Abstract
Background
Growing evidence supports BH3-interacting domain death agonist (Bid) playing a dual role in DNA damage response. However, the effects of Bid on hepatocellular carcinoma (HCC) cell proliferation in response to etoposide-induced DNA damage have not been sufficiently investigated.
Methods
Using a stable Bid-overexpression HCC cell line, Bid/PLC/PRF/5, overexpression of Bid promoted loss of viability in response to etoposide-induced DNA damage. MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]- and BrdU (5′-bromo-2′-deoxyuridine)-labeling assays revealed that etoposide-inhibited HCC cells grew in concentration-and time-dependent manners. The phosphorylations of Akt and mitogen-activated protein kinases (MAPKs) in response to etoposide-induced DNA damage were analyzed by Western blotting.
Results
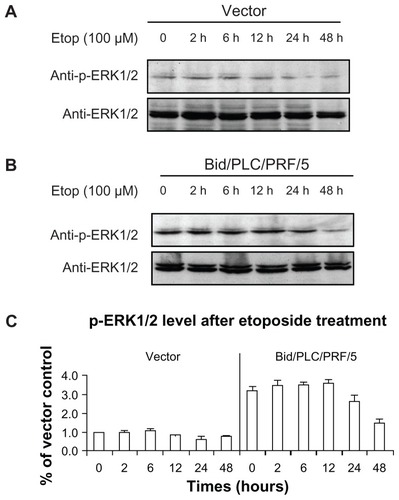
The survival rates of 100 μM etoposide on the cells with control vector and Bid/PLC/PRF/5 at 48 hours amounted to 71% ± 0.75% and 59% ± 0.60% with MTT assay, and similar results of 85% ± 0.08% and 63% ± 0.14% with BrdU-labeling assay respectively. Moreover, overexpression of Bid sensitized the cells to apoptosis at a high dose of etoposide (causing irreparable damage). However, it had little effect on the proliferation at a low dose of etoposide (repairable damage). Furthermore, the phosphorylation status of Akt and MAPKs were investigated. Overexpression of Bid suppressed the activation of Akt with respect to etoposide-induced DNA damage. Similar to Akt, the levels of phosphorylated p38 and phosphorylated c-Jun were attenuated by Bid-overexpression. On the contrary, the level of phosphorylated ERK1/2 was sustained at a high level, especially in Bid/PLC/PRF/5 cells.
Conclusion
Taken together, these results suggest that overexpression of Bid suppressed the activation of Akt, p38, and c-Jun, and promoted the activation of ERK1/2 induced by etoposide, suggesting that the promotion of ERK1/2 activation may have a negative effect on Bid-mediated HCC DNA damage induced by etoposide.
Background
BH3-interacting domain death agonist (Bid), a BH3 domain-only pro-apoptotic molecule, is initially found to be cleaved by caspase-8 in response to death receptor-mediated apoptotic signaling.Citation1,Citation2 The truncated Bid (tBid) is modified by myristoylation and translocated into mitochondria where it activates oligomerization of Bak or Bax and induces cytochrome c release. Cytochrome c in turn activates the apoptotic cascade of caspase-9 and -3, leading to cell death.Citation3,Citation4 Findings from the present authors and others have shown that the cleavage of Bid may not be an absolute requirement for Bid to be pro-apoptotic.Citation5–Citation8 Moreover, upon the administration of a high dose of DNA double-strand break agent causing irreparable damage to hepatocellular carcinoma (HCC), Bid is quickly translocated to the mitochondria to release cytochrome c.Citation9 Thus, it can be seen that the Bid-mediated mitochondrial pathway is critical to apoptotic cell death initiated by external signals from other cells or by internal warnings resulting from cellular stresses.
In addition to facilitating apoptosis with respect to DNA damage, Bid also promotes DNA repair. Upon DNA damage by stimulants such as etoposide or irradiation, DNA damage kinase ATM (ataxia telangiectasia mutated) is activated by phosphorylation, which in turn phosphorylates Bid at serine 61 or serine 78. Phosphorylated Bid participates in cell-cycle arrest and DNA repair.Citation10,Citation11 The pro-apoptotic function of full-length Bid might be affected by such phosphorylation, as it facilitates apoptosis to a lesser extent compared with its truncated form, tBid. However, the functions of Bid with respect to DNA-damage response are controversial.Citation12 Furthermore, recent studies have also demonstrated that the physiological role of Bid in development and cellular homeostasis may be cell-type specific. For example, mice lacking Bid do not enhance tumorigenesis and development of liver cancers following the administration of a chemical carcinogen, diethylnitrosamine (DEN). On the contrary, their tumor development is delayed,Citation13 which could be better explained by the role of Bid in promoting cell proliferation.
Akt and mitogen-activated protein kinase (MAPK) pathways play very important roles in response to extracellular stimuli to regulate several cellular functions including cell proliferation, apoptosis, and survival.Citation14,Citation15 The authors of this present study therefore analyzed the phosphorylation status of Akt and MAPKs with respect to etoposide-induced DNA damage of human HCC cells. As etoposide has been reported to activate Akt and ERK signaling pathways in human gastric cancer cells,Citation16 this study attempted to determine whether etoposide can activate Akt and MAPKs pathways in human HCC cells.
HCC remains one of the most difficult tumors to treat, especially when the tumor is advanced or unresectable. Since there is only a narrow understanding of the molecular, cellular, and environmental mechanisms that drive its disease pathogenesis, there are only limited therapeutic options.Citation17 Previous studies have shown that overexpression of Bid/tBid on HCC cells increase the sensitivity towards chemotherapeutic agents such as staurosporine, camptothecin, 5-fluorouracil (5-FU), doxorubicin (Dox), and etoposide.Citation8,Citation9,Citation18,Citation19 Furthermore, the authors of this present study revealed a novel p53 nucleocytoplasmic shutting function, which plays an important role in assisting Bid translocation from the nucleus to the cytoplasm through interaction with Bid following the stimulation of etoposide.Citation8
In the present study, using a stable Bid- overexpression HCC cell line, Bid/PLC/PRF/5, the effect of Bid- overexpression on the HCC cellular proliferation in response to etoposide-induced DNA damage was examined. The study suggested that overexpression of Bid in HCC cells promoted loss of viability likely through regulating Akt and MAPKs signaling pathways, suggesting Bid could potentially provide a therapeutic option that combines cytotoxic therapy for human HCC.
Materials and methods
Reagents and antibodies
Etoposide was purchased from Calbiochem (USA). MTT [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide] and protease inhibitor cocktail were purchased from Sigma-Aldrich (St Louis, MO). The enhanced chemiluminescence (ECL™) detection system from Amersham Biosciences, Inc (Freiburg, Germany). Protein assay kit for protein quantity analysis was purchased from Bio-Rad Laboratories (Hercules, CA). Lipofectamine 2000 and G418 were purchased from Life Technologies (Carlsbad, CA). The rabbit anti-Bid, anti-actin, anti-phospho-Akt (S473 and T308), anti-Akt, anti-rabbit IgG, and anti-mouse IgG antibodies were provided by Santa Cruz Biotechnology, Inc (Santa Cruz, CA). Mouse anti-phospho-ERK1/2, rabbit anti-ERK1/2, anti-phospho-p38, anti-p38, anti-phospho-c-Jun, and anti-c-Jun were purchased from Cell Signaling Technology (Danvers, MA). All other reagents were of analytical reagent quality.
Cell culture and generation of Bid-producing HCC cells
The human hepatocellular carcinoma cell line PLC/PRF/5 was obtained from American Type Culture Collection (Manassas, VA), and was cultured as previously described.Citation8,Citation9 The construction of Bid/pcDNA3.1 expression plasmid or empty vector was introduced into PLC/PRF/5 cells, and the stable cell clones were obtained by the addition of 500 μg/mL of G418 into the culture medium. The transfection, selection, and culture of the stable cell clones of Bid/PLC/PRF/5 and Vec/PLC/PRF/5 were carried out essentially as previously described.Citation9
MTT assay
The cell viability was measured by MTT method as previously described.Citation20
Brd-U-labeling proliferation assay
Cell proliferation in response to etoposide was confirmed by 5′-bromo-2′-deoxyuridine (Brd-U) incorporation method using an enzyme-linked immunosorbent assay kit procured from Roche Diagnostics Corp (Indianapolis, IN). Briefly, cells were treated in a similar manner as described for the cell viability assay, in 96-well microtiter plates. Six hours prior to the end of the interval of measurement, Brd-U (10 μM) was added. The cells were fixed, lysed, and then treated for 3 minutes at 37°C with peroxidase-conjugated, anti-Brd-U antibody supplied by the manufacturer. The cells were washed three times followed by the addition of substrate solution. Absorbance was measured at 405 nm, with a reference wavelength at 490 nm.
Western blot analysis
Western blot was performed as reported previously.Citation20,Citation21 After harvesting, the experimental cells were washed and lysed in lysis buffer. The total lysate was mixed with loading buffer, and preheated at 95°C for 10 minutes, then loaded onto sodium dodecyl sulfate-polyacrylamine gel. The proteins were transferred onto polyvinylidene fluoride membrane and blocked in 5% milk, and then incubated for 1 hour or overnight at 4°C with primary antibody. After hybridization with primary antibody, the membrane was washed and incubated with horseradish peroxidase-labeled secondary antibody. Final detection was performed with ECL Western blotting reagents. The blot was then stripped at 50°C for 30 minutes. The membranes were re-blotted for detection of other proteins after extensive wash.
Statistic analysis
The data are presented as the mean ± standard deviation for at least three separate determinations for each group. The differences between the groups were examined for statistical significance using the Student’s t-test with SPSS Statistics (IBM Corporation, Somers, NY) software.
Results
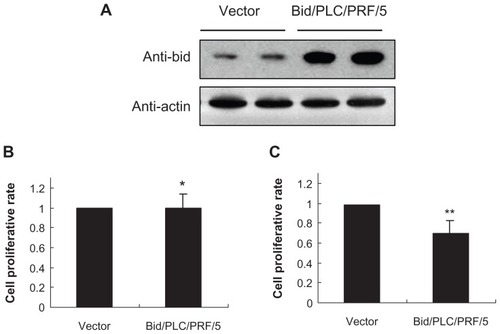
Generation and proliferative characteristics of Bid-producing HCC cells
A previous study showed that the expression of Bid in well and moderately differentiated HCC was lower than in the corresponding nontumorous tissues from the same patient, whereas the Bid expression was notably enhanced in tumorous liver tissues from patients with poorly differentiated tumors.Citation22 To further assess the functional significance of Bid expression during the development of HCC, it was investigated whether stable overexpression of Bid in human HCC cells could alter cell survival in vitro. To this end, a human HCC cell line PLC/PRF/5 was selected since it expresses a low level of endogenous Bid (, left two lanes). Bid DNA construct was transfected into the PLC/PRF/5 cells, and after drug selection, stable cell clones were isolated and verified for Bid expression. The expression of Bid in selected clones was verified with specific antibody against Bid (, right two lanes). Overexpression of Bid in PLC/PRF/5 cells led to an obvious morphological change with an appearance of fibroblast-like cell shape (data not shown), and such a change did not result in an alteration in the rate of cell proliferation under 10% serum conditions in vitro (). However, Bid-producing PLC/PRF/5 cells led to a significant (P < 0.05) reduction in cell proliferation rate under serum-depleted conditions (). These observations suggest that Bid could act to decrease cellular survival for HCC cells by serum starvation-induced cellular stress responses.
Figure 1 Proliferative characteristic of Bid-overexpressing PLC/PRF/5 clones and vector control clones. (A) Analysis of Bid-overexpressing PLC/PRF/5 clones and vector control clones by Western blot. (B and C) The proliferation analysis of cell viability with MTT assay under 10% serum or serum starvation conditions.
Abbreviations: Bid, BH3-interacting domain death agonist; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
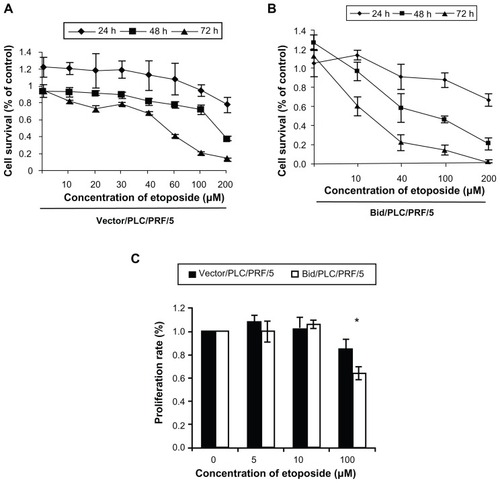
Bid promoted loss of viability in response to etoposide-induced DNA damage
A previous study showed that Bid sensitized apoptosis induced by chemotherapeutic drugs such as staurosporine, camptothecin, 5-FU, and Dox in human HCC cells.Citation18 In this study, two methods were used to further explore the effect of Bid-overexpression on human HCC cell viability in response to etoposide-induced DNA damage. First, cell viability was determined by the MTT assay method. As shown in , etoposide inhibited the HCC cells growth in a concentration- and time-dependent manner. A significant loss of viability was detected in the cells treated with 100 μM or more etoposide for 48 hours. The cell survival rates of 100 μM etoposide on the cells with control vector () and the cells with Bid/PLC/PRF/5 () at 48 hours amounted to 71% ± 0.75% and 59% ± 0.60% respectively. The cell proliferation rate was further examined by the BrdU-labeling assay method. Similar results to the MTT assay were observed. At 100 μM etoposide for 48 hours treatment, the proliferation rates of the cells with control vector and the cells with Bid/PLC/PRF/5 amounted to 85% ± 0.08% and 63% ± 0.14% respectively () (P < 0.05). All these results indicate that overexpression of Bid sensitized the cells to the high concentrations of etoposide treatment. However, it has little effect on the proliferation of the cells treated with low concentrations of etoposide.
Figure 2 Effect of Bid on the PLC/PRF/5 cell viability in the response of etoposide-induced DNA damage. Overexpression of Bid does not alter the rate of cell proliferation under low concentration of etoposide, but Bid-overexpression cells sensitize apoptosis induced by high concentration of etoposide. (A and B) Cell viability was determined by the MTT assay method. (C) Cell proliferation rate was determined by the BrdU-labeling method assay.
Abbreviations: Bid, BH3-interacting domain death agonist; BrdU, 5′-bromo-2′-deoxyuridine; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
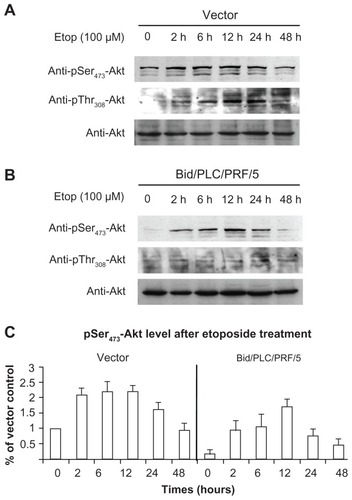
Effects of Bid on the activation of Akt in response to etoposide
Akt and MAPKs signaling pathways are thought to be the pivotal mechanisms of apoptotic signals in malignant cells.Citation14,Citation15 Several extracellular stimuli have been shown to activate Akt and MAPKs via phosphorylation. The phosphorylation status of Akt and MAPKs was therefore analyzed with respect to etoposide-induced DNA damage of the human HCC cells. As shown in , a significant degree of Akt activation was detected using the phosphor-specific Akt (Ser473 and Thr308) antibodies. An increase in phosphorylated Akt occurred at 2 hours after treatment. The maximal level of phosphorylated Akt was recorded at 12 hours after treatment in the cells with Bid overexpression (). At 48 hour after treatment, the levels of phosphorylated Akt gradually declined to the basal level. Moreover, it was observed that the level of phosphorylated Akt in Bid/PLC/PRF/5 cells was lower than that in the control cells (). This result indicates that the overexpression of Bid can suppress the activation of Akt.
Figure 3 Bid-overexpression on the activation of Akt in response to etoposide. Vector control (A) and Bid/PLC/PRF/5 cells (B) were treated with etoposide for different periods, respectively. Then, p-Akt was detected by Western blot analysis. All blots were subsequently stripped and reprobed with antibodies against Akt. The density of pSer473-Akt protein bands was determined (C).
Effects of Bid on the activation of MAPKs in response to etoposide
MAPKs can be activated by several different anticancer drugs. However, their roles in Bid-mediated apoptosis following etoposide treatment remain unclear. The effects of phosphorylated MAPKs in Bid-mediated apoptosis in human HCC cells (–) were therefore determined. Similar to the result of the phosphorylated Akt observed (), the levels of phosphorylated p38 () and phosphorylated c-Jun () were increased in both control vector cells and Bid/PLC/PRF/5 cells. Maximal level of both was detected during the 2–6 hour post-treatment time period, and they gradually declined towards the basal level thereafter. Interestingly, an obvious increase in the phosphorylated ERK was not observed in HCC cells treated with etoposide (). On the contrary, the level of phosphorylated ERK1/2 was sustained at a higher level, especially in Bid/PLC/PRF/5 cells. However, after 24 hours of treatment, the level of phosphorylated ERK declined ().
Figure 4 Effects of Bid-overexpression on the phosphorylation of p38 in response to etoposide. Vector control (A) and Bid/PLC/PRF/5 cells (B) were treated with etoposide for different periods of time, respectively. Then p-p38 was detected by Western blot analysis. All blots were subsequently stripped and reprobed with antibodies against p38. The density of p-p38 protein bands was determined (C).
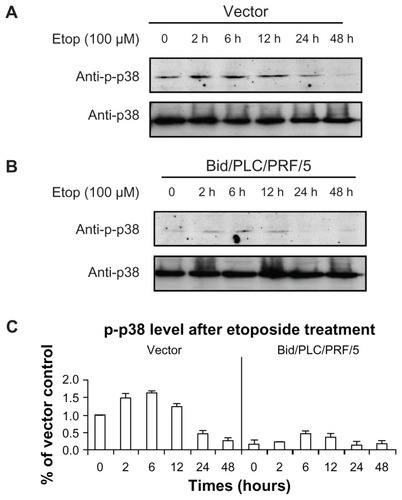
Figure 5 Effects of Bid-overexpression on the phosphorylation of c-Jun in response to etoposide. Vector control (A) and Bid/PLC/PRF/5 cells (B) were treated with etoposide for different periods, respectively. Then, p-c-Jun was detected by Western blot analysis. All blots were subsequently stripped and reprobed with antibodies against c-Jun. The density of p-c-Jun protein bands was determined (C).
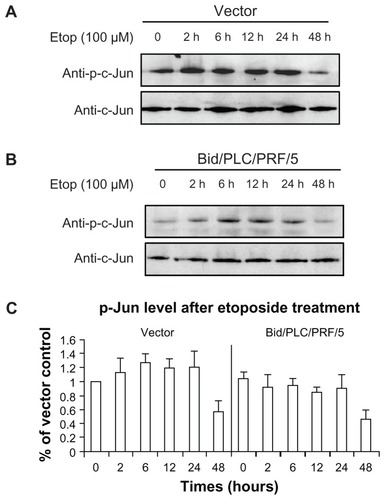
Figure 6 Effects of Bid-overexpression on the phosphorylation of ERK1/2 in response to etoposide. Vector control (A) and Bid/PLC/PRF/5 cells (B) were treated with etoposide for different periods, respectively. Then p-ERK1/2 was detected by Western blot analysis. All blots were subsequently stripped and reprobed with antibodies against ERK1/2. The density of p-ERK1/2 protein bands was determined (C).
Discussion
HCC is the fifth most frequent primary malignant tumor worldwide, particularly in parts of Asia and Africa, and the fourth in annual mortality, causing approximately half a million deaths each year.Citation23 Recently, the rising rate of HCC has been found in some Western countries.Citation24 In addition, the liver is the most frequent site of metastasis, especially from gastrointestinal cancers. Despite its significance, there is only a narrow understanding of the molecular, cellular, and environmental mechanisms that drive disease pathogenesis, and there are only limited therapeutic options.Citation17 HCC remains one of the most difficult tumors to treat, especially when the tumor is advanced or unresectable. Therefore, it is important to develop an efficient treatment for HCC. In this study, Bid was chosen as a potentially therapeutic target that combines cytotoxic therapy for human HCC.
It is well known that the BH3-only pro-apoptotic member, full-length Bid, and its tBid link the extrinsic death receptor pathway and intrinsic mitochondrial apoptotic pathway.Citation2 Etoposide, a well known DNA-damaging agent, is a topoisomerase II inhibitor that has been widely used in several model studies to couple DNA damage with apoptosis.Citation25 Exposure to DNA-damaging agents can cause mutations, developmental abnormalities, or cancer. Cells respond to the presence of DNA damage by activating cell cycle checkpoints and repair mechanisms, and a small part of the cells that cannot be repaired are directed towards the apoptotic pathway, ultimately the damaged cells are eliminated through apoptosis. Differential expressions and functions of several Bcl-2 family members in human HCC have been recently reported, and they were shown to be associated with the effects of anticancer stimuli.Citation26 However, Bid, as an abundant pro-apoptotic protein of the Bcl-2 family, its functions and molecular mechanisms in the progression of human HCC are still not well understood, especially in terms of the response of HCC cells to DNA damage.
To further explore the molecular mechanisms of Bidmediated apoptosis of chemotherapeutic drug-induced DNA damage, a previous study showed that overexpression of Bid/tBid on HCC cells increase the sensitivity towards chemotherapeutic agents such as staurosporine, camptothecin, 5-FU, and Dox.Citation18 The activities of caspase-8, -9, and -3 were increased after a 72-hour treatment of 5-FU. Expression level of tBid was also increased due to accumulation of the truncated counterpart. Compared with the full-length Bid, this truncated form could promote higher sensitivity of HCC cells to 5-FU induced cytotoxicity and cytochrome c release, which can be blocked by overexpression of the pro-survival protein Bcl-xL.Citation18 In addition, adenovirus-mediated overexpression of truncated Bid can also reduce cell viability and promote tumor regression in both p53-sensitive and -resistant HCC cells.Citation19 However, Bid-deficient mice do not have an enhanced development of liver cancers following the administration of a chemical carcinogen, DEN. On the contrary, Bid-deficient livers show significantly less proliferating cells following DEN administration.Citation13 The observations could be better explained by the role of Bid in promoting cell proliferation. In the present investigation, Bid exhibited loss of viability and sensitivity to apoptosis in response to human HCC cells to etoposide-induced DNA damage ().
Initiation of apoptosis is the key mechanism following high DNA damage. Recent reports show that Bid will be cleaved and activated after the external stimulus.Citation2 Bid, in turn, activates Bax and Bak; both trigger apoptosis. The present study also proposed that Bid-overexpression was more sensitive to etoposide-induced high degree of DNA damage than that of the control. At the molecular level, it has been reported that constitutive activation of Akt and MAPKs signaling pathways by chemotherapy drugs including etoposide promotes resistance to chemotherapy in several cancers.Citation16,Citation27,Citation28 The activation of Akt and MAPK signaling pathways are thought to be the pivotal mechanisms of apoptotic signals in malignant cells. To understand the mechanism by which Bid sensitizes HCC cells to apoptosis in response to etoposide-induced DNA damage, the data showed that Bid-overexpression significantly decreased the level of phosphorylated Akt, p38, and c-Jun in HCC cells followed by etoposide-induced DNA damage. Therefore, Bid suppressed the activation of Akt, p38, and c-Jun signaling pathways with the treatment of etoposide, which may be another therapeutic option of chemoresistance in HCC. Bid could promote ERK1/2 phosphorylation, which suggests ERK1/2 activation may have a negative effect on HCC DNA damage induced by etoposide. Furthermore, the levels of Akt and MAPKs phosphorylation decreased gradually to basal levels after 48 hours, and the declinations of Akt and MAPKs phosphorylation in Bid/PLC/PRF/5 cells were coincident with the onset of Bid-overexpression mediated apoptosis. These results again explained that Bid could sensitize apoptosis induced by etoposide.
In summary, the data presented here strongly suggest that Bid-overexpression on human HCC cells promoted loss of viability via mediating different pathways (Akt and MAPKs) in response to different degrees of DNA damage. Elucidation of such intricate mechanisms of the effects of Bid on proliferation could potentially provide a therapeutic option that combines cytotoxic therapy for human HCC.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No 31071187, 81272721), the Fundamental Research Funds for the Central Universities (2010121102), Xiamen Municipal Science and Technology Innovation Fund Project (3502Z20114018), and the Program for New Century Excellent Talents in Fujian Province University.
Disclosure
The authors report no conflicts of interest in this work.
References
- WangKYinXMChaoDTMillimanCLKorsmeyerSJBID: a novel BH3 domain-only death agonistGenes Dev19961022285928698918887
- YinXMBid, a BH3-only multi-functional molecule, is at the cross road of life and deathGene200636971916446060
- LiHZhuHXuCJYuanJCleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosisCell19989444915019727492
- LuoXBudihardjoIZouHSlaughterCWangXBid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptorsCell19989444814909727491
- KonigHGRehmMGudorfDFull length Bid is sufficient to induce apoptosis of cultured rat hippocampal neuronsBMC Cell Biol2007871417326836
- SarigRZaltsmanYMarcellusRCFlavellRMakTWGrossABID-D59A is a potent inducer of apoptosis in primary embryonic fibroblastsJ Biol Chem200327812107071071512519725
- ValentijnAJGilmoreAPTranslocation of full-length Bid to mitochondria during anoikisJ Biol Chem200427931328483285715148322
- SongGChenGGYunJPLaiPBSAssociation of p53 with Bid induces cell death in response to etoposide treatment in hepatocellular carcinomaCurr Cancer Drug Targets20099787188020025574
- SongGChenGGChauDKMiaoJLaiPBSBid exhibits S phase checkpoint activation and plays a pro-apoptotic role in response to etoposide-induced DNA damage in hepatocellular carcinoma cellsApoptosis200813569370118373075
- ZinkelSSHurovKEOngCAbtahiFMGrossAKorsmeyerSJA role for proapoptotic BID in the DNA-damage responseCell2005122457959116122425
- KamerISarigRZaltsmanYProapoptotic BID is an ATM effector in the DNA-damage responseCell2005122459360316122426
- KaufmannTTaiLEkertPGThe BH3-only protein bid is dispensable for DNA damage- and replicative stress-induced apoptosis or cell-cycle arrestCell2007129242343317448999
- BaiLNiHMChenXDifrancescaDYinXMDeletion of bid impedes cell proliferation and hepatic carcinogenesisAm J Pathol200516651523153215855651
- SongGOuyangGBaoSThe activation of Akt/PKB signaling pathway and cell survivalJ Cell Mol Med200591597115784165
- KimEKChoiEJPathological roles of MAPK signaling pathways in human diseasesBiochim Biophys Acta20101802439640520079433
- LiuSQYuJPYuHGLvPChenHLActivation of Akt and ERK signalling pathways induced by etoposide confer chemoresistance in gastric cancer cellsDig Liver Dis200638531031816527552
- Di MaioMDe MaioEPerroneFPignataSDanieleBHepatocellular carcinoma: systemic treatmentsJ Clin Gastroenterol200235Suppl 2S109S11412394214
- MiaoJChenGGChunSYChakECLaiPBBid sensitizes apoptosis induced by chemotherapeutic drugs in hepatocellular carcinomaInt J Oncol200425365165915289866
- MiaoJChenGGChunSYAdenovirus-mediated tBid overexpression results in therapeutic effects on p53-resistant hepatocellular carcinomaInt J Cancer200611981985199316708390
- SongGMingYMaoYBaoSOuyangGOsteopontin prevents curcumin-induced apoptosis and promotes survival through Akt activation via {alpha}v{beta}3 integrins in human gastric cancer cellsExp Biol Med20082331215371545
- SongGOuyangGMaoYBMingYLBaoSHuTHOsteopontin promotes metastatic growth of gastric cancer by augmenting cell survival and invasion through Akt-mediated HIF-1α up-regulation and MMP9 activationJ Cell Mol Med2009138B1706171819602039
- ChenGGLaiPBChakECXuHLeeKMLauWYImmunohistochemical analysis of pro-apoptotic Bid level in chronic hepatitis, hepatocellular carcinoma and liver metastasesCancer Lett20011721758211595132
- KewMCEpidemiology of hepatocellular carcinomaToxicology2002181–1823538
- El-SeragHBHepatocellular carcinoma: an epidemiologic viewJ Clin Gastroenterol2002355 Suppl 2S72S7812394209
- BaldwinELOsheroffNEtoposide, topoisomerase II and cancerCurr Med Chem Anticancer Agents20055436337216101488
- FaraziPADePinhoRAHepatocellular carcinoma pathogenesis: from genes to environmentNat Rev Cancer20066967468716929323
- ClarkASWestKStreicherSDennisPAConstitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cellsMol Cancer Ther20021970771712479367
- KrausACFerberIBachmannSOIn vitro chemo- and radio-resistance in small cell lung cancer correlates with cell adhesion and constitutive activation of AKT and MAP kinase pathwaysOncogene200221578683869512483521