
Abstract
Introduction
Glioblastoma (GBM) is the most commonly diagnosed primary brain tumor in adults. The 14.6 months median survival period of GBM patients is still palliative due to resistance to the first-line chemotherapeutic agent temozolomide (TMZ).
Methods
The cell growth inhibition effect was assessed using the SRB assay. The mRNA expression levels were examined using RT-qPCR. The protein expression levels were determined using Western blot analysis. The DNA repair by non-homologous end-joining (NHEJ) was quantified using NHEJ reporter assay. The TMZ-induced apoptosis was detected by the Caspase 3/7 activity kit. The DNA binding activity in cells was determined using chromatin fractionation assay. The 53BP1 inhibitor was identified using virtual screening followed by Western blot analysis. The synergy between TMZ and 53BP1 inhibitor in vivo was analyzed using a xenograft mouse model.
Results
We found that non-homologous end joining (NHEJ), which is one of the major DNA double-strand break repair pathways, participates in acquired TMZ-resistance in GBM. Canonical NHEJ key factors, XLF and 53BP1, are upregulated in TMZ-resistant GBM cells. Depletion of XLF or 53BP1 in TMZ-resistant cells significantly improve the potency of TMZ against GBM cell growth. Importantly, we identified a small molecule HSU2018 to inhibit 53BP1 at nanomolar concentration. The combination of HSU2018 and TMZ generates excellent synergy for cell growth inhibition in TMZ-resistant GBM cells and xenograft.
Conclusion
Our data suggest that NHEJ is a novel mechanism contributing to TMZ-resistance, and its key factors may serve as potential targets for improving chemotherapy in TMZ-resistant GBM.
Introduction
Glioblastoma (GBM) is a deadly, malignant brain tumor arising from glial cells.Citation1 Patients of GBM show high cellular heterogeneity and complex chromosome aberrations.Citation2,Citation3 GBM is a severe brain tumor with a median survival time of only 12–15 months after the initial diagnosis.Citation4 The conventional therapies for newly diagnosed GBM patients are surgical resection followed by chemotherapy and radiation therapy. Temozolomide (TMZ), which is an alkylating agent, has been applied as the first-line chemotherapeutic regimen since 2005.Citation5 Although TMZ has been contributed to improve life quality and survival time of GBM patients, intrinsic and acquired resistance to TMZ are still the major obstacles for GBM treatment.Citation6,Citation7 TMZ elicits cytotoxicity during replication by methylation at O6 and N7 positions of guanine, and at N3 position of adenine that results in DNA breaks, which eventually leads to cell apoptosis.Citation8 Elevated expression of O6-methylguanine-DNA methyltransferase (MGMT), which directly removes methyl group from O6-methylguanine, has been reported as the major reason for TMZ resistance. However, recent case studies of TMZ resistance reported that a series of TMZ-resistant GBM patients exhibited deficiency of MGMT activity.Citation9 Therefore, it is urgent to understand the TMZ-resistance mechanism independent of MGMT and develop alternative chemotherapy strategies against GBM.
DNA double-strand break (DSB), which is one of the most dangerous and toxic DNA lesions, are generated frequently in human cells.Citation10 Misrepair or unrepair of DSBs results in mutation, chromosomal aberration, carcinogenesis, and cell death.Citation11 To maintain genome stability when DSB occurred, cells developed two major DSB repair pathways: non-homologous recombination (NHEJ) and homologous recombination (HR).Citation12,Citation13 HR is considered as the accurate DSB repair pathway since sister chromatid is incorporated as the template during gap filling. However, this template-dependent feature of HR limits this repair mechanism in the S and G2 phases of the cell cycle, where sister chromatids are available.Citation14,Citation15 NHEJ, on the other hand, is approachable throughout the whole cell cycle and much more tolerant of different forms of broken DNA ends.Citation16–Citation20
Here, we characterize the role of NHEJ key factor XLF and 53BP1 in TMZ-resistant GBM. Both mRNA level and protein level of these two factors are upregulated in TMZ-resistant LN18 and U87 cell lines. Importantly, XLF or 53BP1 deficiency re-sensitizes GBM cells to TMZ by 2–4 folds. We also demonstrated that TMZ treatment induces XLF and 53BP1 expression in TMZ-sensitive GBM cells. Importantly, we identified a potent 53BP1 inhibitor HSU2018 that degrades 53BP1 at 0.5 µM. HSU2018 exhibits excellent synergy with TMZ against GBM in vitro and in vivo. Our results suggest that XLF and 53BP are promising targets to overcome TMZ-resistance in GBM.
Methods And Materials
Cell Lines And Reagents
LN18 (ATCC, CRL-2610) and U87 cells (ATCC, HTB-14) were cultured at 37°C in 5% CO2 atmosphere in DMEM and EMEM with 10% fetal bovine serum (FBS) for less than 6 months, respectively. Resistant cells were generated according to previous studies.Citation22 Briefly, LN18-TR and U87-TR cells were obtained by treating their parental cells with 200 µM TMZ for 6 hrs and released in drug-free media for 2 weeks. TMZ concentration was increased every 2 weeks up to 1 mM. TMZ (Sigma, T2577) was dissolved in DMSO and diluted in certain media.
Cell Viability Assay
GBM cells were seed at 4×103 cells/well and cultured for overnight. Cells were incubated with TMZ for 72 hrs and the cell viability was detected by using Sulforhodamine B (SRB) assay.Citation23 Cells were fixed by 100 µL/well of 10% trichloroacetic acid at 4°C for 1 hr. Plate was washed 4 times with slow running tap water and air-dried for 1 hr at room temperature (RT) or 20 mins in fume hood. Cells were stained by 100 µL/well 0.02% SRB in 1% acetate acid for 1 hr at RT. Plates were washed for 3 times with 200 µL/well 1% acetate acid and air dried. 200 µL/well of 10 mM Tris-HCl, pH 10.5 was added in each well to extract SRB with 1 hr shaking on an orbital shaker. The absorbance was measured at 510 nm by microplate reader (BioTek).
Quantitative Reverse Transcription-Polymerase Chain Reaction
Total RNA was isolated using the PureLink RNA Mini Kit (Invitrogen #12183018A). Complementary DNA was synthesized using the iScript™ cDNA Synthesis Kit (Bio-Rad #1708891) according to the manufacturer’s instructions. Fluorescence real-time RT-PCR was performed with SsoAdvanced™ Universal SYBR® Green Supermix following the instruction manual (Bio-Rad #172-5274) and detected by CFX96 Dx System (Bio-Rad #1708891). The experiment was performed in triplicate and repeated at least three times.
Primers: Ku80 (Left: cgacaggtgtttgctgagaa, Right: tcacatccatgctcacgatt), DNA-PKcs (Left: tctgaaggggttgtcctcac, Right: tgggcacaccactttaacaa), Ligase IV (Left: agcctgacctggagaacaga, Right: catgcaggcttgacaacatc), XLF (Left: tctctggcctccccttctat, Right: gatctcgaatcagcgtagcc), XRCC4 (Left: cattgttgtcaggagcagga, Right: tctgcaggtgctcatttttg), MRE11 (Left: gccttcccgaaatgtcacta, Right: ttcaaaatcaacccctttcg), 53BP1 (Left: tgacagcacagcccagtaag, Right: cctcaggctctggtgacttc), RAD51 (Left: ctcagcctcccgagtagttg, Right: catcactgccagagagacca), RPA32 (Left: ccagtgggttgacacagatg, Right: tgataggtgctctccctgct), BRAC1 (Left: ctcaaggaaccagggatgaa, Right: gctgtaatgagctggcatga), EXO1 (Left: gctgtcagaagatgacctgttg, Right: gcaccactttcttcatttttcc), Polδ (POLD3) (Left: agaaagccatgctaaaggacag, Right: catgtgaagatgtgactgctca), GAPDH (intrinsic control) (Left: cgagatccctccaaaatcaa, Right: ggtgctaagcagttggtggt)
Western Blot Analysis
Method has been described in previous works.Citation24 Briefly, protein samples were resuspended in SDS-PAGE sample buffer, and boiled for 5 mins at 95°C. The samples were loaded and separated on an 12% polyacrylamide gel (29:1) at 120 V for 1.5 hr on the electrophoresis apparatus (BioRad). Separated samples were transferred to nitrocellulose membrane at 100 V and 4°C in cold room for 45 mins. Membrane was blocked by 3% BSA diluted in PBS with 0.1% Tween 20 and probed by relevant antibody followed by HRP-conjugated rabbit secondary antibody. The protein signal was developed by SuperSignal™ west Femto Maximum Sensitivity Substrate (ThermoFisher Scientific #34096) and detected by ChemiDoc™ (BioRad). Antibodies: anti-XLF: abcam #33499; anti-53BP1: abcam #21083; anti-actin: abcam #8227.
NHEJ Reporter Assay
NHEJ reporter assay was previously described.Citation25 Briefly, 40 μg of NHEJ reporter plasmid were linearized by NheI in 50 μL for 6 hrs at 37°C. Linearized DNA was then extracted by using Qiagen gel extraction kit (Qiagen #28704). 2 μg of linearized DNA was transfected into LN18 or U87 cells by using Lipofectamine 3000 as previously described.Citation26 Cells with chromosomally integrated reporter constructs were selected by incubating in media with 1 mg/mL geneticin 24h after transfection for 2 weeks. Plasmid-integrated cells were seeded at 3×105 cells/mL in a 6-well plate and cultured for 24 hrs to allow adhere. 2µg/well of I-SceI was transfected into the cell by lipofectamine 3000 to recognize I-SceI site located in reporter plasmid and generate DSB. Cells were then incubated for 48 hrs to allow DSB repair by NHEJ. Repaired cells, which express GPF, were harvested by using trypsin and resuspend in PBS followed by quantification using flow cytometry (Beckman Coulter) to indicate NHEJ efficiency.
Chromatin Fractionation Assay
1 × 107 cells were harvested and washed twice in ice-cold PBS, and extracted twice in CSK buffer (100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 0.7% Triton X-100, 10 mM PIPES, pH 7.0) supplemented with 1× protease inhibitor cocktail (Roche, 4693159001) and 0.4 mg/mL RNase A (Thermo Fisher, 12091021) at 4°C for 10 mins. Cells were then washed 3 times in ice-cold PBS and harvested in 2× SDS loading buffer for analysis by SDS-PAGE and Western blotting assay.
Transfection
For siRNA transfection, 50 nM of siXLF or si53BP1 was transfected in GBM cells by using Lipofectamine RNAiMAX (ThermoFisher) according to the manufacturer’s instruction. SiGL2 was used as a negative control. Cell viability assay or reporter assay was performed 48 hrs after siRNA transfection. si53BP1-UTR: AAAUGUGUCUUGUGUGUAA (Sigma). siXLF-UTR (Dharmacon #A-014446-16-0005). siGL2 (intrinsic control): CGUACGCGGAAUACUUCGAUU (Sigma).
For ectopic expression, XLF and 53BP1 (Addgene #52507) plasmids were transfected in GBM cells by using Lipofectamine 3000 (ThermoFisher) 24 hrs after siRNA transfection. cDNA for XLF were cloned into pcDNA3 carrying a FLAG epitope (Invitrogen) using standard DNA cloning methods.
Caspase‑3/7 Activity Assay
Caspase‑3/7 activity was examined using SensoLyte® Homogeneous AMC Caspase‑3/7 Assay kit (AnaSpec Inc., no. 7118) in LN18-TR and U87-TR cells according to the manufacturer’s instructions.
Drug Screening
Virtual screening was performed by docking small molecule structure (ZINK database) to the available crystal structure of XLF (PDB: 2R9A) and 53BP1 (PDB: 1GZH and 2IG0) with Glide software 2.5 (Schrödinger, Inc, USA). The hydrogen bond acceptor, donor, hydrophobic and ring aromatic were adopted for calculations of all chemical feature. Estimated binding energy was used to rank inhibitor candidates. The small molecule with lowest binding energy was then further modified to achieve higher docking score. Top candidates were analyzed by using Western blotting assay for protein inhibition activity.
LN18-TR Xenograft
Thirty-two female BALB/c nude mice (5–6 weeks old, body weight 18–22 g) were randomly assigned to four groups, 8 mice per group. Note that 5 × 106 LN18-TR cells were injected subcutaneously into mice. Mice were treated intraperitoneally with control (DMSO), TMZ (20 mg/kg/3 day), HSU2018 (2mg/kg/3 days) and Combo (20 mg/kg/3 day of TMZ + 2 mg/kg/3 day of HSU2018) for 2 weeks. The tumor volume was measured starting from day 10 after injection at a frequency of three times a week. Tumor volume, expressed in mm3, was calculated using the following formula, V= (length × width2)/2. Experiments were approved by Shandong University.
Statistical Analysis
Data are presented as the mean±SD of three independent experiments. Statistical evaluation was performed using the Student’s t-test or one-way ANOVA. Differences were considered to be statistically significant when P<0.05. All statistical analyses were performed using Prism7 software (GraphPad Software, La Jolla, CA, USA).
Results
XLF And 53BP1 Are Upregulated In TMZ-Resistant Cells
To investigate the mechanism of TMZ-resistance in GBM, we first generated two TMZ-resistant GBM cell lines by incubating the LN18-wild type (LN18-WT) and U87-wild type (U87-WT) cells with TMZ for 4 months. As shown in , LN18-TMZ-resistant (LN18-TR) and U87-TMZ-resistant (U87-TR) cell line showed 4.47-fold and 4.76-fold increase of IC50, respectively, as compared to their parental cell lines.
Figure 1 XLF and 53BP1 are upregulated in TMZ-resistant cells. (A) Cell viability of TMZ-sensitive and TMZ-resistant LN18 cell line. TMZ concentrations are 0 mM, 0.25 mM, 0.5 mM, 1 mM and 2 mM. LN18-TR: LN18-TMZ-resistant cell line. (B) Cell viability of TMZ-sensitive and TMZ-resistant U87 cell line. TMZ concentrations are 0 mM, 0.5 mM, 1 mM, 2 mM and 4 mM. U87-TR: U87-TMZ-resistant cell line. (C) mRNA expression of Ku80, DNAPKcs, ligase IV, XLF, XRCC4, MRE11 and 53BP1 in LN18 cell lines. (D) mRNA expression of Ku80, DNAPKcs, ligase IV, XLF, XRCC4, MRE11 and 53BP1 in U87 cell lines. (E) mRNA expression of RAD511, RPA32, BRCA1, EXO1, and Pol δ in LN18 cell lines. (F) mRNA expression of RAD511, RPA32, BRCA1, EXO1, and Pol δ in U87 cell lines. **p<0.01, ***p<0.001.
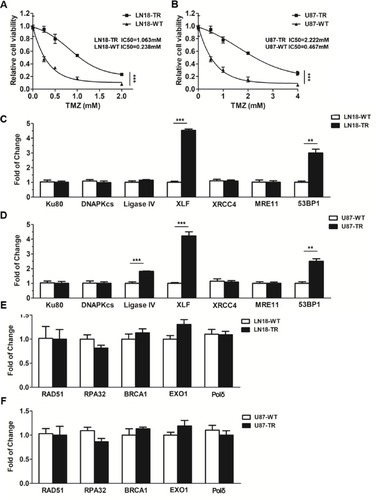
To determine whether DSB pathways participate in TMZ-resistance, we used qPCT to examine the mRNA expression of canonical factors that are required for NHEJ and HR. Among NHEJ factors, including Ku80, DNA-PKcs, Ligase IV, XRCC4, XLF, MRE11 and 53BP1, XLF and 53BP1 were upregulated in LN18-TR cells by 4.5 and 3.1 folds, respectively (). To further confirm the upregulation of XLF and 53BP1 are consistent in GBM cell lines, we evaluated the mRNA levels of these factors in U87 cell lines. The mRNA expression levels of XLF and 53BP1 in U87-TR cells were about 4.2- and 2.5-fold higher than in U87-WT cells, respectively (). XLF is essential for NHEJ efficiency, therefore, our data suggest NHEJ pathway contributes to TMZ-resistance. Interestingly, 53BP1 participates in pathways choice between NHEJ and HR. Since the expression of 53BP1 was upregulated in TMZ-resistant cells, we next examined the mRNA expression levels of HR factors, including RAD51, RPA32, BRAC1, EXO1, and Polδ, in LN18 and U87 cell lines. As shown in , mRNA expression of HR key factors did not show a significant difference between TR and WT cells. These data indicate that NHEJ pathway participates in the TMZ-resistance mechanism.
TMZ Induces XLF And 53BP1 Expression In TMZ-Sensitive Cells
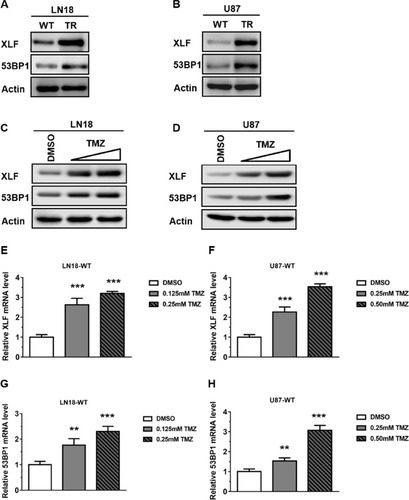
To further examine protein expression levels of XLF and 53BP1, we extracted whole cell lysate and compared the protein expression levels between TMZ sensitive and resistant cell lines. Using Western Blotting assay, we found that protein expression of both XLF and 53BP1 indeed increased in TMZ-resistant cells (; Supplementary Figure 1A and B). These data are consistent with the mRNA results as shown in . Since TMZ-resistant GBM cells were generated by incubating WT cells with TMZ, we hypothesize that the upregulation of XLF and 53BP1 in the TR cells result from response to TMZ treatment. To test our hypothesis, we incubated the TMZ-sensitive cells with different concentrations of TMZ for 48 hrs and evaluated protein expression levels of XLF and 53BP1. As shown in , XLF and 53BP1 expressions were increased in both LN18-WT and U87-WT cells treated with TMZ in a dose-dependent manner (Supplementary Figure 1C and D). Upregulated mRNA levels of XLF and 53BP1 in response to TMZ treatment were furthermore confirmed using qPCR (–). These results suggest that XLF and 53BP1 may contribute to TMZ resistance in GBM cells.
Figure 2 TMZ induces XLF and 53BP1 expression in TMZ-sensitive cells. (A) Immunoblotting of XLF and 53BP1 protein expression in LN18 cell lines. (B) Immunoblotting of XLF and 53BP1 protein expression in U87 cell lines. (C) Immunoblotting of XLF and 53BP1 protein expression in LN18-WT treated with TMZ. LN18-WT was treated with 0.125 mM and 0.25 mM of TMZ. (D) Immunoblotting of XLF and 53BP1 protein expression in U87-WT treated with TMZ. U87-WT was treated with 0.25 mM and 0.5 mM of TMZ. (E) Gene expression levels of XLF were determined by qRT-PCR in TMZ treated LN18-WT cells. (F) Gene expression levels of XLF were determined by qRT-PCR in TMZ treated U87-WT cells. (G) Gene expression levels of 53BP1 were determined by qRT-PCR in TMZ treated LN18-WT cells. (H) Gene expression levels of 53BP1 were determined by qRT-PCR in TMZ treated U87-WT cells. Results represent the mean ± SD of three independent experiments. **P<0.01, ***P<0.001.
XLF Or 53BP1 Depletion Improves TMZ Sensitivity In GBM Cells
Given that XLF and 53BP1 are upregulated at both translational and transcriptional levels in TMZ-resistant cells, it is encouraging to investigate whether XLF or 53BP1 deficiency re-sensitize TR cells to TMZ. We used siRNA to knockdown XLF or 53BP1 in LN18-TR cells and evaluated cell viability against TMZ. Significantly, XLF and 53BP1 depletion-sensitized LN18-TR cells to TMZ by 4.2 and 2.3 folds, respectively (). In support to this result, compromised XLF and 53BP1 expression also contributed to sensitize U87-TR cells to TMZ by 2.8 and 2 folds, respectively (). To exclude the possibility that the increased sensitivity to TMZ could be masked by siRNA off target effect, we complemented XLF and 53BP1 into XLF and 53BP1 knockdown cells, respectively. Since the siRNAs of XLF and 53BP1 target 5ʹUTR region of the genes, ectopic expression of these proteins was not affected (Supplementary Figure 2A and B). We found that TMZ resistance can be successfully rescued by ectopic expression of 53BP1 and XLF ().
Figure 3 XLF or 53BP1 depletion improves TMZ sensitivity in GBM cells. (A) Cell viability in LN18-TR+siGL2, LN18-TR+si53BP1, LN18-TR+siXLF, LN18-TR+si53BP1 with 53BP1 ectopic expressed and LN18-TR+siXLF with XLF ectopic expressed cell lines. TMZ concentrations are 0 mM, 0.25 mM, 0.5 mM, 1 mM and 2 mM. IC50s of TMZ was indicated. (B) Cell viability in U87-TR+siGL2, U87-TR+si53BP1, U87-TR+siXLF, U87-TR+si53BP1 with 53BP1 ectopic expressed and U87-TR+siXLF with XLF ectopic expressed cell lines. TMZ concentrations are 0 mM, 0.5 mM, 1 mM, 2 mM and 4 mM. IC50s of TMZ was indicated. (C) NHEJ efficiency in LN18-TR+siGL2, LN18-TR+si53BP1, LN18-TR+siXLF, LN18-TR+si53BP1 with 53BP1 ectopic expressed and LN18-TR+siXLF with XLF ectopic expressed cell lines. NHEJ efficiency of LN18-TR+siGL2 is normalized to 100%. (D) NHEJ efficiency in U87-TR+siGL2, U87-TR+si53BP1, U87-TR+siXLF, U87-TR+si53BP1 with 53BP1 ectopic expressed and U87-TR+siXLF with XLF ectopic expressed cell lines. NHEJ efficiency of U87-TR+siGL2 is normalized to 100%. (E) Caspase‑3/7 activity in LN18-TR and (F) U87-TR cells was assessed using the SensoLyte Homogeneous AMC Caspase‑3/7 assay kit. Results represent the mean ± SD of three independent experiments. ***P<0.001.
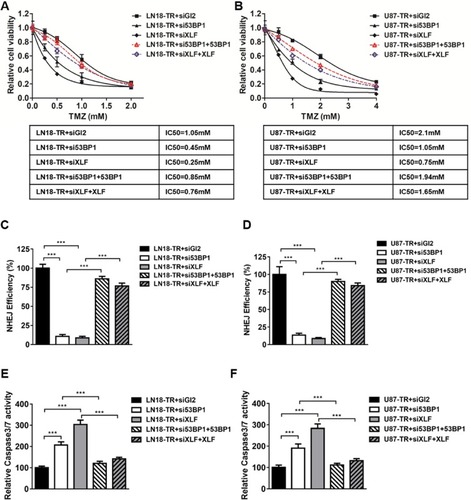
To determine whether the improved sensitivity to TMZ, which was generated by XLF or 53BP1 in TR cells, is mediated by NHEJ efficiency, we incorporated NHEJ reporter assay to measure NHEJ efficiency in XLF or 53BP1 deficient cells. We first knocked down XLF or 53BP1 in LN18-TR and U87-TR cells, which was stably transfected with NHEJ reporter. DSBs were then introduced by using I-SceI restriction enzyme and cells were incubated for another 48 hrs to allow DSB repair by NHEJ. Repaired cells express GPF that can be quantified by flow cytometry to indicate NHEJ efficiency. As shown in , both 53BP1 and XLF deficiency led to significant decrease of NHEJ efficiency in TMZ-resistant cells. Interestingly, NHEJ efficiency is elevated in TMZ-resistant cells as compared to their parental cells, indicating that inhibition of XLF or 53BP1 sensitizes GBM cells to TMZ via compromising NHEJ efficiency (Supplementary Figure 3A and B). To the end of investigating the mechanism by which XLF and 53BP1 deficiency increase cell growth inhibition, we found that caspase3/7 activity was profoundly promoted in cells lack of XLF or 53BP1 (). These results indicate that inhibition of NHEJ by depleting XLF or 53BP1 sensitzes GBM cells to TMZ via promoting apoptosis.
TMZ Induces DNA Binding Activity Of XLF And 53BP1 In GBM Cells
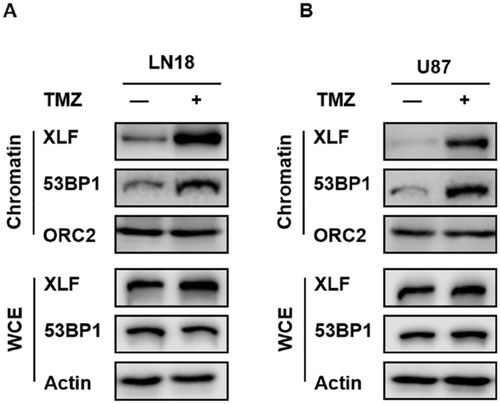
Although XLF does not have enzymatic activity, it facilitates NHEJ through tethering DNA breaks.Citation21 In addition, 53BP1 is also a well-established DNA binding protein. Therefore, we investigated whether XLF- or 53BP1-DNA interaction is affected by TMZ treatment. We used the chromatin fractionation assay to examine their DNA binding activities after acute treatment in LN18-WT cells with 5 mM TMZ for 2 hrs. As shown in and Supplementary Figure 4A, chromatin-bound XLF and 53BP1 were significantly increased after TMZ treatment while the total levels of these proteins were not affected within 2 hrs. We also observed the similar results using U87-WT cells (; Supplementary Figure 4B). Our data suggest TMZ induces DNA binding activity of both XLF and 53BP1, and increased NHEJ efficiency in TMZ resistance cells may be due to more efficient interaction between DNA and NHEJ factors.
Figure 4 TMZ induces DNA binding activity of XLF and 53BP1 in GBM cells. (A) Chromatin fractionation of XLF and 53BP1 in LN18-WT treated with TMZ. LN18-WT was treated with 5 mM of TMZ for 2 hrs. WCE: whole-cell extract. (B) Chromatin fractionation of XLF and 53BP1 in U87-WT treated with TMZ. U87-WT was treated with 10 mM of TMZ for 2 hrs. WCE: whole-cell extract.
53BP1 Inhibitor Re-Sensitizes GBM Cells To TMZ
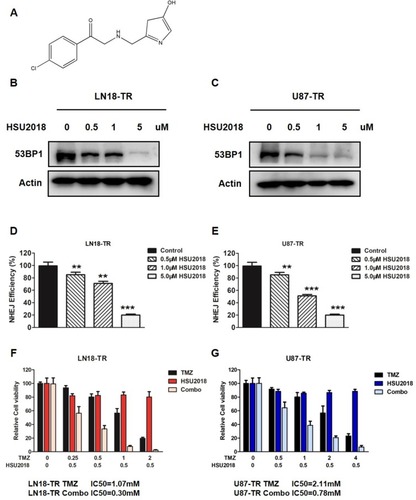
Given that both XLF and 53BP1 contribute to TMZ resistance in GBM, our group was interested in developing XLF and 53BP1 inhibitors. In a screening to identify small molecules that target XLF or 53BP1, we found that HSU2018 () interacts with 53BP1 and markedly degrades 53BP1 in both LN18-TR and U87-TR cells by using Western blotting assay (; Supplementary Figure 5A and B). Significantly, HSU2018 generated 53BP1 degradation at 0.5 µM in both TMZ-resistant cell lines, indicating that HSU2018 is a very potent 53BP1 inhibitor. Since 53BP1 is one of the key factors of NHEJ regulation, we then evaluated NHEJ efficiency in the presence of HSU2018. As we expected, as a 53BP1 inhibitor, HSU2018 impaired NHEJ efficiency in a dose-dependent fashion in TMZ-resistant GBM cells (). To determine whether HSU2018 can improve TMZ sensitivity in GBM cells, we treated TMZ-resistant cells with 0.5 µM HSU2018 for 48 hrs and combined with 0 to 2 mM of TMZ for another 72 hrs. By using cell viability assay, we found that HSU2018 decreased IC50 of TMZ by 3.6 and 2.7 folds in LN18-TR and U87-TR cells, respectively (). Importantly, 0.5 µM of HSU2018 did not result in severe cell death, indicating that HSU2018 is a promising TMZ sensitizer with low toxicity at an effective dose.
Figure 5 53BP1 inhibitor re-sensitizes GBM cells to TMZ. (A) The chemical structure of HSU2018. (B) LN18-TR and (C) U87-TR cells were treated with HSU2018 for 48 hrs. 53BP1 protein expression was analyzed by using Western blotting assay against the anti-53BP1 antibody. (D) NHEJ efficiency in LN18-TR and (E) U87-TR cells treated with HSU2018. HSU2018 was added to the cells 48 hrs before I-SceI transfection. **P<0.01, ***P<0.001. (F) HSU2018 sensitizes TMZ-resistant LN18 and (G) U87 cells to TMZ. 0.5 µM HSU was added to TR cells 48 hrs before TMZ treatment.
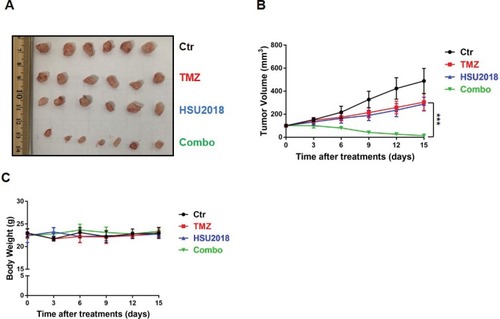
To further evaluate the potency of HSU2018 in combine with TMZ in vivo, we subcutaneously implanted LN18-TR cells into 5–6 weeks old female nude mice to form TMZ-resistant GBM tumors. The mice were then treated with control (DMSO), HSU2018, TMZ or combination of both via intraperitoneal injection. We found that, similar to the result of in vitro cell survival assay, the combinational chemotherapy with TMZ and HSU2018 exhibited an excellent synergy in reducing tumor growth as compared to TMZ or HSU2018 treatment alone (–). Together our results suggest that inhibition of 53BP1 is a promising therapeutic approach to overcome TMZ-resistance in GBM.
Figure 6 HSU2018 restores sensitivity to TMZ in LN18-TR xenograft model. (A) Representative images of xenograft tumor. LN18-TR xenograft mice treated with control (DMSO), TMZ (20 mg/kg/3day intraperitoneally), HSU2018 (2 mg/kg/3days), and Combo (20 mg/kg/3day of TMZ +2 mg/kg/3day of HSU2018). (B) Tumor volume was reported in mm3 as the mean ± SD and statistically compared between TMZ and Combo tumors. n ≥6 tumors/group. ***P < 0.001. (C) Body weight was measured twice a week as the mean ± SD. n ≥6 tumors/group.
Discussion
In this study, we identified two NHEJ factors that contribute to acquired TMZ-resistant GBM. Depletion of XLF or 53BP1 significantly improved therapeutic effect of TMZ, suggesting a role of NHEJ pathway in chemoresistance in GBM. Importantly, we identified a very potent 53BP1 inhibitor HSU2018 that inhibits 53BP1 protein expression. This small molecule generates great synergy with TMZ at 0.5 µM in reducing cell survival and tumor growth.
DNA has been targeted in various types of cancers to generate DNA lesions that eventually lead to cell death since 1940s.Citation27 However, efficient DNA repair could impede the therapeutic effect and contribute to chemoresistance. Therefore, determine whether certain DNA repair pathway participates in TMZ-resistance may provide promising ways to overcome the recurrence of GBM after chemotherapy. Although mismatch repair and upregulated MGMT are most studied TMZ-resistance mechanism, emerging evidences indicate that DSB repair pathways may also prevent cell death in GBM.Citation28,Citation29 NHEJ is the predominant DSB repair pathway in human that can be used in the whole cell cycle. Importantly, NHEJ repairs most of the DSBs generated by ionizing radiation. Therefore, inhibition of NHEJ may sensitize glioma cells to radiotherapy, which is commonly used for GBM treatment, to overcome radioresistance.
The DSB ends constructions are important for the pathway choice between HR and NHEJ. HR requires DNA end resection to initiate the repair, while NHEJ is inhibited when the gap of two broken ends reaches 3–4 nucleotides.Citation30 Therefore, 53BP1, which prevents DNA end resection, is considered to facilitate the transition from HR to NHEJ.Citation31 NHEJ is initiated with the interaction between Ku70/80 heterodimer and DSB in a sequence-independent manner.Citation32 Once Ku-DNA complex formed, Ku recruits other NHEJ required factors including DNA-dependent protein kinase catalytic subunit (DNA-PKcs), the X-ray repair cross-complementing 4 (XRCC4)-ligase IV complex and the XRCC4-like factor protein (XLF, also as known as Cernunnos).Citation33–Citation35 Although NHEJ is well recognized, the mechanism of how NHEJ factors assemble is not thoroughly studied. One of the theories of NHEJ assembly demonstrates that XLF and XRCC4 form long filaments wrapping around broken DNA ends to promote ends approximation.Citation21,Citation36 XLF is a relative new NHEJ key factor discovered by two laboratories in 2006.Citation37,Citation38 XLF functions as homodimer and facilitates ligation in NHEJ.Citation39 However, enzymatic activity of XLF has not been found. XLF may serve as scaffold protein by interacting with XRCC4 to tether DNA ends. It is interesting to observe that TMZ-induced chromatin binding of XLF and 53BP1. Given that NHEJ efficiency is elevated in TMZ-resistant cells, acquired TMZ-resistance may result from improved NHEJ achieved by more efficient DNA binding and tethering.
Since the alternative chemotherapies of GBM are very limited, identification of new targets to improve sensitivity to TMZ is critical to prolong the survival time of GBM patients. Here, we suggest that both XLF and 53BP1 have the potential to be targeted. HSU2018 may provide a treatment option for patients that are less sensitive to TMZ. In addition, standard GBM treatments include radiation that generates DNA damages. These DNA damages majorly rely on NHEJ for repair. Therefore, elevated NHEJ efficiency could also impair radiation therapy. As an inhibitor of NHEJ key factor, we hypothesize that HSU2018 can also sensitize GBM cells to radiation.
Abbreviations
GBM, glioblastoma; TMZ, temozolomide; NHEJ, non-homologous end joining; HR, homologous recombination; DSB, DNA double-strand break; DNA-PKcs, DNA-dependent protein kinase catalytic subunit; XRCC4, X-ray repair cross-complementing 4; XLF, XRCC4-like factor protein.
Availability Of Data And Materials
All data generated or analyzed during this study are included in this published article.
Disclosure
The authors declare that they have no competing interests.
References
- Inskip PD, Linet MS, Heineman EF. Etiology of brain tumors in adults. Epidemiol Rev. 1995;17(2):382–414. doi:10.1093/oxfordjournals.epirev.a0362008654518
- Beroukhim R, Getz G, Nghiemphu L, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A. 2007;104(50):20007–20012. doi:10.1073/pnas.071005210418077431
- Patel AP, Tirosh I, Trombetta JJ, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–1401. doi:10.1126/science.125425724925914
- Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. doi:10.1016/S1470-2045(09)70025-719269895
- Nanegrungsunk D, Onchan W, Chattipakorn N, Chattipakorn SC. Current evidence of temozolomide and bevacizumab in treatment of gliomas. Neurol Res. 2015;37(2):167–183. doi:10.1179/1743132814Y.000000042325033940
- Chamberlain MC. Temozolomide: therapeutic limitations in the treatment of adult high-grade gliomas. Expert Rev Neurother. 2010;10(10):1537–1544. doi:10.1586/ern.10.3220925470
- Patil SA, Hosni-Ahmed A, Jones TS, Patil R, Pfeffer LM, Miller DD. Novel approaches to glioma drug design and drug screening. Expert Opin Drug Discov. 2013;8(9):1135–1151. doi:10.1517/17460441.2013.80724823738794
- Kanzawa T, Bedwell J, Kondo Y, Kondo S, Germano IM. Inhibition of DNA repair for sensitizing resistant glioma cells to temozolomide. J Neurosurg. 2003;99(6):1047–1052. doi:10.3171/jns.2003.99.6.104714705733
- Atkins RJ, Ng W, Stylli SS, Hovens CM, Kaye AH. Repair mechanisms help glioblastoma resist treatment. J Clin Neurosci. 2015;22(1):14–20. doi:10.1016/j.jocn.2014.09.00325444993
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi:10.1146/annurev.biochem.052308.09313120192759
- Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol. 2017;18(8):495–506. doi:10.1038/nrm.2017.4828512351
- Burma S, Chen BP, Chen DJ. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair (Amst). 2006;5(9–10):1042–1048. doi:10.1016/j.dnarep.2006.05.02616822724
- Jasin M, Rothstein R. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol. 2013;5(11):a012740. doi:10.1101/cshperspect.a01274024097900
- Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol. 2003;23(16):5706–5715. doi:10.1128/MCB.23.16.5706-5715.200312897142
- Hinz JM, Yamada NA, Salazar EP, Tebbs RS, Thompson LH. Influence of double-strand-break repair pathways on radiosensitivity throughout the cell cycle in CHO cells. DNA Repair (Amst). 2005;4(7):782–792. doi:10.1016/j.dnarep.2005.03.00515951249
- Jasin M. Genetic manipulation of genomes with rare-cutting endonucleases. Trends Genet. 1996;12(6):224–228. doi:10.1016/0168-9525(96)10019-68928227
- Interthal H, Chen HJ, Kehl-Fie TE, Zotzmann J, Leppard JB, Champoux JJ. SCAN1 mutant Tdp1 accumulates the enzyme–DNA intermediate and causes camptothecin hypersensitivity. Embo J. 2005;24(12):2224–2233. doi:10.1038/sj.emboj.760069415920477
- Li J, Summerlin M, Nitiss KC, Nitiss JL, Hanakahi LA. TDP1 is required for efficient non-homologous end joining in human cells. DNA Repair (Amst). 2017;60:40–49. doi:10.1016/j.dnarep.2017.10.00329078113
- Heo J, Li J, Summerlin M, et al. TDP1 promotes assembly of non-homologous end joining protein complexes on DNA. DNA Repair (Amst). 2015;30:28–37. doi:10.1016/j.dnarep.2015.03.00325841101
- Yannone SM, Khan IS, Zhou RZ, Zhou T, Valerie K, Povirk LF. Coordinate 5ʹ and 3ʹ endonucleolytic trimming of terminally blocked blunt DNA double-strand break ends by Artemis nuclease and DNA-dependent protein kinase. Nucleic Acids Res. 2008;36(10):3354–3365. doi:10.1093/nar/gkn20518440975
- Mahaney BL, Hammel M, Meek K, Tainer JA, Lees-Miller SP. XRCC4 and XLF form long helical protein filaments suitable for DNA end protection and alignment to facilitate DNA double strand break repair. Biochem Cell Biol. 2013;91(1):31–41. doi:10.1139/bcb-2012-005823442139
- Meng Y, Chen CW, Yung MMH, et al. DUOXA1-mediated ROS production promotes cisplatin resistance by activating ATR-Chk1 pathway in ovarian cancer. Cancer Lett. 2018;428:104–116. doi:10.1016/j.canlet.2018.04.02929704517
- Chen CW, Li Y, Hu S, et al. DHS (trans-4,4ʹ-dihydroxystilbene) suppresses DNA replication and tumor growth by inhibiting RRM2 (ribonucleotide reductase regulatory subunit M2). Oncogene. 2018.
- Sun J, Guo Y, Fu X, et al. Dendrobium candidum inhibits MCF-7 cells proliferation by inducing cell cycle arrest at G2/M phase and regulating key biomarkers. Onco Targets Ther. 2016;9:21–30. doi:10.2147/OTT.S9330526730200
- Seluanov A, Mao Z, Gorbunova V. Analysis of DNA double-strand break (DSB) repair in mammalian cells. J Vis Exp. 2010;(43):2002.20864925
- Guo Y, Fu X, Jin Y, et al. Histone demethylase LSD1-mediated repression of GATA-2 is critical for erythroid differentiation. Drug Des Devel Ther. 2015;9:3153–3162. doi:10.2147/DDDT.S81911
- Chabner BA, Roberts TG Jr. Timeline: chemotherapy and the war on cancer. Nat Rev Cancer. 2005;5(1):65–72. doi:10.1038/nrc152915630416
- Liu X, Li P, Hirayama R, et al. Genistein sensitizes glioblastoma cells to carbon ions via inhibiting DNA-PKcs phosphorylation and subsequently repressing NHEJ and delaying HR repair pathways. Radiother Oncol. 2018;129(1):84–94. doi:10.1016/j.radonc.2018.04.00529685705
- Gil Del Alcazar CR, Todorova PK, Habib AA, Mukherjee B, Burma S. Augmented HR repair mediates acquired temozolomide resistance in glioblastoma. Mol Cancer Res. 2016;14(10):928–940. doi:10.1158/1541-7786.MCR-16-012527358111
- Daley JM, Laan RL, Suresh A, Wilson TE. DNA joint dependence of pol X family polymerase action in nonhomologous end joining. J Biol Chem. 2005;280(32):29030–29037. doi:10.1074/jbc.M50527720015964833
- Dimitrova N, Chen YC, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456(7221):524–528. doi:10.1038/nature0743318931659
- Mazzarelli P, Parrella P, Seripa D, et al. DNA end binding activity and Ku70/80 heterodimer expression in human colorectal tumor. World J Gastroenterol. 2005;11(42):6694–6700. doi:10.3748/wjg.v11.i42.669416425368
- Costantini S, Woodbine L, Andreoli L, Jeggo PA, Vindigni A. Interaction of the Ku heterodimer with the DNA ligase IV/Xrcc4 complex and its regulation by DNA-PK. DNA Repair (Amst). 2007;6(6):712–722. doi:10.1016/j.dnarep.2006.12.00717241822
- Nick McElhinny SA, Snowden CM, McCarville J, Ramsden DA. Ku recruits the XRCC4-ligase IV complex to DNA ends. Mol Cell Biol. 2000;20(9):2996–3003. doi:10.1128/MCB.20.9.2996-3003.200010757784
- Uematsu N, Weterings E, Yano K, et al. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J Cell Biol. 2007;177(2):219–229. doi:10.1083/jcb.20060807717438073
- Hammel M, Yu Y, Fang S, Lees-Miller SP, Tainer JA. XLF regulates filament architecture of the XRCC4.ligase IV complex. Structure. 2010;18(11):1431–1442. doi:10.1016/j.str.2010.09.00921070942
- Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124(2):301–313. doi:10.1016/j.cell.2005.12.03116439205
- Buck D, Moshous D, de Chasseval R, et al. Severe combined immunodeficiency and microcephaly in siblings with hypomorphic mutations in DNA ligase IV. Eur J Immunol. 2006;36(1):224–235. doi:10.1002/eji.20053540116358361
- Riballo E, Woodbine L, Stiff T, Walker SA, Goodarzi AA, Jeggo PA. XLF-Cernunnos promotes DNA ligase IV-XRCC4 re-adenylation following ligation. Nucleic Acids Res. 2009;37(2):482–492. doi:10.1093/nar/gkn95719056826