoma.Citation35 In response to activated RTKs, the PI3K phosphorylates phosphatidylinositol-4,5- biphosphate to phosphatidylinositol-3,4,5-triphosphate (PIP3), recruiting other proteins to the plasma membrane and leading to activation of the major downstream effector of the PI3K pathway, AKT.Citation36 Once active, AKT phosphorylates a number of substrates that promote cell survival, proliferation, and invasion.Citation36 One substrate is the mTOR, a serine threonine kinase that modulates protein synthesis, angiogenesis, and cell cycle progression and validated therapeutic target in renal cell carcinoma, neuroendocrine tumors, and lymphoma.
Although activating mutations in PI3K are rare, downstream effectors PTEN and AKT, are altered in the majority of melanomas.Citation37,Citation38 The tumor suppressor gene PTEN encodes a lipid and protein phosphatase that negatively regulates the PI3K cascade through dephosphorylation of PIP3.Citation39,Citation40 Decreased or loss of PTEN expression or function through epigenetic silencing, inactivating mutation or deletion is observed in 30%–60% of primary melanomas.Citation37,Citation41–Citation43 Interestingly, somatic mutations of PTEN gene occur in association with BRAF, but not NRAS mutations suggesting the dual pathway activation can be accomplished by alterations in NRAS alone or the combination of PTEN and BRAF.Citation25,Citation44 In contrast, AKT is an oncogene that encodes a kinase that is frequently activated in human cancers.Citation45 There are three AKT isoforms and AKT3 is the major isoform deregulated in melanoma.Citation37 Overexpression of phospho-AKT, typically associated with increased gene copy number, is seen in a greater proportion of melanomas and melanoma metastases than neviCitation37,Citation45 and may be associated with earlier progression and shorter survival of patients with melanoma.Citation46 Inhibition of AKT3 using small interfering RNA or increased activation of PTEN stimulated apoptosis of melanoma cell lines, indicating a prosurvival function of AKT in melanoma.Citation37 In vitro cell line and in vivo mouse xenograft studies of simultaneously targeting AKT3 and V600E BRAF with small interfering RNAs led to increasing cellular apoptosis and decreasing proliferation supporting the relevance of both pathways in melanoma.Citation47
c-KIT
c-KIT (also known as CD117) is a RTK that also contributes to the pathogenesis of a subset of melanomas that do not harbor NRAS or BRAF mutations. c-KIT activates several signaling cascades, including the MAPK, PI3K/AKT/ mTOR, and microphthalmia associated transcription factor pathways.Citation48,Citation49 c-KIT signaling is necessary for differentiation, proliferation, and migration of normal melanocytes. c-KIT mutations and/or copy number increases were identified in the melanomas from mucosa, acral and chronic sun-induced damaged skin.Citation50 Point mutations in this gene result in constitutive activation of downstream effectors of key signaling pathways in melanoma cells.Citation51,Citation52 Although the frequency of c-KIT alterations is reported to be lower in Chinese, Korean, and Australian cohortsCitation53–Citation55 compared to Western patients, such aberrations have been identified in Chinese patients with nonchronic sun-damaged skin and melanomas of unknown primary.Citation54 Geographical variability in predisposing risk factors, small study sample sizes, incomplete testing of the c-KIT gene, and differing criteria for solar elastosis, the marker of chronic sun-damage may explain the apparent variation on KIT mutation frequencies between populations.
The above laboratory and clinical studies support the importance of activating c-KIT and components of the PI3K and MAPK pathways for melanoma pathogenesis. Based on this evidence, activation may occur through NRAS mutations that activate both the PI3K and MAPK pathways, or concurrent BRAF mutations with altered function of either PTEN or AKT, or through c-KIT mutations. Although these are not the sole alterations that contribute to melanoma pathogenesis, the clinical and laboratory results provide the initial rationale for testing specific agents and combinations in melanoma patients.
Potential targeted treatments for patients with advanced melanoma
Agents targeting the MAPK pathway
Agents targeting the MAPK pathway are the first small molecule kinase inhibitors to demonstrate clinical benefit for melanoma patients. Agents that have entered into clinical testing can be grouped into those that are relatively specific for BRAF and/or mutant BRAF, nonspecific RAF inhibitors, and MEK inhibitors. Clinical trials of agents within these classes show varying activity and toxicity that may be due to the pharmacology of the individual agents (ie, specificity and potency of target inhibition) and to the specific tumor molecular profile of patients. Pharmacodynamic studies suggest that >80% inhibition of ERK is needed for clinical activity.Citation56 Common adverse events are rash, fatigue, nausea, and diarrhea. Specific toxicities include the development of cutaneous keratoacanthoma or squamous-cell carcinoma with specific BRAF inhibitors, and retinal toxicity (central serous retinopathy and retinal vein occlusion) with MEK inhibitors.
Laboratory results suggest there are feedback loops within the MAPK system of biological and clinical relevance and that genotype may determine the activity of specific agents. For instance, the development of borderline and malignant skin tumors such as keratoacanthomas and squamous-cell carcinomas associated with BRAF inhibitors, is thought to arise due to paradoxical MAPK signaling in cells with wild-type BRAF.Citation57 Clinical studies show that inhibition of wild-type RAF leads to upregulation of RAS signaling and ERK activation. Moreover, the results of three studies suggest that inhibitors thought to be selective for mutant BRAF can also activate CRAF through the formation of dimeric RAF complexes – a process that is enhanced by the presence of an oncogenic RAS mutation.Citation58–Citation61 These studies support the conclusion that mutant-specific BRAF inhibitors should be used for treating cancers caused by BRAF mutants (such as BRAF V600E-associated melanomas), but should not be used as single agents in melanomas (and other cancers) with RAS mutations as they may promote tumor genesis. Such mechanistic insight into both normal and pathogenic MAPK signaling suggest that certain agents and combinations may be preferable in certain molecular contexts and lead to the development of effective new anticancer therapies.
Selective mutant BRAF-inhibitor vemurafenib
Vemurafenib (PLX-4032, RG7204) is an inhibitor of a mutant form of the BRAF kinase and the second agent, after ipilimumab, to improve OS in patients with advanced melanoma.Citation15 In the Phase III study, named BRIM 3 (BRAF Inhibitor in Melanoma 3), previously untreated patients with advanced melanoma that harbored the V600E mutation, were randomized to either vemurafenib or dacarbazine. A significant improvement in 6-month OS was observed in the vemurafenib group versus the dacarbazine group (84% versus 64%, respectively). In the interim analysis for OS and final analysis for progression-free survival (PFS), vemurafenib was associated with a significant relative reduction of 63% in the risk of death and of 74% in the risk of either death or disease progression compared with dacarbazine. Common adverse events associated with vemurafenib were arthralgia, rash, fatigue, alopecia, photosensitivity, nausea, and diarrhea. Cutaneous squamous-cell carcinoma, keratoacanthoma, or both developed in 18% of patients. All lesions were treated by simple excision. This trial was the first to show that a rationally targeted agent improved survival by inhibiting an aberrantly overactive signaling pathway in melanoma.
Resistance to vemurafenib
Despite the benefits observed with vemurafenib, both intrinsic and acquired resistance has been observed in patients with advanced melanoma. Emerging evidence suggests that resistance to vemurafenib is complex and multifactorial. Preclinical studies suggest that PTEN loss, C-RAF mutations, or Cot-1 mutations may confer intrinsic resistance to vemurafenib.Citation62,Citation63 Vemurafenib-mediated apoptosis was significantly impaired in PTEN negative melanoma cell lines.Citation63 Moreover, screening for kinases that prevented vemurafenib-mediated cell growth arrest identified both CRAF and Cot1 as potential drivers of resistance.Citation62 Both CRAF and Cot 1 bypass BRAF signaling, activating the MAPK pathway. Although these studies may make mechanistic sense, no study has demonstrated that pre-existing alterations in PTEN, CRAF, or Cot-1 predict for intrinsic resistance to vemurafenib.
In attempts to identify the genetic changes that lead to acquired resistance to vemurafenib, researchers have compared matched tumor samples from patients prior to treatment and after treatment failure. Cot-1 mRNA levels, and insulinlike growth factor recptor-1 and platelet-derived growth factor receptor (PDGFR) immunohistochemistry staining have all been found to be elevated in post-treatment samples when compared to pretreatment biopsies.Citation62,Citation64,Citation65 Moreover, RAS or MEK activating mutations, that were not present in the pretreatment tumor, have been identified in tumors from patients progressing on vemurafenib treatment.Citation64,Citation66 A common theme in these mechanisms of resistance is the restoration of MAPK signaling and/or an increase in PI3K/AKT/mTOR signaling, emphasizing the importance of these pathways on melanoma progression. By understanding the molecular events that lead to resistance, rational combinations or subsequent treatments can be designed.
Other selective BRAF inhibitors
Selective inhibitors of BRAF are in development including SB90885, GDC-0879, and GSK2118436 (GSK436). The most detailed clinical information is available for the latter and is summarized in the next column.
GSK436
GSK436 is a highly potent and selective adenosine-5′-triphosphate (ATP)-competitive BRAF inhibitor. The selectivity of this agent for mutant BRAF is over 100 times greater than the wild-type protein.Citation67 It displays dose-dependent inhibition of MEK and ERK phosphorylation in mutant BRAF cell lines and tumor regression in xenograft models.Citation67 In the first-in-human Phase I study, 61 patients (57 with BRAF mutations) had been accrued and the maximum tolerated dose had not yet been determined by the time the study was reported (see ).Citation68 In patients with mutant BRAF melanoma, a response rate of 63% (10 of 16 patients) was observed in the patients receiving doses ≥150 mg twice a day. Cohorts receiving lower doses had a response rate of 39% (26 of 41 patients, one CR, and 25 partial responses [PRs]). The probability of response to GSK436 directly correlated with the presence of a BRAF mutation and inversely with the presence of a PTEN alteration. Due to its high response rate and reasonable toxicity profile, GSK436 is being evaluated in combination with the MEK inhibitor GSK1120212 in an attempt to enhance the clinical activity (discussed further below).
Table 2 Clinical trials of single-agent kinase inhibitors in melanoma
Nonselective BRAF inhibitors
Sorafenib
Sorafenib is a small molecule inhibitor of multiple kinases, including wild-type BRAF, V600E BRAF, and CRAF as well as vascular endothelial growth factor receptors (VEGFR) and PDGFR.Citation69,Citation70 Inhibition of the MAPK pathway has been demonstrated in vitro and in vivo with this agent. Sorafenib has limited single agent activity in patients with melanoma.Citation71,Citation72 Although interesting clinical activity was reported from a Phase II trial of sorafenib combined with carboplatin and paclitaxel,Citation73 the Phase III trials testing this combination in the first- and second-line settings did not show improvements in response rate, PFS, or OS ().Citation74,Citation75 Currently, clinical investigations are focused on combining sorafenib with other targeted, immunomodulatory, and chemotherapy agents (see ).
Table 3 Clinical trials of combinations in melanoma
RAF265
RAF265 is also a multitargeted small molecule inhibitor of both the V600E BRAF mutant and VEGFR. A Phase I trial treating advanced melanoma patients with RAF265 demonstrated an overall response rate of 16% (6 of 37 patients) for BRAF mutation-positive melanoma patients and 13% (4 of 30) for wild-type or BRAF mutation status unknown patients (see ).Citation76 Because of dose limiting hematologic toxicity, an intermittent schedule will be explored in an effort to improve the therapeutic index. Whether this agent offers particular advantage over more selective and effective BRAF inhibitors, such as vemurafenib and GSK2118436, will be determined in future clinical trials.
MEK inhibitors
MEK is an attractive therapeutic target as it is downstream of both activated BRAF and NRAS. Preclinical studies suggested that melanoma cell lines with mutant BRAF were more sensitive to MEK inhibition than those harboring activating NRAS mutations and wild-type BRAF genes.Citation77 Small-molecule inhibitors of MEK completely abrogated tumor growth in BRAF mutant xenografts, whereas RAS mutant tumors were only partially inhibited.Citation77 Agents that entered clinical development include CI-1040, PD-0325901, GSK1120212, and AZD6244. The activity of first- and second-generation MEK inhibitors CI-1040 and PD-0325901 has been limited by pharmacologic and toxicity issues. Cl-1040 was an agent that was generally tolerated well, but it failed to demonstrate sufficient anticancer activity to warrant further development. Citation78 PD-0325901 is structurally similar to Cl-1040, but is more potent at MEK inhibition and has greater systemic exposure. In Phase I trials, melanoma patients achieved objective responses, and suppression of phosphorylated ERK in melanoma specimens.Citation78,Citation79 However, clinical development was terminated because of unexpected high incidence of musculoskeletal and neurological adverse events (see ). Newer MEK inhibitors such as GSK1120212 appear to have greater potency and have demonstrated promising clinical activity, particularly in tumors with BRAF mutations.
GSK1120212
GSK1120212 (GSK212) is a reversible, selective allosteric inhibitor of both MEK1 and MEK2. Preclinical studies have demonstrated that it can inhibit the phosphorylation of ERK and had growth inhibition in a variety of cancer cell lines.Citation80 The first-in-human Phase I study of GSK212 determined the recommended Phase II dose to be 2 mg daily (see ).Citation81 Common toxicities were grade 1 and 2 rash and diarrhea. There were three cases of reversible central serous retinopathy. Seventy-two of 162 patients enrolled in the study had advanced melanoma. Of the patients with melanoma, 24 tumors harbored BRAF mutations, 24 had a wild-type BRAF gene, and 22 had an unknown BRAF status. There were two CRs and six PRs among 20 evaluable patients with BRAF mutant melanoma for a preliminary objective response rate of 40%. In contrast, two of 22 evaluable patients with BRAF wild-type melanomas had PRs. These results suggest that response correlates with BRAF mutation status. Activity in some BRAF wild-type melanoma tumors suggest that these are also dependent on the MAPK pathway by mechanisms unrelated to most common BRAF mutations. Trials of drug combinations are currently being tested to improve the efficacy of this agent.
AZD6244
AZD6244 (formerly called ARRY-142886) is a selective non-ATP competitive inhibitor of MEK1 and MEK2.Citation82 In a Phase II trial, 210 patients with advanced melanoma were randomly allocated to AZD6244 or temozolomide (see ).Citation83 Although there was no significant difference in the primary endpoint of PFS between the two arms, five of six patients with PRs from AZD6244 had BRAF V600Emutated tumors. The activity observed warrants further investigation of this agent in combination with other drugs in selected patients.
The activity of MEK inhibitors in some patients with BRAF wild-type melanomas seen with GSK212 and AZD6244 suggest that there are other mechanisms determining sensitivity to MEK inhibition. Results from laboratory studies evaluating inhibitors across multiple cell line panels indicate that MEK inhibitor activity does not absolutely correlate with the mutational or phosphorylation status of BRAF, MEK, RAS, or PI3K.Citation84 This lack of correlation has fueled efforts to develop expression profiles of MEK activation and sensitivity. An 18-gene expression signature of MEK activation and inhibition derived from multiple tumor cell lines and xenografts may correlate with sensitivity and be used as a marker of pharmacodynamics response to MEK inhibition.Citation85 A 13-gene signature was also identified that was predictive of resistance to a MEK inhibitor in cancer cells, despite these cells having functional MEK activity. These signatures potentially may be used to select patients sensitive and resistant to MEK inhibition.Citation84
Agents targeting the PI3K/AKT/mTOR pathway
In contrast to the clinical benefits seen with agents targeting the MAPK pathway tested in melanoma patients, the results from clinical trials evaluating agents targeting the PI3K/AKT/ mTOR pathway have been disappointing. There are a number of potential explanations for the lack of activity: (1) suboptimal pharmacology and target modulation of the agents at maximum tolerable doses, (2) relative lack of importance of the pathway as a driver of melanoma cell proliferation or survival, (3) intrinsic or rapid onset of acquired resistance to target inhibition, or (4) failure to enrich enrollment into trials for patients likely to benefit. Unfortunately, not all trials have incorporated pharmacodynamics assessment of target inhibition in tumor or surrogate tissue nor employed patient enrichment strategies, thus further evaluation of emerging agents targeting the pathway with better pharmacology is warranted. Descriptions of agents and results from clinical trials are provided in the following sections.
AKT inhibitors
PI3K/AKT/mTOR pathway is another signaling transduction pathway that is aberrantly activated in melanoma and pharmacological inhibition may also benefit patients with melanoma.Citation35 Although a number of PI3K inhibitors are in clinical development and may have activity in PTEN mutant tumors, inhibiting downstream targets AKT or mTOR may be preferable due to the frequency of AKT amplification in melanomas. Despite this finding, the activity of AKT inhibitors perifosine and GSK2141795 as single agents in the treatment of advanced melanoma has been disappointing. These initial results may be due to issues of pharmacology of the individual agents and lack of optimal target inhibition in tumor due to off-target or target-specific toxicity-limiting doses.
Perifosine
Perifosine is an AKT and PI3K inhibitor and structurally related to miltefosine. It interferes with the recruitment of proteins with the pleckstrin homology domain, like AKT, to the plasma membrane. A Phase II study of perifosine treatment in previously untreated patients with metastatic melanoma was conducted and of 14 evaluable patients, none achieved an objective response (see ).Citation86 The extent of AKT or MAPK pathway inhibition was not evaluated in this study. The authors recommended that no further development of single agent perifosine was required in metastatic melanoma patients.
GSK2141795
GSK2141795 (GSK795) is a reversible, selective ATPcompetitive inhibitor of all AKT isoforms. Preclinical studies had demonstrated that GSK795 could inhibit proliferation of cancer cell lines. A Phase I trial has been conducted with GSK795 to determine the recommended Phase II dose, pharmacokinetics, pharmacodynamics, and safety of the drug.Citation87 In total, 76 patients were enrolled. In the 54 patients evaluable for response, one PR was observed in a patient with metastatic anal cancer, two patients with endometrial cancer had prolonged stable disease (SD) and minor tumor responses in a population selected for molecularly defined predictive signatures (PTEN loss or PIK3CA mutations). No responses were observed in the patients with melanoma, although few were enrolled in this study.
mTOR inhibitors
Inactivating PTEN mutations are identified in 50% of melanomas.Citation88 The resulting increase in activity of mTOR is believed to derail the normal control of nutrients leading to excessive cell growth and proliferation.Citation89 However, the results from early Phase clinical trials testing mTOR inhibitors and rapamycin derivatives, temsirolimus and everolimus, in melanoma patients have also shown limited single agent activity.
Temsirolimus
Temsirolimus is an intravenous mTOR kinase inhibitor and ester derivative of rapamycin. When bound to FK506 binding protein, it interacts and inhibits the mTOR kinase activity, resulting in inhibition of the cell cycle.Citation90 Preclinical studies of temsirolimus demonstrated that it could inhibit tumor activity in a variety of cancers including melanoma cell lines and animal models. A Phase II trial of temsirolimus was conducted in patients with metastatic melanoma (see ).Citation91 Thirty-three patients were treated with only one patient experiencing a PR lasting 2 months. The median time to disease progression was 10 weeks. Of note, the dose/ schedule used in this trial is not the maximum tolerated dose and target inhibition in tumor was not evaluated in this trial. Whether antitumor activity would be greater with higher doses is an unanswered question.
Everolimus
Everolimus is an oral mTOR inhibitor. A two-stage Phase II trial was conducted in 29 patients with metastatic melanoma (see ).Citation92 By the first interim analysis, the activity of everolimus looked favorable with 35% of patients experiencing SD at 16 weeks. The median PFS and OS were just over 3 months and 12 months, respectively. To further investigate the activity of everolimus, the enrollment of a second cohort treated with a higher dose was conducted. An interim analysis of the second cohort found the median PFS and OS was just under 2 months and 9.5 months respectively, tempering the enthusiasm generated by the activity observed in the first cohort. Of the 53 patients enrolled in total, two patients had unconfirmed PRs. The authors concluded that everolimus as a single agent had insufficient activity in metastatic melanoma patients. The reason as to why patients treated at the lower dose had longer survival compared with those treated at higher doses is unknown.
c-KIT inhibitors
Three commercially available, orally administered small molecule tyrosine kinases inhibitors, imatinib, dasatinib, and nilotinib, are under evaluation in melanoma. While all three agents inhibit c-KIT, PDGFR, and BCR-ABL, they differ in potency of inhibition, cellular uptake, mechanism interaction with kinase, and overall inhibitory profile for other cellular kinases.Citation67,Citation93,Citation94 The three agents were initially approved for the treatment of patients with chronic myelogenous leukemia which results from the translocation product BCR-ABL.Citation95–Citation97 Both imatinib and dasatinib have demonstrated activity in gastrointestinal stromal tumors, which harbors activating mutations in c-KIT or PDGFR.Citation98,Citation99 Results suggest that these agents may have activity in c-KIT mutated and, possibly, c-KIT amplified melanomas.
Imatinib mesylate
Five Phase II trials of single agent imatinib have been conducted (see ) and the results of these trials demonstrate the importance of patient selection based on the presence of mutations in tumors.Citation100–Citation104 Across three trials conducted in patients with mostly nonchronically sun-damaged melanomas and not requiring c-KIT mutation for enrollment, only one of 65 patients achieved a PR, and that patient had a c-KIT mutation-positive acral melanoma.Citation102–Citation104 In contrast, two published Phase II studies that required patients to harbor melanomas with c-KIT mutations reported dramatically different results.Citation100,Citation101 Across these two studies, there were two CRs and 14 PRs among 65 evaluable patients, for a response rate of almost 25%. Responses were seen in melanomas with c-KIT mutations with known functional relevance. Mutations in c-KIT exons 11 and 13 seemed to correlate better with response than either amplifications or c-KIT overexpression assessed by immunohistochemistry.Citation67 These promising results with imatinib in this molecular subgroup of melanoma patients warrant confirmation in a prospective randomized trial.
Dasatinib
A Phase II study in a molecularly unselected population of advanced melanoma patients has recently been published (see ).Citation105 Of the 36 patients evaluable for response, two had a PR lasting 24 weeks or more. One of these patients was found to have a c-KIT mutation. The other responder had a wild-type c-KIT gene, but amplification of c-KIT was not tested in this study. The activity of the drug in patients with melanomas likely to carry c-KIT aberrations is currently being evaluated.Citation106
Nilotinib
Nilotinib is a second-generation tyrosine kinase inhibitor of c-KIT, PDGFR, and BCR-ABL. Nilotinib has similar potencies against c-KIT and PDGFR compared to imatinib. Citation67 However, the agents differ with respect to cellular transport, where imatinib is transported actively and nilotinib is transported passively. Due to this passive diffusion, nilotinib may achieve higher intracellular concentrations and be subject to less extrinsic resistance mechanism as compared with imatinib.Citation67 Several Phase II studies are currently recruiting patients with melanomas from acral, mucosal, and chronically sun-damaged skin or melanomas that harbor c-KIT aberrations.Citation107–Citation111
New treatment paradigm for advanced melanoma
Ipilimumab and vemurafenib are poised to become the therapy of choice for patients with previously untreated advanced melanoma. The initial treatment for patients with tumors that harbor the BRAF V600E mutation will be vemurafenib, while those patients harboring wild-type BRAF tumors will be treated with ipilimumab. However, despite the advances observed with these agents, both intrinsic and acquired resistance has been observed and no standard second-line treatment has been established. Hence, further research into the molecular predictors and mechanisms contributing to resistance will aid in the design of rational treatment combinations and regimens to employ after treatment failure. Research into the molecular pathogenesis of melanoma and its dependence on particular signaling cascades will aid in these efforts.
Combinations of targeted agents in melanoma
Intrinsic and acquired resistance to novel targeted agents is likely due, in part, to the cross talk that occurs between the pathways required for melanoma development and progression. Coordinated blockade of multiple pathways with combinations of targeted agents will hopefully lead to improved treatment efficacy. With the goal of overcoming resistance and enhancing clinical efficacy, a Phase I/II trial is opening soon evaluating the combination of ipilimumab and vemurafenib in subjects with metastatic melanoma and the mutant form of BRAF (clinicaltrials.gov identifier: NCT1400451). Other combinations of targeted agents are currently in varying phases of development. Combination strategies have focused on: (1) inhibiting additional targets within the same pathway or (2) inhibiting a different pathway or cellular process that are involved in the pathogenesis or drug resistance of melanoma. Trials combining CTLA4 and other immunomodulatory antibodies, and/or vaccines are also underway. In general, vaccine development strategies may benefit most from combinations with novel immunomodulating monoclonal antibodies. These strategies are based on our current understanding of melanoma targets/pathways, availability of the agents, and limited preclinical experiments that suggest at least additive efficacy of the combination. Results to date suggest that activity of a combination is related to obtaining effective inhibition of the individual targets without inducing significant additional normal tissue toxicity.
Inhibiting targets in parallel pathways
Sorafenib and temsirolimus
As single agents, sorafenib and temsirolimus have modest antimelanoma activity; however, these agents inhibit targets within the parallel signaling pathways and have limited overlapping toxicities. Hence the combination of sorafenib and temsirolimus was expected to produce at least additive activity and have a tolerable side effect profile. The results of a Phase I study of a combination of sorafenib and temsirolimus in patients with advanced melanoma did not meet these expectations (see ).Citation112 Dose escalations of the individual agents were impeded by higher than expected rates of toxicity. There were no responses among 23 evaluable patients, although ten patients had SD of 8–24 weeks. The combination did have effects on target modulation, as both phosphorylated MEK and AKT decreased from pre- and on-treatment tumor biopsies. However, there was no trend in the change of phosphorylated ERK with the combination treatment. The reason for the decrease in phosphorylated MEK and not ERK is unknown, but probably speaks to the complexity of crosstalk amongst these pathways and challenges of assessing pharmacodynamics markers in clinical trials.
Sorafenib and tipifarnib
The frequency of activating RAS mutations in melanoma and most cancers suggest that it would be an ideal target for inhibition. Unfortunately, no direct inhibitor of RAS has been developed, as it lacks an accessible small molecule binding site. One strategy indirectly inactivates RAS function by inhibiting the post-translational modifications required for the full biological activity of RAS. RAS undergoes farnesylation to associate with the plasma membrane and is subsequently activated.Citation113 Inhibitors targeting farnesyl transferase, the enzyme that catalyzes this modification, have been developed and tested in patients with advanced melanoma. The majority of farnesyl transferase inhibitors have limited activity as single agents and in combination with chemotherapy in melanoma patients.Citation114–Citation117 However, several trials combining farnesyl transferase inhibitors with other targeted agents in patients with melanoma are underway. A Phase I trial evaluating the combination of the farnesyl transferase inhibitor, tipifarnib with sorafenib in patients with advanced malignancies has been reported (see ).Citation118 Evidence of target modulation was seen in a quarter of patients with a 50% or more reduction in farnesyl transferase levels. Three of seven melanoma patients had SD. One patient with prolonged SD had a PDGFR mutation. It is unknown whether the observed response was due to sorafenib, which is known to inhibit PDGFR, or the combination of both agents.
Another randomized Phase II trial in molecularly unselected metastatic melanoma patients tested the combinations of sorafenib and temsirolimus (Arm A) or sorafenib and tipifarnib (Arm B) (see ).Citation119 Significant toxicity limiting dose escalation was seen with both combinations. Among 66 evaluable patients in Arm A, there were three PRs (5%) and 24 SD (36%). Of the 42 evaluable patients in Arm B, there was one PR (2%) and ten SDs (24%). Based on these results, neither combination appear to show sufficient activity for further evaluation, possibly due to poor pathway inhibition at tolerated doses.
Bevacizumab and everolimus
Vascular endothelial growth factor (VEGF) is frequently overexpressed in melanoma and high levels of VEGF may represent an adverse prognostic feature.Citation120 Aberrant activity of the VEGF pathway results in tumor angiogenesis and monocyte/macrophage migration. Concurrent inhibition of tumor and endothelial cell proliferation by combining a VEGF inhibitor with an mTOR inhibitor like everolimus, which also down regulates the VEGF receptor may be efficacious. The combination of the VEGF monoclonal antibody bevacizumab and everolimus has activity in metastatic clear cell renal carcinoma.Citation121 A Phase II study involving 57 patients with metastatic melanoma evaluated the efficacy and safety of bevacizumab and everolimus. Seven patients (12%) experienced objective responses (1CR, 6PR) and 33 had SD (58%) (see ).Citation122 The median PFS and OS were 4 months and 8.6 months, respectively. This activity is greater than expected for the individual agents and the researchers recommended further exploration of these agents in other combinations.
Ipilimumab and bevacizumab
The activity of immunological therapies may in part be related to damage to the tumor vasculature. Ipilimumab induces a hemorrhagic necrosis of tumors that is likely due in part to immune mediated vasculopathy.Citation123 Similar changes are observed in metastases of patients with durable clinical responses to CTLA-4 antibody blockade and vaccination with irradiated autologous tumor cells engineered to secrete GM-CSF.Citation124 In these patients, high titer antibodies are produced against VEGF, suggesting ipilimumab and other immunotherapy may induce host responses that target angiogenic networks in the tumor microenvironment. In addition, VEGF not only affects tumor angiogenesis, it also potently inhibits the maturation of antigen presenting cells and thus may contribute to the ability of tumors to evade the host immune response. Based on these observations, the combination of ipilimumab and bevacizumab was studied in a Phase I study (see ).Citation123 Among 22 patients evaluable for response, six (27%) PRs, one (5%) CR, and seven (32%) durable SDs were observed. Serial perfusion computed tomography scans showed a persistent decreased tumor blood flow, and post-treatment biopsies in twelve patients revealed activated vessel endothelium with extensive T-cell trafficking and nonproductive central angiogenesis. Clinical activity and correlative studies suggest additive to synergistic effects with the combination of ipilimumab and bevacizumab and that this combination merits further exploration.
Inhibiting targets within the same pathway
Combined BRAF and MEK inhibition
The most promising combination strategy under evaluation is the simultaneous inhibition of BRAF and MEK. Promising evidence derived from a BRAF V600E human melanoma xenograft model, illustrated that combined administration of BRAF inhibitor GSK2118436 (GSK436) and MEK inhibitor GSK1120212 (GSK212) lead to greater antitumor activity compared to either monotherapy and decreased incidence of the BRAF inhibitor induced hyper-proliferative skin lesions.Citation125 In fact, a Phase I/II trial has been conducted with the combination of GSK436 and GSK212 in patients with tumors harboring V600 BRAF mutations, 93% (n = 101 patients) with advanced melanoma (see ).Citation125 Full monotherapy doses were tolerated in combination and the common adverse events included: pyrexia, rash, chills, nausea, vomiting, diarrhea, and fatigue. Grade 3 neutropenia and leukopenia were also observed in only three and two patients respectively. As predicted by the xenograft model the co-administration of a MEK inhibitor lowered the incidence of rash and hyper-proliferative skin lesions, presumably by GSK212 blocking the BRAF inhibitor-induced activation of the MAPK pathway in cells with a wild-type BRAF gene. Forty-one of 71 patients with advanced melanoma and no prior exposure to a BRAF inhibitor had objective responses. Of 24 patients that had previous BRAF inhibitor therapy, three had PRs. These results exemplify the fact that combination treatments targeting the same pathway may be tolerable, reduce mechanism-based toxicity, and potentially enhance activity.
Conclusion
Improvements in the understanding of the molecular underpinnings of melanoma have finally translated into targeted therapies that improved survival for patients with advanced melanoma. Ipilimumab and vemurafenib are now the first-line treatment options for patients. CTLA4, other immune modulatory antibodies, and vaccines may play a crucial role in initiating and maintaining a melanoma specific immune response. Vaccine development strategies may benefit most from combinations with novel immune modulating monoclonal antibodies. In early Phase clinical trials of small molecule kinase inhibitors, there has been some significant antitumor activity identified in unique molecular subsets of melanoma patients. Research is currently focused on understanding the mechanisms of intrinsic and acquired resistance to these agents. These advances will lead to rationally designed combinations of first- and secondline therapies that will hopefully improve efficacy and tolerability in selected groups of melanoma patients.
Disclosure
The authors report no conflicts of interest in this work.
References
- American Cancer SocietyCancer Facts and Figures20112011160 Available from: http://www.cancer.org/Research/CancerFactsFigures/CancerFactsFigures/cancer-facts-figures-2011Accessed January 16, 2012
- Canadian Cancer SocietyCanadian Cancer Statistics201120111133 Available from: http://www.cancer.caAccessed January 16, 2012
- BalchCMBuzaidACSoongSJFinal version of the American Joint Committee on Cancer staging system for cutaneous melanomaJ Clin Oncol200119163635364811504745
- De VitaVHellmanSRosenbergSBiologic Therapy of Cancer: MelanomaPhiladelphia, PALippincott1995388411
- AtkinsMBKunkelLSznolMRosenbergSAHigh-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival updateCancer J Sci Am20006Suppl 1S11S1410685652
- CipponiAWieersGvan BarenNCouliePGTumor-infiltrating lymphocytes: apparently good for melanoma patients. But why?Cancer Immunol Immunother20116081153116021553145
- HadrupSRBakkerAHShuCJParallel detection of antigenspecific T-cell responses by multidimensional encoding of MHC multimersNat Methods20096752052619543285
- KalialisLVDrzewieckiKTKlyverHSpontaneous regression of metastases from melanoma: review of the literatureMelanoma Res200919527528219633580
- AhmadMReesRCAliSAEscape from immunotherapy: possible mechanisms that influence tumor regression/progressionCancer Immunol Immunother2004531084485415197495
- KrasagakisKThölkeDFarthmannBElevated plasma levels of transforming growth factor (TGF)-beta1 and TGF-beta2 in patients with disseminated malignant melanomaBr J Cancer1998779149214949652767
- HunderNNWallenHCaoJTreatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1N Eng J Med20083582526982703
- HodiFSO’DaySJMcDermottDFImproved survival with ipilimumab in patients with metastatic melanomaN Engl J Med2010363871172320525992
- RobertCThomasLBondarenkoIIpilimumab plus dacarbazine for previously untreated metastatic melanomaN Eng J Med20113642625172526
- HarrisNLRoncheseFThe role of B7 costimulation in T-cell immunityImmunol Cell Biol199977430431110457196
- ChapmanPBHauschildARobertCImproved survival with vemurafenib in melanoma with BRAF V600E mutationN Engd J Med20113642625072516
- RamanMChenWCobbMHDifferential regulation and properties of MAPKsOncogene200726223100311217496909
- GarnettMJMaraisRGuilty as charged: B-RAF is a human oncogeneCancer Cell20046431331915488754
- Lange-CarterCAJohnsonGLRas-dependent growth factor regulation of MEK kinase in PC12 cellsScience19942655177145814618073291
- MaraisRLightYPatersonHFMarshallCJRas recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylationEMBO J19951413313631457542586
- MindenALinAMcMahonMDifferential activation of ERK and JNK mitogen-activated protein kinases by Raf-1 and MEKKScience19942665191171917237992057
- MaraisRLightYPatersonHFMasonCSMarshallCJDifferential regulation of Raf-1, A-Raf, and B-Raf by oncogenic ras and tyrosine kinasesJ Biol Chem19972727437843839020159
- MasonCSSpringerCJCooperRGSerine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activationEMBO J19991882137214810205168
- XuSRobbinsDFrostJMEKK1 phosphorylates MEK1 and MEK2 but does not cause activation of mitogen-activated protein kinasePro Nat Acad Sci U S A1995921568086812
- CohenCZavala-PompaASequeiraJHMitogen-actived protein kinase activation is an early event in melanoma progressionClin Cancer Res20028123728373312473582
- DaviesHBignellGRCoxCMutations of the BRAF gene in human cancerNature2002417689294995412068308
- OmholtKKarsbergSPlatzAScreening of N-ras codon 61 mutations in paired primary and metastatic cutaneous melanomas: mutations occur early and persist throughout tumor progressionClin Cancer Res20028113468347412429636
- PollockPMHarperULHansenKSHigh frequency of BRAF mutations in neviNat Genet2003331192012447372
- EmussVGarnettMMasonCMaraisRMutations of C-RAF are rare in human cancer because C-RAF has a low basal kinase activity compared with B-RAFCancer Res200565219719972616266992
- PritchardCASamuelsMLBoschEMcMahonMConditionally oncogenic forms of the A-Raf and B-Raf protein kinases display different biological and biochemical properties in NIH 3T3 cellsMol Cell Biol19951511643064427565795
- TsaiJLeeJTWangWDiscovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activityPro Nat Acad Sci U S A2008105830413046
- BarbacidMras genesAnnu Rev Biochem1987567798273304147
- WhitwamTVanbrocklinMWRussoMEDifferential oncogenic potential of activated RAS isoforms in melanocytesOncogene200726314563457017297468
- BallNJYohnJJMorelliJGRas mutations in human melanoma: a marker of malignant progressionJ Invest Dermatol199410232852908120410
- van ElsasAZerpSvan der FlierSAnalysis of N-ras mutations in human cutaneous melanoma: tumor heterogeneity detected by polymerase chain reaction/single-stranded conformation polymorphism analysisRecent Results Cancer Res199513957677597312
- CullyMYouHLevineAJMakTWBeyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesisNat Rev Cancer20066318419216453012
- RobertsonGPFunctional and therapeutic signif icance of Akt deregulation in malignant melanomaCancer Metastasis Rev200524227328515986137
- StahlJMSharmaACheungMDeregulated Akt3 activity promotes development of malignant melanomaCancer Res200464197002701015466193
- ZhouXPGimmOHampelHEpigenetic PTEN silencing in malignant melanomas without PTEN mutationAm J Pathol200015741123112811021816
- YamadaKMArakiMTumor suppressor PTEN: modulator of cell signaling, growth, migration and apoptosisJ Cell Sci2001114Pt 132375238211559746
- HennessyBTSmithDLRamPTLuYMillsGBExploiting the PI3K/AKT pathway for cancer drug discoveryNat Rev Drug Discov2005412988100416341064
- TsaoHMihmMCJrSheehanCPTEN expression in normal skin, acquired melanocytic nevi, and cutaneous melanomaJ Am A Dermatol2003495865872
- WhitemanDCZhouXPCummingsMCNuclear PTEN expression and clinicopathologic features in a population-based series of primary cutaneous melanomaInt J Cancer2002991636711948493
- WuHGoelVHaluskaFGPTEN signaling pathways in melanomaOncogene200322203113312212789288
- CurtinJAFridlyandJKageshitaTDistinct sets of genetic alterations in melanomaNew Eng J Med2005353202135214716291983
- SlipicevicAHolmRNguyenMTExpression of activated Akt and PTEN in malignant melanomas: relationship with clinical outcomeAm J Clin Pathol2005124452853616146807
- MeierFSchittekBBuschSThe RAS/RAF/MEK/ERK and PI3K/ AKT signaling pathways present molecular targets for the effective treatment of advanced melanomaFront Biosci2005102986300115970553
- CheungMSharmaAMadhunapantulaSVRobertsonGPAkt3 and mutant V600E B-Raf cooperate to promote early melanoma developmentCancer Res20086893429343918451171
- HemesathTJPriceERTakemotoCBadalianTFisherDEMAP kinase links the transcription factor Microphthalmia to c-Kit signalling in melanocytesNature199839166642983019440696
- RoskoskiRJrStructure and regulation of Kit protein-tyrosine kinase – the stem cell factor receptorBiochem Biophys Res Commun200533831307131516226710
- CurtinJAFridlyandJKageshitaTDistinct sets of genetic alterations in melanomaN Engl J Med2005353202135214716291983
- AshidaATakataMMurataHKidoKSaidaTPathological activation of KIT in metastatic tumors of acral and mucosal melanomasInt J Cancer2009124486286819035443
- JiangXZhouJYuenNKImatinib targeting of KIT-mutant oncoprotein in melanomaClin Cancer Res200814237726773219047099
- HandoliasDSalemiRMurrayWMutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposurePigment Cell Melanoma Res201023221021520088873
- KongYSiLZhuYLarge-scale analysis of KIT aberrations in Chinese patients with melanomaClin Cancer Res20111771684169121325067
- YunJLeeJJangJKIT amplification and gene mutations in acral/mucosal melanoma in KoreaAPMIS2011119633033521569090
- FlahertyKTPuzanovIKimKBInhibition of mutated, activated BRAF in metastatic melanomaN Engl J Med2010363980981920818844
- RobertCArnaultJPMateusCRAF inhibition and induction of cutaneous squamous cell carcinomaCur Opin Oncol2011232177182
- ArnaultJPWechslerJEscudierBKeratoacanthomas and squamous cell carcinomas in patients receiving sorafenibJ Clin Oncol20092723e59e6119597016
- HeidornSJMilagreCWhittakerSKinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAFCell2010140220922120141835
- HatzivassiliouGSongKYenIRAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growthNature2010464728743143520130576
- PoulikakosPIZhangCBollagGShokatKMRosenNRAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAFNature2010464728742743020179705
- JohannessenCMBoehmJSKimSYCOT drives resistance to RAF inhibition through MAP kinase pathway reactivationNature2010468732696897221107320
- ParaisoKHXiangYRebeccaVWPTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expressionCancer Res20117172750276021317224
- NazarianRShiHWangQMelanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulationNature2010468732697397721107323
- VillanuevaJVulturALeeJTAcquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3KCancer Cell201018668369521156289
- WagleNEmeryCBergerMFDissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profilingJ Clin Oncol201129223085309621383288
- RomanoESchwartzGKChapmanPBWolchockJDCarvajalRDTreatment implications of the emerging molecular classification system for melanomaLancet Oncol201112991392221349766
- KeffordRArkenauHBrownMPPhase I/II study of GSK2118436, a selective inhibitor of oncogenic mutant BRAF kinase, in patients with metastatic melanoma and other solid tumors [abstract]ASCO Annual Meeting20108503
- WilhelmSMCarterCTangLBAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesisCancer Research200464197099710915466206
- AdnaneLTrailPATaylorIWilhelmSMSorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculatureMethods Enzymol200640759761216757355
- AhmadTMaraisRPyleLBAY 43-9006 in patients with advanced melanoma: The Royal Marsden experience [abstract]ASCO Annual Meeting20047506
- EisenTAhmadTFlahertyKTSorafenib in advanced melanoma: a Phase II randomised discontinuation trial analysisBr J Cancer200695558158616880785
- FlahertyKBroseMSchuchterLPhase I/II trial of BAY 43-9006, carboplatin (C) and paclitaxel (P) demonstrates preliminary antitumor activity in the expansion cohort of patients with metastatic melanoma [abstract]ASCO Annual Meeting20047507
- FlahertyKTLeeSJSchuchterLMFinal results of E2603: A double-blind, randomized phase III trial comparing carboplatin (C)/paclitaxel (P) with or without sorafenib (S) in metastatic melanoma [abstract]ASCO Annual Meeting20108511
- HauschildAAgarwalaSSTrefzerUResults of a phase III, randomized, placebo-controlled study of sorafenib in combination with carboplatin and paclitaxel as second-line treatment in patients with unresectable stage III or stage IV melanomaJ Clin Oncol200927172823283019349552
- SharfmanWHHodiFSLawrenceDPResults from the first-inhuman (FIH) phase I study of the oral RAF inhibitor RAF265 administered daily to patients with advanced cutaneous melanoma [abstract]ASCO Annual Meeting20118508
- SolitDBGarrawayLAPratilasCABRAF mutation predicts sensitivity to MEK inhibitionNature2006439707435836216273091
- LoRussoPMKrishnamurthiSSRinehartJJPhase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancersClin Cancer Res20101661924193720215549
- BoasbergPDRedfernCHDanielsGAPilot study of PD-0325901 in previously treated patients with advanced melanoma, breast cancer, and colon cancerCancer Chemother Pharmacol201168254755221516509
- GilmartinAGBleamMRGroyAGSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibitionClin Cancer Res2011175989100021245089
- InfanteJRFecherLANallapareddySSafety and efficacy results from the first-in-human study of the oral MEK 1/2 inhibitor GSK1120212 [abstract]ASCO Annual Meeting20102503
- YehTCMarshVBernatBABiological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogenactivated protein kinase kinase 1/2 inhibitorClin Cancer Res20071351576158317332304
- DummerRRobertCChapmanPBAZD6244 (ARRY-142886) vs temozolomide (TMZ) in patients (pts) with advanced melanoma: An open-label, randomized, multicenter, phase II study [abstract]ASCO Annual Meeting20089033
- DryJRPaveySPratilasCaTranscriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244)Cancer Res20107062264227320215513
- PratilasCATaylorBSYeQ(V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathwayProc Nat Acad Sci U S A20091061145194524
- ErnstDSEisenhauerEWainmanNPhase II study of perifosine in previously untreated patients with metastatic melanomaInvest New Drugs200523656957516034524
- BurrisHASiuLLInfanteJRSafety, pharmacokinetics (PK), pharmacodynamics (PD), and clinical activity of the oral AKT inhibitor GSK2141795 (GSK795) in a phase I first-in-human study [abstract]Asco Annual Meeting20113003
- NeshatMSMellinghoffIKTranCEnhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTORProc Nat Acad Sci U S A200198181031410319
- SchmelzleTHallMNTOR, a central controller of cell growthCell2000103225326211057898
- GeraJFMellinghoffIKShiYAKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating cyclin D1 and c-myc expressionJ Bio Chem200427942737274614576155
- MargolinKLongmateJBarattaTCCI-779 in metastatic melanoma: a phase II trial of the California Cancer ConsortiumCancer200510451045104816007689
- RaoRDWindschitlHEAllredJBPhase II trial of the mTOR inhibitor everolimus (RAD-001) in metastatic melanoma [abstract]ASCO Annual Meeting20068043
- FecherLACummingsSDKeefeMJAlaniRMToward a molecular classification of melanomaJ Clin Oncol200725121606162017443002
- LombardoLJLeeFYChenPDiscovery of N-(2-chloro-6-methylphenyl)- 2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin- 4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assaysJ Med Chem200447276658666115615512
- DrukerBJTalpazMRestaDJEfficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemiaN Engl J Med2001344141031103711287972
- PavlùJMarinDDasatinib and chronic myeloid leukemia: twoyear follow-up in eight clinical trialsClin Lymphoma Myeloma20099641742419951880
- KantarjianHMHochhausASaglioGNilotinib versus imatinib for the treatment of patients with newly diagnosed chronic phase, Philadelphia chromosome-positive, chronic myeloid leukaemia: 24-month minimum follow-up of the phase 3 randomised ENESTnd trialLancet Oncol201112984185121856226
- VerweijJCasaliPGZalcbergJProgression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trialLancet200436494401127113415451219
- von MehrenMBeyond imatinib: second generation c-KIT inhibitors for the management of gastrointestinal stromal tumorsClin Colorectal Cancer20066Suppl 1S30S3417419150
- CarvajalRDAntonescuCRWolchokJDKIT as a therapeutic target in metastatic melanomaJAMA2011305222327233421642685
- GuoJSiLKongYPhase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplificationJ Clin Oncol201129212904290921690468
- KimKBEtonODavisDWPhase II trial of imatinib mesylate in patients with metastatic melanomaBr J Cancer200899573474018728664
- UgurelSHildenbrandRZimpferALack of clinical efficacy of imatinib in metastatic melanomaBr J Cancer20059281398140515846297
- WymanKAtkinsMBPrietoVMulticenter Phase II trial of high-dose imatinib mesylate in metastatic melanoma: significant toxicity with no clinical efficacyCancer200610692005201116565971
- KlugerHMDudekAZMcCannCA phase 2 trial of dasatinib in advanced melanomaCancer2011117102202220821523734
- KalinskyKLeeSJLawrenceDPKirkwoodJMA phase II trial of dasatinib in patients with unresectable locally advanced or stage IV mucosal, acral, and solar melanomas: An Eastern Cooperative Oncology Group study (E2607) [abstract]ASCO Annual Meeting2010TPS312
- Assistance Publique – Hôpitaux de Paris Efficacy of nilotinib in first or second line treatment of primary melanomas stage III unresectable melanomas (NILOMEL)ClinicalTrialsgov [website on the Internet]Bethseda, MDUS National Library of Medicine2010 [updated February 7, 2011]. Available from: http://clinicaltrials.gov/ct2/show/NCT01168050. NLM identifier: NCT01168050Accessed February 8, 2012
- Dana-Farber Cancer InstituteNilotinib in TKI resistant or intolerant patients with metastatic mucosal, acral, or chronically sun damaged melanomaClinicalTrialsgov [website on the Internet]Bethseda, MDUS National Library of Medicine2008 [updated October 27, 2011]. Available from: http://clinicaltrials.gov/ct2/show/NCT00788775. NLM identifier: NCT00788775Accessed February 8, 2012
- Institute of Cancer Research United KingdomA trial looking at nilotinib to treat acral and mucosal melanoma skin cancer that has spread (NICAM)ClinicalTrialsgov [website on the Internet]Bethseda, MDUS National Library of Medicine2011 [updated July 14, 2011]. Available from: http://clinicaltrials.gov/ct2/show/NCT01395121. NLM identifier: NCT01395121Accessed February 8, 2012
- Novartis PharmaceuticalsA study of AMNN107 in the treatment of metastatic and/or inoperable melanoma harboring a c-Kit mutation (TEAM)ClinicalTrialsgov [website on the Internet]Bethseda, MDUS National Library of Medicine2009 [updated August 11, 2011]. Available from: http://clinicaltrials.gov/ct2/show/NCT01028222. NLM identifier: NCT01028222Accessed February 8, 2012
- Samsung Medical CenterStudy of nilotinib in metastatic melanoma with KIT aberrationsClinicalTrialsgov [website on the Internet]Bethseda, MDUS National Library of Medicine2010 [updated January 10, 2012]. Available from: http://clinicaltrials.gov/ct2/show/NCT01099514. NLM identifier: NCT01099514Accessed February 8, 2012
- KimKBDaviesMAPapadopoulosNEPhase I/II study of the combination of sorafenib and temsirolimus in patients with metastatic melanoma [abstract]ASCO Annual Meeting20099026
- SebtiSMAdjeiAAFarnesyltransferase inhibitorsSemin Oncol2004311 Suppl 1S28S39
- ChowLQEckhardtSGO’BryantCLA phase I safety, pharmacological, and biological study of the farnesyl protein transferase inhibitor, lonafarnib (SCH 663366), in combination with cisplatin and gemcitabine in patients with advanced solid tumorsCancer Chemother Pharmacol200862463164618058098
- GajewskiTFNiedzwieckiDJohnsonJPhase II study of the farnesyltransferase inhibitor R115777 in advanced melanoma: CALGB 500104 [abstract]ASCO Annual Meeting20068014
- LiptonAReadyNBukowskiRMPhase I study of continuous oral lonafarnib plus weekly paclitaxel for advanced cancer [abstract]ASCO Annual Meeting2002364
- ZujewskiJHorakIDBolCJPhase I and pharmacokinetic study of farnesyl protein transferase inhibitor R115777 in advanced cancerJ Clin Oncol200018492794110673536
- HongDSSebtiSMNewmanRAPhase I trial of a combination of the multikinase inhibitor sorafenib and the farnesyltransferase inhibitor tipifarnib in advanced malignanciesClin Cancer Res200915227061706819903778
- MargolinKAMoonJFlahertyLERandomized phase II trial of sorafenib (SO) with temsirolimus (TEM) or tipifarnib (TIPI) in metastatic melanoma: Southwest Oncology Group Trial S0438 [abstract]ASCO Annual Meeting20108502
- UgurelSRapplGTilgenWReinholdUIncreased serum concentration of angiogenic factors in malignant melanoma patients correlates with tumor progression and survivalJ Clin Oncol200119257758311208853
- HainsworthJDSpigelDRBurrisHA3rdPhase II trial of bevacizumab and everolimus in patients with advanced renal cell carcinomaJ Clin Oncol201028132131213620368560
- HainsworthJDInfanteJRSpigelDRBevacizumab and everolimus in the treatment of patients with metastatic melanoma: a phase 2 trial of the Sarah Cannon Oncology Research ConsortiumCancer2010116174122412920564157
- HodiFSFriedlanderPAAtkinsMBA phase I trial of ipilimumab plus bevacizumab in patients with unresectable stage III or stage IV melanoma [abstract]ASCO Annual Meeting20118511
- SchoenfeldJJinushiMNakazakiYActive immunotherapy induces antibody responses that target tumor angiogenesisCancer Res20107024101501016021159637
- InfanteJRFalchookGSLawrenceDPPhase I/II study to assess safety, pharmacokinetics, and efficacy of the oral MEK 1/2 inhibitor GSK1120212 (GSK212) dosed in combination with the oral BRAF inhibitor GSK2118436 (GSK436) [abstract]ASCO Annual Meeting2011CRA8503
- RibasAHauschildAKeffordRPhase III, open-label, randomized, comparative study of tremelimumab (CP-675,206) and chemotherapy (temozolomide [TMZ] or dacarbazine [DTIC]) in patients with advanced melanoma [abstract]ASCO Annual Meeting2008LBA9011
- BrahmerJRDrakeCGWollnerIPhase I study of singleagent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlatesJ Clin Oncol201028193167317520516446
- SznolMHodiFSMargolinKPhase I study of BMS-663513, a fully human anti-CD137 agonist monoclonal antibody, in patients (pts) with advanced cancer (CA) [absract]ASCO Annual Meeting20083007
- KruitWHSuciuSDrenoBImmunization with recombinant MAGE-A3 protein combined with adjuvant systems AS15 or AS02B in patients with unresectable and progressive metastatic cutaneous melanoma: A randomized open-label phase II study of the EORTC Melanoma Group (16032-18031) [abstract]ASCO Annual Meeting20089065
- SchwartzentruberDJLawsonDHRichardsJMGp100 peptide vaccine and interleukin-2 in patients with advanced melanomaN Engl J Med2011364222119212721631324
- SenzerNNKaufmanHLAmatrudaTPhase II clinical trial of a granulocyte-macrophage colony-stimulating factorencoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanomaJ Clin Oncol200927345763577119884534
- KaufmanHLKimDWDeRaffeleGLocal and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanomaAnn Surg Oncol201017371873019915919
- EscudierBLassauNAngevinEPhase I trial of sorafenib in combination with IFN alpha-2a in patients with unresectable and/or metastatic renal cell carcinoma or malignant melanomaClin Cancer Res20071361801180917363536
- HauschildAGutzmerRUgurelSSorafenib and pegylated interferon-alpha-2b in advanced metastatic melanoma: a multicenter phase II DeCOG trialAnn Oncol20112271667167421220519
- McDermottDFSosmanJAGonzalezRDouble-blind randomized phase II study of the combination of sorafenib and dacarbazine in patients with advanced melanoma: a report from the 11715 Study GroupJ Clin Oncol200826132178218518445842
- EisenTMaraisRAffolterAAn open-label phase II study of sorafenib and dacarbazine as first-line therapy in patients with advanced melanoma [abstract]ASCO Annual Meeting20078529