Abstract
Emerging laboratory and clinical investigations demonstrate that Hedgehog signaling (Hh) represents a novel therapeutic target in various human cancers. This conserved signaling pathway precisely regulates self-renewal and terminal differentiation in embryonic development, but is typically silenced in adult tissues, with reactivation usually only during tissue repair. Aberrant Hh pathway signaling has been implicated in the pathogenesis, self-renewal, and chemotherapy resistance of a growing number of solid and hematologic malignancies. Major components of the Hh pathway include the Hh ligands (Sonic, Desert, and Indian), the transmembrane receptor Patched, the signal transducer Smoothened (Smo), and transcription factors Gli1–3 which regulate the transcription of Hh target genes. Mutations in Hh pathway genes, increased Hh signaling in tumor stroma, and Hh overexpression in self-renewing cells (cancer stem cells) have been described, and these different modes of Hh signaling have implications for the design of Hh pathway inhibitors and their integration into conventional treatment regimens. Discovery of a naturally-occurring Smo inhibitor, cyclopamine, and the identification of Hh pathway mutations and over expression in cancer cells prompted the development of several cyclopamine derivatives. Encouraging laboratory and in vivo data has resulted in Phase I and II clinical trials of Smo inhibitors. In this review, we will discuss the current understanding of Hh pathway signaling in malignancy and Smo antagonists in development. Recent data with these agents shows that they are well-tolerated and may be effective for subsets of patients. Challenges remain for appropriate patient selection and the optimal combination and sequence of these targeted therapies into current treatment paradigms.
Introduction
A growing body of evidence demonstrates a role for conserved embryonic signaling pathways such as Hedgehog (Hh), Wingless, and Notch in the development of human cancers.Citation1,Citation2 Preclinical data in various tumor types suggests a role for Hh signaling in cancers of the skin,Citation3,Citation4 brain,Citation5,Citation6 lung,Citation7 breast,Citation8 prostate,Citation9,Citation10 colon,Citation11 as well as hematologic malignancies including leukemia,Citation12–Citation15 lymphoma,Citation16–Citation19 and multiple myeloma (MM).Citation16,Citation20 Laboratory data suggests that Hh signaling regulates multiple pathogenic processes including tumor growth, self-renewal, and resistance to chemotherapy. Given that Hh signaling is implicated in a wide range of malignancies, there is much interest in the development of Hh pathway inhibitors as novel anticancer therapy. In this review, we will discuss the Hh pathway in cancer, mechanisms of its aberrant activation in different tumor types, and its role in the self-renewing cell population, or cancer stem cells (CSCs). We will then review published and preliminary data regarding Hh pathway inhibitors clinical trials.
The Hh pathway
The Hh signaling pathway regulates cell differentiation and self-renewal in the developing embryo and is typically silenced in adult tissues.Citation21 Aberrant Hh signaling may result from mutations in pathway genes or overexpression of signaling through other mechanisms in either tumor cells themselves or cells in the supportive tumor microenvironment.Citation22–Citation26 Much of our knowledge of the Hh pathway comes from studies in Drosophila, and although several major components of the pathway are well described, some details remain poorly understood. In mammals, one of three Hh pathway ligands (Desert, Indian, and Sonic) binds to the transmembrane receptor Patched (Ptch) to initiate pathway signaling. In the inactive state, Ptch exerts an inhibitory effect on the signal transducer Smoothened (Smo), and no downstream signaling occurs. When Hh ligand binds to Ptch, the inhibition on Smo is released and downstream signaling occurs, regulating the expression of the transcription factors Gli1–3 (). Primary cilia present on most cells during interphase are involved in signal transduction, and Hh pathway components translocate during activation. In the inactive state, when Hh ligand is not present, Ptch is located in the primary cilia but Smo is not. When ligand binds and Ptch inhibition of Smo is released, Ptch moves out of the primary cilia and Smo moves in to facilitate interaction with Glis and associated proteins. They subsequently enter the nucleus and regulate expression of Hh target genes.Citation27–Citation32
Figure 1 Hh signaling pathway. In the absence of Hh ligand, Ptch exerts an inhibitory effect on Smo, and no downstream signaling occurs. In the presence of Hh ligand binding to Ptch, the suppression of Smo is released. Smo interacts with Suppressor of fused (SUFU), which promotes the activation and nuclear translocation of Gli1. Gli1 translocation results in the transcription of Hh target genes.
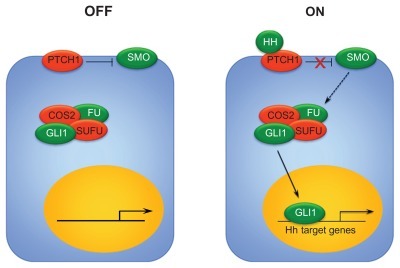
Hh expression is precisely regulated through both positive and negative feedback loops which may be interrupted by mutations in Hh pathway genes themselves or epigenetic changes. Increased transcription of Hh target genes results in increased cell proliferation and survival, induction of stem cell markers, as well as promotion of bone metastases.Citation33 Aberrant Hh signaling has also been associated with chemotherapy-resistance in gliomas,Citation34 pancreatic cancer,Citation35 leukemia,Citation36,Citation37 lymphoma,Citation17,Citation38 and MM.Citation39 Interactions with other signaling pathways, including Notch, PI3K, RAS-MEK/AKT, and NF-κB, to promote cancer growth, recurrence, and chemotherapy resistance have also been described.Citation40–Citation43
Several Smo inhibitors are in clinical development for the treatment of human cancers. Recently, emerging clinical data have demonstrated the potential activity of these agents in several diseases, particularly medulloblastoma, basal cell carcinoma (BCC), pancreatic cancer, and hematologic malignancies. Ongoing trials will evaluate the role for Smo inhibitors as single agents, as well as in combination with traditional chemotherapy. This review will discuss the mechanisms of Hh signaling in malignancy and the evidence for Hh signaling in CSCs. These preclinical studies provide the rationale for human trials of Hh inhibition in various malignancies, and we will review the progress and challenges of translating the laboratory investigations of Smo inhibitors into meaningful clinical results for patients.
The Hh pathway in cancer
Similar to its role in normal development, dysregulated Hh signaling results in the expression of a number of genes responsible for cell proliferation, survival, and self-renewal. Aberrant Hh signaling is associated with the development of cancer, as demonstrated by the Gorlin syndrome, caused by an autosomal dominant germline mutation in the PTCH1 gene.Citation4,Citation44 This resultant mutated Ptch is unable to exert its tonic inhibition of Smo, resulting in hyperactivation of the pathway. Patients with Gorlin syndrome are predisposed to various malignancies, most commonly BCC and medulloblastoma.Citation45 These observations led to the discovery of Hh activation in the majority of the more common sporadic form of BCC, with mutations in the PTCH1 allele occurring in up to 30% of casesCitation3 and SMO mutations in approximately 10%.Citation46 In addition, mutations in Hh pathway genes have been implicated in the pathogenesis of up to 30% of sporadic medulloblastoma.Citation47
Mechanisms of Hh signaling in cancer
Although Hh pathway gene mutations lead to inappropriate Hh signaling in BCC and medulloblastoma, a greater number of cancers are driven by Hh signaling through other mechanisms, either in the bulk population of cells or specifically within the CSC population. We will briefly discuss the different mechanisms of Hh signaling, and for a complete review, the reader is referred to Reference Citation8.Citation26 In both BCC and medulloblastoma, Hh pathway activation results from specific gene mutations and is independent of the presence of Hh ligand binding to Ptch. This mechanism of Hh activation, which is ligand-independent and driven by specific Hh gene mutations within the tumor cells, is termed Type I Hh signaling ().Citation26 Hh inhibitors which are antagonists to Hh ligand will not be effective in overcoming this mechanism of aberrant signaling because it occurs downstream and independent of ligand due to the mutation. The other mechanisms of Hh signaling observed in cancer rely upon Hh ligand initiation of the signaling, and vary by source and recipient cells of ligand secretion.
Figure 2 Modes of Hh pathway signaling. (A) Type I Hh signaling is activated by specific mutations within pathway genes within tumor cells, resulting in ligand-independent constitutive activation. (B) Type II Hh signaling results from autocrine signaling from tumor cell to tumor cell. (C) Type IIIa activation results from secretion of Hh ligand by tumor cells, resulting in pathway activation in surrounding tumor stroma. (D) Type IIIb Hh signaling results from Hh ligand secretion by tumor stroma, resulting in activation of the pathway within tumor cells themselves.
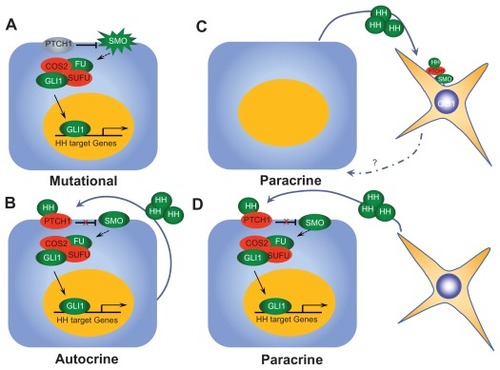
In Type II signaling, activation of the pathway is ligand-dependent and autocrine, meaning it originates and is received by the tumor cells (or neighboring cells). Most data for Type II Hh signaling comes from in vitro studies in various cancers including lung,Citation48,Citation49 prostate,Citation50 glioblastoma,Citation51,Citation52 gastrointestinal,Citation11,Citation53 breast,Citation54 and leukemia.Citation13,Citation15 These studies observed Hh expression in tumor cells and growth inhibition with Hh blockade by cyclopamine in models absent of tumor stroma. This data supports the premise that Hh ligand originates within the tumor cells and that pathway activation also occurs within tumor cells (either the same cells or neighboring cells). Several authors remain unconvinced that Type II signaling actually exists in vivo because much of this data is based on studies with higher doses of cyclopamine which exhibit some non-specific cytotoxicity.Citation25,Citation26,Citation46,Citation55 However, in our group’s report of Hh signaling in acute lymphocytic leukemia (ALL), we demonstrated findings of increased Hh pathway expression in human ALL cell lines and clinical samples. Using a luceriferase reporter assay, we observed decreased Gli1 expression in ALL cell lines following treatment with 5E1, antagonist to Hh ligand, cyclopamine, or IPI-926 (Infinity Pharmaceuticals, Cambridge, MA), a semi-synthetic Smo inhibitor at doses which did not result in apoptosis or growth inhibition. Treatment with these Hh inhibitors resulted in decreased self-renewal when cells were treated alone without the presence of stromal cells both in in vitro clonogenic assays, as well as in serial transplantation models in mice. Although there is likely a contributory effect of stromally-mediated Hh signaling in ALL, we believe that our data also supports a role for autocrine, Type II Hh signaling in ALL.Citation15 Tumors characterized by Type II signaling may be susceptible to Hh inhibition at either the level of Hh ligand binding or further downstream.
A growing body of data confirms the importance of Type III Hh signaling which is ligand dependent and paracrine; that is, ligand is secreted by one type of cell (either tumor or stroma) and Hh pathway activation occurs in another (tumor or stroma). Ligand secretion by tumor cells resulting in Hh signaling in supportive stromal cells is termed Type IIIa signaling, whereas ligand secretion by stromal cells resulting in Hh signaling in the tumor cells is termed reverse paracrine signaling or Type IIIb. Tumor types in which paracrine signaling has been described include prostate,Citation9 pancreas, and metastatic colon.Citation25 Human prostate cancer cell lines showed enhanced growth in vivo with addition of Hh ligand while no differences were seen when cells were grown alone in vitro in absence of stroma, suggesting a role for stromally mediated Hh signaling in promoting tumor growth. RT-PCR and in situ hybridization confirmed that increased tumor ligand expression correlated with increased mouse Gli1, Gli2, and Ptch1 from stromal cells.Citation9 Yauch et al demonstrated similar findings of increased mouse Gli1 expression in response to human Hh ligand expression in pancreatic cancer and metastatic colon cancer in xenografts from human cell lines and primary tumors.Citation25 Importantly, these findings from mouse models were also seen upon examination of human clinical samples comprised of tumor cells and infiltrating stromal cells in prostate, pancreatic, and metastatic colon cancer.Citation9,Citation56,Citation57 Type IIIb signaling has only been described in B-cell malignancies, including leukemia, MM, and non-Hodgkin’s lymphoma. Citation16 Hh ligands secreted by supportive stroma in lymph nodes, spleen, and bone marrow activated Hh signaling in tumor cells. Hh inhibition resulted in increased apoptosis associated with down regulated Bcl2 expression.
Hh inhibition as a CSC-targeted strategy
CSC theory states that tumors are comprised of two distinct populations of cells: a majority population of differentiated tumor cells which phenotypically characterize the disease; and a second population of rare, CSC or tumor-initiating cells, with properties of self-renewal and differentiation, responsible for disease maintenance and relapse.Citation58,Citation59 CSC theory attempts to explain the common clinical scenario of complete response to initial chemotherapy followed by relapsed disease propagated by a small population of residual cancer cells which were undetectable following initial therapy. For many cancers, conventional chemotherapy is effective against the bulk, differentiated tumor cells. Novel strategies targeting the residual CSCs responsible for disease recurrence are needed to prolong remissions, eradicate the tumor-initiating cells, and result in long-term cure. Hh signaling has been identified as a potential CSC-specific target in various cancers.Citation6,Citation13,Citation15,Citation20,Citation51,Citation60–Citation77
Techniques used to isolate and characterize CSC in vitro include aldehyde dehydrogenase expression, phenotypic markers, side population by Hoechst dye exclusion, and colony-forming assays. To date, the “gold standard” for CSC identification has been the ability of this rare population of cells to regenerate tumor consisting of both phenotypic populations, differentiated cells, and CSC in animal models.Citation78 A detailed discussion of the in vitro and in vivo methods used to characterize CSC in various tumor types is beyond the scope of this review. The reader is referred to Reference Citation78 for further details of these techniques, as well to the publications cited below concerning Hh signaling and CSC. CSC theory remains controversial due to the varying techniques for identification and the discrepancies in CSC numbers identified in primary samples and required to recreate tumors in mice by different researchers with varying techniques. Also, the inherent heterogeneity among different tumor types as well as within specific cancer types themselves. Regardless of the exact criteria for identifying CSC or properties of “stem-ness” or whether the CSC is a primitive progenitor or a de-differentiated cell, the clinical observation holds that rare populations of cancer cells persist following initial therapy and cause disease recurrence, and novel strategies to target these resistant, persistent cells are needed. In fact, CSC from different cancers may share similar targets, even more so than the CSC and differentiated cell of the same cancer type. Hh signaling has been implicated in the bulk, differentiated cell populations of many human cancers, and here we review the preclinical evidence for Hh signaling in the CSC or self-renewing cells.
Our group demonstrated a role for Hh signaling in the self-renewing cells of MM and ALL. Flow cytometric analysis of MM cell lines following treatment with the Smo inhibitor cyclopamine resulted in decreases in the CD138-negative cell fraction (the CSC population) as well as in side population (SP) cells. To assess the effects of Hh inhibition on self-renewal potential, an in vitro clonogenic assay was performed. In this assay, cells were treated with an anti-Shh antibody 5E1 to block ligand-dependent Hh signaling or the Smo inhibitor cyclopamine and then assayed for the ability to form tumor colonies in semi-solid media. Compared to a vehicle control, both 5E1 or cyclopamine significantly inhibited colony-formation by MM cell lines and primarily clinical samples.Citation20 Furthermore, this inhibitory effect was maintained during serial replating indicative of self-renewal potential. Similarly, in ALL, treatment of human ALL cell lines with the Smo inhibitors cyclopamine or IPI-926 resulted in decreased ALDH+ cells, decreased in vitro colony-formation, and decreased serial transplantation in mice, all reflective of effects on the self-renewing cell population.Citation15 In chronic myeloid leukemia (CML) mouse models, Smo −/− mice had longer latency to develop CML when mouse bone marrow transduced with BCR-ABL was transplanted. A decrease in the number of phenotypically primitive cells (Lin- Sca-1- c-Kit+), a surrogate for CSC, was also observed in the Smo −/− mice as compared to Smo +/+ mice.Citation67 Similarly, Dierks et al transduced Smo −/− fetal liver cells with BCR-ABL and observed more rapid development of CML in transplanted mice. Secondary recipients of Smo −/− cells had no development of CML at 9+ months of observation. CML stem cells were isolated by flow cytometry and treated with the Smo inhibitor cyclopamine or Hh ligand blocking antibody 5E1. Decreased colony-formation in vitro was observed with both treatments, demonstrating a role for ligand-dependent Hh signaling in mediating self-renewal in CML. Mice transplanted with BCR-ABL infected hematopoietic stem cells were treated with cyclopamine. All untreated animals rapidly developed CML and died within 4 weeks, but cyclopamine-treated mice had a 60% survival at 7 weeks. In addition, CML from cyclopamine treated mice was unable to be serially transplanted, consistent with an observed decrease in the number of CML stem cells persisting after treatment.Citation13
In glioblastoma multiforme, elevated Gli1 expression was seen in five of 19 primary samples and four of seven cell lines tested. Treatment with cyclopamine depleted the putative CSC population as identified by increased ALDH expression and SP. In vitro, glioblastoma multiforme neurospheres were unable to produce new neurospheres after exposure to cyclopamine, and in vivo injection of cyclopamine treated glioblastoma multiforme cells did not result in tumor formation versus untreated cells, demonstrating that Hh inhibition with cyclopamine eliminated the self-renewal capacity of these cells.Citation52 Tanaka et al demonstrated in human breast cancer cells that the CSC population, marked by either markers CD44+ CD24−/low or SP had increased Hh expression and that proliferation was limited following treatment with siRNA against Gli1.Citation79 Similarly, Liu and colleagues isolated human breast tumor CSC by flow sorting for cells with markers CD44+ CD24−/low Lin−. Cyclopamine or siRNA against Gli1 or Gli2 inhibited Hh signaling, resulting in diminished self-renewal capacity, mediated by BMI-1, a known regulator of normal stem cell self-renewal.Citation65 Tian et al looked at the effects of Hh inhibition with the synthetic Smo inhibitor GDC-0449 (Genentech, South San Francisco, CA; Curis, Lexington, MA; Roche, Basel, Switzerland) in lung cancer cell lines and used SP technique to isolate and enrich for CSC. They observed expression of Smo in the SP fraction, clonogenicity restricted to the SP fraction, and that treatment with GDC-0449 reduced the number of SP cells.Citation73 Pancreatic CSCs as characterized by ALDH expression were preferentially reduced in number as compared to differentiated tumor cells when treated with cyclopamine.Citation62 Similarly, Singh et al showed that Hh inhibition with GDC-0449 resulted in increased apoptosis of pancreatic cell line CSC as well as decreased transcription of Hh target genes.Citation70 Other groups have described Hh signaling in the CSC of cancers of the prostate,Citation60 stomach,Citation71 colon,Citation63 and ovary.Citation80
Targeting Hh signaling in cancer
The identification of a naturally-occurring Hh pathway antagonist, cyclopamine, led to the subsequent development of synthetic and semi-synthetic derivatives of cyclopamine with increased potency and bioavailability. In addition, a commonly used anti-fungal agent, itraconazole, has also been found to have Hh inhibitory activity. These agents are now moving from the laboratory into clinical trials. Similar to challenges encountered in translating other targeted therapies, questions remain including the optimal way to integrate them into regimens of conventional chemotherapy. In addition, our growing understanding of mechanisms of Hh signaling in different malignancies raises the issue of whether there should be different approaches to using these agents which vary by the mechanism of Hh signaling in each disease. Currently, all of the Hh inhibitors in clinical development are at the level of Smo, but other agents with distinct mechanisms of action have been identified and it is possible that these may be more or less effective depending on the mode of signaling. Below, we will discuss cyclopamine, itraconazole, and Smo inhibitors in clinical trials.
Cyclopamine
In the 1960s, teratogenic effects of ingestion of the corn lily Veratrum californicum were observed, resulting in one-eyed offspring (cyclopia) in lambs.Citation81 It was subsequently discovered that the active agent, cyclopamine, exerted its effects through Hh pathway inhibition,Citation82 specifically acting at the level of Smo.Citation83 Cyclopamine displayed anti-tumor activity in vitro and in vivo, but poor oral bioavailability and acid sensitivity has prevented further clinical development. One clinical report of four patients with BCC, one of whom had Gorlin syndrome, described dramatic, rapid clinical regression of the lesions in response to topical cyclopamine application versus placebo. In addition, histological and immunohistochemical analysis showed apoptosis and increased markers of differentiation in response to Hh inhibition.Citation84 Although no longer in clinical development due to increased potency and bioavailability of cyclopamine derivatives, cyclopamine remains an important agent in preclinical models of Hh inhibition.
Itraconazole
Interestingly, the anti-fungal agent itraconazole was found to have Hh inhibitory properties on a drug screen of known compounds. Kim et al showed that commonly used doses of itraconazole suppressed Hh pathway activity and inhibited growth of medulloblastoma in vivo. It appears to act on Smo, as does cyclopamine and its synthetic derivatives, but its mechanism of Smo antagonism is distinct from that of cyclopamine and appears to limit the ciliary accumulation of Smo.Citation85 Itraconazole is currently in studies of patients with BCC, metastatic prostate cancer, and recurrent non-small cell lung cancer (see ).
Table 1 Smoothened inhibitors currently in clinical trials for cancer
Synthetic and semi-synthetic cyclopamine derivatives
All of the Hh inhibitors in clinical trials currently act at the level of Smo (see for a list of currently open clinical trials and for a summary of findings of Smo inhibitors in clinical trials). Due to the decreased oral bioavailability and acid sensitivity of cyclopamine, semi-synthetic and synthetic derivatives have been developed. These derivatives appear to have increased potency and all are oral agents. Smo inhibitors currently under investigation appear to inhibit Smo through binding at the same portion of the transmembrane segment 6.Citation86,Citation87 Here, we will review the published or presented data regarding Smo inhibitors in clinical trials for cancer.
Table 2 Summary of clinical findings from Phase I trials of Smoothened inhibitors in cancer
Vismodegib
Early results from patients with medulloblastoma and BCC on the initial Phase I study of GDC-0449 were published in the New England Journal of Medicine in 2009 demonstrating a role for Smo inhibitors in cancers known to be driven by Hh pathway mutations.Citation88,Citation89 In the first report, one patient with heavily pre-treated medulloblastoma had clinical and radiographic regression of widespread systemic metastases on vismodegib. These results were short-lived, however, with measurable increases in tumor size at 3 months of therapy and subsequent identification of a single amino acid substitution conferring resistance.Citation89 A recent update of the entire Phase I cohort demonstrated that patients with BCC on vismodegib had a response rate of 58%. Twelve of 33 advanced BCC subjects had been on therapy from 8.5 months to 26.5 months, with median duration of response of 12.8 months (as of January 2010, with several patients remaining on therapy and with continued response).Citation90 Responses were only observed in the patients with medulloblastoma or BCC. At the American Society of Clinical Oncology (ASCO) Annual Meeting in 2010, preliminary data was presented from eleven patients enrolled in a Phase I study of vismodegib in medulloblastoma coordinated through the Pediatric Brain Tumor Consortium. The limited data presented suggested that the drug was well-tolerated as a single agent in patients ranging from age 4–20 years. Magnetic resonance imaging scans of the knees will monitor for changes in bone development, as this was a significant side effect observed in mouse studies with Hh inhibition.Citation91
More recently, encouraging Phase II results with vismodegib in 99 patients with locally advanced or metastatic BCC were presented during the European Multidisciplinary Cancer Congress in 2011. In patients with metastatic disease, disease stabilization occurred in 63% with an overall response rate of 30%. In locally advanced BCC, the overall response rate was 43% with stable disease in 40% of patients. Median duration of progression-free survival was 9.5 months.Citation92 The plenary session at the American Association for Cancer Research Annual Meeting in 2011 featured results from 36 patients on a Phase II study of vismodegib in patients with Gorlin Syndrome/Basal Cell Nevus Syndrome. This randomized, double-blind, placebo-controlled trial was designed to evaluate the efficacy of vismodegib in preventing the development of new BCC. The data safety monitoring board discontinued the placebo arm due to statistically significant differences in the number of new BCC (0.07 new BCC/month in treated patients vs 1.74 new BCC/month for those on placebo) and the change in size of existing BCC (decrease of 24 cm in vismodegib arm vs −3 cm in placebo arm).Citation93 The 2011 ASCO Annual Meeting featured two reports on vismodegib in combination with chemotherapy. A Phase I trial examined the combination of vismodegib with erlotinib and gemcitabine for unresectable pancreatic cancer. Observed dose-limiting toxicities of the combination included rash, nausea, infection, visual disturbances, and thrombocytopenia. Transient responses with stable disease were seen in two dose cohorts, and a dose was determined for Phase II testing.Citation94 An NCI-sponsored, randomized, double-blind placebo-controlled trial of chemotherapy with 5-fluorouracil and oxaliplatin (FOLFOX) with or without vismodegib in previously untreated patients with advanced gastric or gastroesophageal junction cancer was reviewed at the Trials in Progress poster session at the meeting. The study plans to enroll 116 patients in order to detect a 69% increase in progression-free survival. Laboratory correlative studies to look at effects on the tumor microenvironment are also planned.Citation95
Across these early studies, there were no dose-limiting toxicities, and common side effects were mild and included muscle spasms, dysgeusia (taste alteration), fatigue, alopecia, and nausea. Grade 3 adverse events included reversible hyponatremia, abdominal pain, fatigue, and muscle cramps. Mild to moderate side effects included dysgeusia, muscle cramps, and weight loss, with two grade 3–4 adverse events in the treatment arm (muscle cramps and suicide attempt). In the trial of vismodegib in patients with basal cell nevus syndrome, the drug was discontinued in 20% of patients due to adverse effects.Citation92 Therefore, it appears that vismodegib is well tolerated.
IPI-926
IPI-926 (Infinity Pharmaceuticals, Cambridge, MA) is the only Smo inhibitor in development which is a semi-synthetic derivative of cyclopamine. Early phase clinical trial results were presented at the ASCO Annual Meeting in 2011. In the Phase I study of patients with locally advanced or metastatic solid tumors, partial responses were seen in patients with BCC, and several patients continued on therapy for more than 1 year.Citation96 Preliminary findings from a Phase Ib/II study of IPI-926 in combination with gemcitabine in patients with untreated metastatic pancreatic cancer were also presented. Median progression free survival was approximately 5.5 months with five of 16 patients demonstrating a partial response to the combination.Citation97 In both studies, the drug was well-tolerated with mild effects of nausea, fatigue, muscle spasms, and dysgeusia observed. There was also asymptomatic and reversible elevation of liver function tests.
LDE225
Two recent reports comment on the clinical experience with LDE225 (Novartis, Basel, Switzerland). Preliminary findings from a Phase I dose escalation study of LDE225 were reported at the ASCO Annual Meeting in 2010. One patient with medulloblastoma maintained a partial response for 4 months and five additional patients with various malignancies including lung cancer and BCC have tolerated therapy for more than 4 months.Citation98 A topical formulation of LDE225 was also recently tested in patients with Gorlin syndrome and BCC. In this double-blind, randomized, vehicle-controlled intra-individual study, 27 BCC in eight patients were treated with LDE225 cream or vehicle twice daily for 4 weeks. In twelve of 13 BCC, there were three complete responses and nine partial responses compared to only one partial response in 14 vehicle treated BCC.Citation99 Similar to other Smo inhibitors, the oral drug was well-tolerated with side effects of fatigue, nausea, muscle cramps, and dysgeusia being most common. The topical formulation was well-tolerated with no skin irritation.
BMS-833923 (XL139)
BMS-833923 (XL139) (Bristol Myers Squibb, New York, NY; Exelixis, South San Francisco, CA) is also being studied in early phase trials as a single agent in advanced and metastatic solid tumors. There are ongoing clinical trials looking at this drug in combination with standard chemotherapy in MM, gastric and esophageal cancers, and small cell lung cancer. Data was presented at the American Association for Cancer Research-National Cancer Institute-European Organization for Research and Treatment of Cancer Annual Meeting in 2009. One patient with medulloblastoma continues on study for more than 10 months with stable disease. A second subject with Gorlin syndrome had a partial response to therapy which was ongoing at the time of the report.Citation100 At the 2011 American Society of Hematology meeting, Huff et al presented Phase I data on BMS-833293 in patients with MM. BMS-833923 was given either as monotherapy, or in combination with lenalidomide plus dexamethasone, or with bortezomib in patients with relapsed or refractory MM. No clinical response data had been analyzed to date.Citation101 In both trials, the drug was well-tolerated with the most common side effects being dysgeusia, muscle spasms, and nausea. There was one episode of grade 2 lipase elevation and pancreatitis.
PF-04449913
Preliminary data was presented in abstract form with PF-04449913 (Pfizer, Manhattan, NY) at the 2011 American Society of Hematology Annual Meeting. The drug was administered as a single agent in escalating doses in a Phase Ia study of 32 patients with various hematologic malignancies including acute myeloid leukemia, CML in lymphoid blast crisis, myelodysplastic syndrome, myelofibrosis, and chronic myelomonocytic leukemia. Some evidence of response was seen in all disease subsets, and one patient with acute myeloid leukemia evolved from chronic myelomonocytic leukemia achieved a complete remission with incomplete blood count recovery. Five patients with acute myeloid leukemia had significant reduction in circulating blast counts, and one patient with T315I mutation CML in lymphoid blast crisis achieved a major cytogenetic response with loss of the T315I mutation. Other patients had hematologic improvement or disease stabilization. The drug was relatively well-tolerated with the most common side effects being dysgeusia, alopecia, nausea, and arthralgias.Citation102 An expanded multicenter Phase Ib/II study is planned in combination with chemotherapy in hematologic malignancies as well.
Discussion
Preclinical in vitro and in vivo data with Hh signaling demonstrate a role for the pathway in the pathogenesis, self-renewal, and chemotherapy resistance in a variety of human cancers. Years of laboratory investigations have led to Phase I trials of several Smo inhibitors which appear to be relatively well-tolerated either as single agents or in combination regimens with conventional chemotherapy. Modest side effects include nausea, dysgeusia, muscle cramps, and fatigue, with rare grade 3 adverse events observed. It is unclear, however, what side effects may occur in children, as permanent defects in bone growth have been seen in mouse models of Hh inhibitorsCitation91 which may complicate their clinical development for pediatric tumors such as medulloblastoma. The known teratogenic effects of cyclopamine caution for the need to prevent pregnancy in treated subjects. Encouraging preliminary results have led to ongoing clinical trials in adults in hematologic malignancies, pancreatic cancer, glioblastoma, gastrointestinal tumors, lung cancer, and other advanced solid tumors. These early trials may suggest how to incorporate Smo inhibitors into treatment regimens with conventional chemotherapy. Interestingly, many of these trials incorporate prolonged post-chemotherapy maintenance therapy with the Smo inhibitor, reflecting an understanding of the potential role of Hh signaling in maintaining the CSC population which persists after initial therapy.
Despite early enthusiasm and preclinical successes, challenges remain in the development of Smo inhibitors. Drug resistance is an important concern. Preclinical models suggest several potential mechanisms of resistance, but the most compelling data comes from one clinical example, the first patient treated with the Smo inhibitor vismodegib for refractory medulloblastoma. Despite an initial dramatic response to therapy, resistant disease emerged after 3 months with relapse at multiple sites.Citation89 Tumor biopsies taken both before and after vismodegib provided important insights into the mechanisms of resistance to Smo inhibitors. Yauch and colleagues were able to identify a single amino acid substitution in a conserved aspartate acid residue of Smo. This mutation retained Smo activity but interfered with vismodegib binding, preventing the drug effect. Whether this mutation arose in the setting of vismodegib therapy or was present at levels too low to be detected pre-treatment remains unclear. In mouse models of medulloblastoma, the same mutation was identified in a tumor resistant to vismodegib as well.Citation103 Similar to the development of BCR-ABL tyrosine kinase inhibitors which retain activity against mutations conferring resistance to imatinib, studies are underway to develop second-generation Smo inhibitors which remain effective in the face of known Smo mutations conferring drug resistance.Citation104 Other mechanisms of resistance that have been identified in preclinical models include amplification of Hh signaling molecules downstream of Smo (cf, Gli2),Citation104 amplification of Hh target genes,Citation105 and upregulation of signaling pathways which interact with Hh, such as PI3K.Citation41 In addition, aberrant Hh signaling may result from pathway activation downstream of Smo. Hh inhibitors which act at the level of the Gli transcription factors are also under investigation in the laboratory,Citation106 and may prove effective in the setting of Smo inhibitor resistance or in combination. For example, arsenic trioxide, used in the treatment of acute promyelocytic leukemia, has been shown to inhibit Gli proteins.Citation107,Citation108
Preclinical testing of Smo inhibitors has identified tumor types which appear dependent on Hh signaling for their growth, self-renewal, and chemotherapy resistance. Hh signaling does not appear consistent throughout any tumor type, including those with specific activating mutations such as BCC and medulloblastoma, in which a majority of tumors may be dependent on the pathway. At this time, testing to know which particular patients and tumors are dependent on Hh signaling is not available. Efficacy results from early phase trials of these agents should be interpreted cautiously, as the effects may be more pronounced in only specific subsets of patients, and we are currently unable to prospectively identify them. The potential to profile an individual’s own tumor for dependence on Hh or other signaling pathways would better direct patients into clinical trials and suggest potential synergistic combinations of inhibitors. However, current techniques do not permit this level of personalized medicine.
The development of Smo inhibitors represents an exciting advance in cancer therapy. Hh signaling is a cancer-specific target in adults, and likely a CSC-specific target in several diseases, with the potential to have efficacy as single agents and in order to sensitize tumors to chemotherapy. As these clinical trials provide tumor samples and outcomes suggesting which patients may benefit, additional questions will need to be answered through laboratory correlative studies to better identify which patients and tumor types are most likely to be responsive to these interventions. Clinical trials of CSC-targeted therapies such as Hh inhibition may not produce early measurable clinical responses, but may instead prolong survival as the CSC are extinguished over time. In cancers where the role of Hh signaling may be due to a tumor-stromal interaction, Hh inhibition alone may result in a cytostatic effect with no measurable benefit radiographically but an improvement in survival. These factors must be considered as clinical trials incorporate Smo inhibitors with traditional chemotherapy, and overall survival represents the ideal endpoint to assess efficacy of a treatment such as this with potential CSC- or stroma-specific effects. A better understanding of the role of Hh signaling in the response of tumors to traditional therapy, as well as the interactions of the Hh pathway with other signaling pathways, may suggest combinations of Smo inhibitors with other pathway inhibitors for further study.
Disclosure
The authors report no conflicts of interest in this work.
References
- BeachyPAKarhadkarSSBermanDMTissue repair and stem cell renewal in carcinogenesisNature2004432701532433115549094
- TaipaleJBeachyPThe Hedgehog and wnt signalling pathways in cancerNature2001411683534935411357142
- GailaniMRStåhle-BäckdahlMLeffellDJThe role of the human homologue of Drosophila patched in sporadic basal cell carcinomasNat Genet199614178818782823
- HahnHWickingCZaphiropoulousPGMutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndromeCell19968568418518681379
- ZurawelRHAllenCChiappaSAnalysis of PTCH/SMO/SHH pathway genes in medulloblastomaGenes Chromosomes Cancer2000271445110564585
- BarEEChaudhryALinACyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastomaStem Cells200725102524253317628016
- WatkinsDNBermanDMBaylinSBHedgehog signaling: progenitor phenotype in small-cell lung cancerCell Cycle20032319619812734424
- VorechovskýIBenediktssonKPToftgårdRThe patched/hedgehog/Smoothened signalling pathway in human breast cancer: no evidence for H133Y SHH, PTCH and SMO mutationsEur J Cancer199935571171310505029
- FanLPepicelliCDibbleCHedgehog signaling promotes prostate xenograft tumor growthEndocrinology200414583961397015132968
- KarhadkarSSBovaGSAbdallahNHedgehog signalling in prostate regeneration, neoplasia and metastasisNature2004431700970771215361885
- BermanDKarhadkarSMaitraAWidespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumoursNature2003425696084685114520411
- BaiLChiuCLinCDifferential expression of Sonic hedgehog and Gli1 in hematological malignanciesLeukemia200822122622817928882
- DierksCBeigiRGuoGExpansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activationCancer Cell200814323824918772113
- WarzechaJBonkeLKoehlUThe hedgehog inhibitor cyclopamine induces apoptosis in leukemic cells in vitroLeuk Lymphoma200849122383238619052992
- LinTLWangQHBrownPSelf-renewal of acute lymphocytic leukemia cells is limited by the Hedgehog pathway inhibitors cyclopamine and IPI-926PLoS One2010512e1526221203400
- DierksCGrbicJZirlikKEssential role of stromally induced hedgehog signaling in B-cell malignanciesNat Med200713894495117632527
- HegdeGMungerCEmanuelKTargeting of sonic hedgehog-GLI signaling: a potential strategy to improve therapy for mantle cell lymphomaMol Cancer Ther2008761450146018524848
- KawaharaTKawaguchi-IharaNOkuhashiYItohMNaraNTohdaSCyclopamine and quercetin suppress the growth of leukemia and lymphoma cellsAnticancer Res200929114629463220032413
- SinghRKimJDavuluriYHedgehog signaling pathway is activated in diffuse large B-cell lymphoma and contributes to tumor cell survival and proliferationLeukemia20102451025103620200556
- PeacockCDWangQGesellGSHedgehog signaling maintains a tumor stem cell compartment in multiple myelomaProc Natl Acad Sci U S A2007104104048405317360475
- InghamPMcMahonAHedgehog signaling in animal development: paradigms and principlesGenes Dev200115233059308711731473
- WickingCMcGlinnEThe role of hedgehog signalling in tumorigenesisCancer Lett200117311711578802
- Ruiz i AltabaASánchezPDahmaneNGli and hedgehog in cancer: tumours, embryos and stem cellsNat Rev Cancer20022536137212044012
- Ruiz i AltabaATherapeutic inhibition of Hedgehog-GLI signaling in cancer: epithelial, stromal, or stem cell targets?Cancer Cell200814428128318835029
- YauchRGouldSScalesSA paracrine requirement for hedgehog signalling in cancerNature2008455721140641018754008
- ScalesSde SauvageFMechanisms of Hedgehog pathway activation in cancer and implications for therapyTrends Pharmacol Sci200930630331219443052
- CorbitKCAanstadPSinglaVNormanARStainierDYReiterJFVertebrate Smoothened functions at the primary ciliumNature200543770611018102116136078
- HaycraftCJBanizsBAydin-SonYZhangQMichaudEJYoderBKGli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and functionPLoS Genet200514e5316254602
- RohatgiRMilenkovicLScottMPPatched1 regulates hedgehog signaling at the primary ciliumScience2007317583637237617641202
- KimJKatoMBeachyPAGli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleusProc Natl Acad Sci U S A200910651216662167119996169
- WilsonCWChenMHChuangPTSmoothened adopts multiple active and inactive conformations capable of trafficking to the primary ciliumPLoS One200944e518219365551
- RubinLLde SauvageFJTargeting the Hedgehog pathway in cancerNat Rev Drug Discov20065121026103317139287
- KatohYKatohMHedgehog target genes: mechanisms of carcino-genesis induced by aberrant hedgehog signaling activationCurr Mol Med20099787388619860666
- CuiDXuQWangKCheXGli1 is a potential target for alleviating multidrug resistance of gliomasJ Neurol Sci20102881–215616619818966
- OliveKPJacobetzMADavidsonCJInhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancerScience200932459331457146119460966
- KobuneMTakimotoRMuraseKDrug resistance is dramatically restored by hedgehog inhibitors in CD34+ leukemic cellsCancer Sci2009100594895519245435
- QueirozKCRuela-de-SousaRRFuhlerGMHedgehog signaling maintains chemoresistance in myeloid leukemic cellsOncogene201029486314632220802532
- SinghRRKunkallaKQuCABCG2 is a direct transcriptional target of hedgehog signaling and involved in stroma-induced drug tolerance in diffuse large B-cell lymphomaOncogene201130494874488621625222
- MatsuiWWangQBarberJClonogenic multiple myeloma progenitors, stem cell properties, and drug resistanceCancer Res200868119019718172311
- SchreckKCTaylorPMarchionniLThe Notch target Hes1 directly modulates Gli1 expression and Hedgehog signaling: a potential mechanism of therapeutic resistanceClin Cancer Res201016246060607021169257
- BuonamiciSWilliamsJMorrisseyMInterfering with resistance to Smoothened antagonists by inhibition of the PI3K pathway in medulloblastomaSci Transl Med201025151ra70
- KasperczykHBaumannBDebatinKMFuldaSCharacterization of sonic hedgehog as a novel NF-kappaB target gene that promotes NF-kappaB-mediated apoptosis resistance and tumor growth in vivoFASEB J2009231213318772349
- SteccaBMasCClementVMelanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RASMEK/AKT pathwaysProc Natl Acad Sci U S A2007104145895590017392427
- JohnsonRRothmanAXieJHuman homolog of patched, a candidate gene for the basal cell nevus syndromeScience19962725268166816718658145
- GorlinRJNevoid basal-cell carcinoma syndromeMedicine (Baltimore)1987662981133547011
- XieJMuroneMLuohSMActivating Smoothened mutations in sporadic basal-cell carcinomaNature1998391666290929422511
- ThompsonMCFullerCHoggTLGenomics identifies medulloblastoma subgroups that are enriched for specific genetic alterationsJ Clin Oncol200624121924193116567768
- WatkinsDBermanDBurkholderSWangBBeachyPBaylinSHedgehog signalling within airway epithelial progenitors and in smallcell lung cancerNature2003422692931331712629553
- YuanZGoetzJASinghSFrequent requirement of hedgehog signaling in non-small cell lung carcinomaOncogene20072671046105516909105
- KarhadkarSBovaGAbdallahNHedgehog signalling in prostate regeneration, neoplasia and metastasisNature2004431700970771215361885
- ClementVSanchezPde TriboletNRadovanovicIRuiz i AltabaAHEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicityCurr Biol200717216517217196391
- BarEChaudhryALinACyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastomaStem Cells200725102524253317628016
- VarnatFDuquetAMalerbaMHuman colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansionEMBO Mol Med200916–733835120049737
- MukherjeeSFrolovaNSadlonovaAHedgehog signaling and response to cyclopamine differ in epithelial and stromal cells in benign breast and breast cancerCancer Biol Ther20065667468316855373
- ZhangJLipinskiRShawAGippJBushmanWLack of demonstrable autocrine hedgehog signaling in human prostate cancer cell linesJ Urol200717731179118517296441
- TianHCallahanCDuPreeKHedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesisProc Natl Acad Sci U S A2009106114254425919246386
- BaileyJMMohrAMHollingsworthMASonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancerOncogene200928403513352519633682
- ReyaTMorrisonSClarkeMWeissmanIStem cells, cancer, and cancer stem cellsNature2001414685910511111689955
- O’BrienCAKresoAJamiesonCHCancer stem cells and self-renewalClin Cancer Res201016123113312020530701
- ChangHHChenBYWuCYHedgehog overexpression leads to the formation of prostate cancer stem cells with metastatic property irrespective of androgen receptor expression in the mouse modelJ Biomed Sci201118621241512
- Enguita-GermánMSchiapparelliPReyJACastresanaJSCD133+ cells from medulloblastoma and PNET cell lines are more resistant to cyclopamine inhibition of the sonic hedgehog signaling pathway than CD133-cellsTumour Biol201031538139020480407
- FeldmannGDharaSFendrichVBlockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancersCancer Res20076752187219617332349
- GulinoAFerrettiEDe SmaeleEHedgehog signalling in colon cancer and stem cellsEMBO Mol Med200916–730030220049733
- Kawaguchi-IharaNOkuhashiYItohMMurohashiINaraNTohdaSPromotion of the self-renewal capacity of human leukemia cells by sonic hedgehog proteinAnticancer Res201131378178421498696
- LiuSDontuGMantleIDHedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cellsCancer Res200666126063607116778178
- MaoLXiaYPZhouYNA critical role of Sonic Hedgehog signaling in maintaining the tumorigenicity of neuroblastoma cellsCancer Sci2009100101848185519622100
- ZhaoCChenAJamiesonCHHedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemiaNature2009458723977677919169242
- ZhangCLiCHeFCaiYYangHIdentification of CD44+CD24+ gastric cancer stem cellsJ Cancer Res Clin Oncol2011137111679168621882047
- SchiapparelliPShahiMHEnguita-GermánMInhibition of the sonic hedgehog pathway by cyplopamine reduces the CD133+/CD15+ cell compartment and the in vitro tumorigenic capability of neuroblastoma cellsCancer Lett2011310222223121803487
- SinghBNFuJSrivastavaRKShankarSHedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: molecular mechanismsPLoS One2011611e2730622087285
- SongZYueWWeiBSonic hedgehog pathway is essential for maintenance of cancer stem-like cells in human gastric cancerPLoS One201163e1768721394208
- TakebeNWarrenRQIvySPBreast cancer growth and metastasis: interplay between cancer stem cells, embryonic signaling pathways and epithelial-to-mesenchymal transitionBreast Cancer Res201113321121672282
- TianFMysliwietzJEllwartJGamarraFHuberRMBergnerAEffects of the Hedgehog pathway inhibitor GDC-0449 on lung cancer cell lines are mediated by side populationsClin Exp Med2011426 [Epub ahead of print]
- TangSNFuJNallDRodovaMShankarSSrivastavaRKInhibition of sonic hedgehog pathway and pluripotency maintaining factors regulate human pancreatic cancer stem cell characteristicsInt J Cancer2011727 [Epub ahead of print]
- UchidaHAritaKYunoueSRole of sonic hedgehog signaling in migration of cell lines established from CD133-positive malignant glioma cellsJ Neurooncol2011104369770421380601
- WangXVenugopalCManoranjanBSonic hedgehog regulates Bmi1 in human medulloblastoma brain tumor-initiating cellsOncogene201231218719921685941
- WatkinsDNBermanDMBurkholderSGWangBBeachyPABaylinSBHedgehog signalling within airway epithelial progenitors and in small-cell lung cancerNature2003422692931331712629553
- ClarkeMFDickJEDirksPBCancer stem cells – perspectives on current status and future directions: AACR Workshop on cancer stem cellsCancer Res200666199339934416990346
- TanakaHNakamuraMKamedaCThe Hedgehog signaling pathway plays an essential role in maintaining the CD44+CD24–/low subpopulation and the side population of breast cancer cellsAnticancer Res20092962147215719528475
- RayAMengEReedEShevdeLARocconiRPHedgehog signaling pathway regulates the growth of ovarian cancer spheroid forming cellsInt J Oncol201139479780421701772
- BinnsWJamesLFShupeJLEverettGA congenital cyclopian-type malformation in lambs induced by maternal ingestion of a range plant, veratrum californicumAm J Vet Res1963241164117514081451
- CooperMKPorterJAYoungKEBeachyPATeratogen-mediated inhibition of target tissue response to Shh signalingScience19982805369160316079616123
- ChenJKTaipaleJCooperMKBeachyPAInhibition of Hedgehog signaling by direct binding of cyclopamine to SmoothenedGenes Dev200216212743274812414725
- TabsSAvciOInduction of the differentiation and apoptosis of tumor cells in vivo with efficiency and selectivityEur J Dermatol20041429610215196999
- KimJTangJYGongRItraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growthCancer Cell201017438839920385363
- ChenJKTaipaleJYoungKEMaitiTBeachyPASmall molecule modulation of Smoothened activityProc Natl Acad Sci U S A20029922140711407612391318
- RomingerCMBeeWLCopelandRAEvidence for allosteric interactions of antagonist binding to the smoothened receptorJ Pharmacol Exp Ther20093293995100519304771
- Von HoffDDLoRussoPMRudinCMInhibition of the hedgehog pathway in advanced basal-cell carcinomaN Engl J Med2009361121164117219726763
- RudinCMHannCLLaterraJTreatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449N Engl J Med2009361121173117819726761
- LorussoPMJimenoADyGPharmacokinetic dose-scheduling study of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with locally advanced or metastatic solid tumorsClin Cancer Res201117175774578221753154
- KimuraHNgJMCurranTTransient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structureCancer Cell200813324926018328428
- DirixLMigdenMOroAA pivotal multicenter trial evaluating efficacy and safety of the Hedgehog pathway inhibitor (HPI) vismodegib in patients with advanced basal cell carcinoma (BCC) [abstract]2011 European Multidisciplinary Cancer CongressSeptember 23–24, 2011Stockholm, Sweden20111BA
- TangJMackay-WigganJAszterbaumMAn investigator-initiated, phase II randomized, double-blind, placebo-controlled trial of GDC-0449 for prevention of BCCs in basal cell nevus syndrome (BCNS) patients (abstract)American Association for Cancer Research 102 nd Annual MeetingApril 2–6, 2011Orlando, FL2011LB-1
- PalmerSErlichmanCFernandez-ZapicoMPhase I trial erlotinib, gemcitabine, and the hedgehog inhibitor, GDC-0449 [abstract]J Clin Oncol2011293092
- CohenDLiebesLXuRA randomized, double-blind placebo-controlled phase II study of FOLFOX with or without GDC-0449 (vismodegib) in patients with advanced gastric or gastroesophageal junction carcinoma (NCI 8376) [abstract]J Clin Oncol201129TPS173
- RudinCJimenoAMillerWA phase I study of IPI-926, a novel hedgehog pathway inhibitor, in patients with advanced or metastatic solid tumors [abstract]J Clin Oncol2011293014
- StephensonJRichardsDWolpinBThe safety of IPI-926, a novel hedgehog pathway inhibitor, in combination with gemcitabine in patients with metastatic pancreatic cancer [abstract]J Clin Oncol2011294114
- Rodon AhnertJBaselgaJTawbiHA phase I dose-escalation study of LDE225, a smoothened (Smo) antagonist, in patients with advanced solid tumors [abstract]J Clin Oncol20102815s2500
- SkvaraHKalthoffFMeingassnerJGTopical treatment of Basal cell carcinomas in nevoid Basal cell carcinoma syndrome with a smoothened inhibitorJ Invest Dermatol201113181735174421430703
- SiuLPapadapoulosKAlbertsSA first-in-human phase 1study of an oral hedgehog pathway antagonist, BMS-833923 (XL139), in subjects with advanced or metastatic solid tumorsAACR-NCI-EORTC International Conference on Molecular Targets and Cancer TherapeuticsNovember 15–19, 2009Boston, MA2009
- HuffCAPadmanabhanSKellyKRA phase I study of an oral hedgehog pathway antagonist, BMS-833923, in patients with relapsed or refractory multiple myelomaBlood20111183993
- JamiesonCCortesJEOehlerVPhase 1 dose-escalation study of PF-04449913, an oral hedgehog (Hh) inhibitor, in patients with select hematologic malignanciesBlood2011118424
- YauchRLDijkgraafGJAlickeBSmoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastomaScience2009326595257257419726788
- DijkgraafGJAlickeBWeinmannLSmall molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistanceCancer Res201171243544421123452
- PogorilerJMillenKUtsetMDuWLoss of cyclin D1 impairs cerebellar development and suppresses medulloblastoma formationDevelopment2006133193929393716943274
- LauthMBergströmAShimokawaTToftgårdRInhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonistsProc Natl Acad Sci U S A2007104208455846017494766
- KimJLeeJJGardnerDBeachyPAArsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effectorProc Natl Acad Sci U S A201010730134321343720624968
- BeauchampEMRingerLBulutGArsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathwayJ Clin Invest2011121114816021183792