
Abstract
Photodynamic therapy (PDT) is a groundbreaking approach involving the induction of cytotoxic reactive oxygen species (ROS) within tumors through visible light activation of photosensitizers (PS) in the presence of molecular oxygen. This innovative therapy has demonstrated success in treating various cancers. While PDT proves highly effective in most solid tumors, there are indications that certain cancers exhibit resistance, and some initially responsive cancers may develop intrinsic or acquired resistance to PDT. The molecular mechanisms underlying this resistance are not fully understood. Recent evidence suggests that, akin to other traditional cancer treatments, the activation of survival pathways, such as the KEAP1/Nrf2 signaling pathway, is emerging as an important mechanism of post-PDT resistance in many cancers. This article explores the dual role of Nrf2, highlighting evidence linking aberrant Nrf2 expression to treatment resistance across a range of cancers. Additionally, it delves into the specific role of Nrf2 in the context of photodynamic therapy for cancers, emphasizing evidence that suggests Nrf2-mediated upregulation of antioxidant responses and induction of drug efflux transporters are potential mechanisms of resistance to PDT in diverse cancer types. Therefore, understanding the specific role(s) of Nrf2 in PDT resistance may pave the way for the development of more effective cancer treatments using PDT.
Introduction
Cancer is presently ranked as the second-leading cause of morbidity and mortality globally; however, it has been predicted that it will become the leading cause of death by the year 2060 because of the increase in its incidence rate.Citation1 The 2020 GLOBOCAN statistics on cancer demonstrated that around 19.3 million new cases of cancer and 10 million cancer-related deaths occurred in the year 2020.Citation2 The World Health Organization (WHO) reported that when assessing Disability-Adjusted Life Years (DALYs) specific to causes across all human diseases, cancer imposed the greatest social and economic burden on a global scale, accounting for 244.6 million DALYs in both men and women.Citation1,Citation3 Multiple factors are reported to be responsible for the worldwide increase in cancer incidence rate and mortality, including lifestyle modification (unhealthy diet, alcohol, smoking and sedentary lifestyle), exposure to carcinogens and pollutants, aging and growth of populations, exposure to infectious agents and socioeconomic development status.Citation2,Citation4
Although the spectrum of treatment for combating cancers has been broadened and improved in recent decades, surgery, followed by adjuvant radiotherapy, chemotherapy, immunotherapy, and targeted therapy are the most common treatment protocol for most cancers. These treatment protocols, however, have not always been totally effective due to poor response by patients, severe side effects of the therapies, and development of intrinsic and acquired resistance to the therapies, leading to relapse and recurrence. Recurrence due to drug resistance is widespread in cancers, with evidence indicating that the rate of recurrence varies in patients with non-small cell lung cancer (30–55%), soft tissue carcinoma (15–40%), ovarian cancer (85%), pancreatic cancer (37%) and glioblastoma (>55%).Citation5–9 It is well known that tumor cells are able to induce several survival mechanisms to drive resistance to various treatments. These mechanisms include alteration in drug target, increased efflux of therapeutic drug, DNA damage repair, altered metabolism, intra-tumor heterogeneity, and activation of signaling pathways that promote tumor cells survival and suppress tumor cell death among others.Citation10–12
Post-operative adjuvant treatment for most cancers such as radiotherapy, chemotherapy and immunotherapy is based on the formation of reactive oxygen species (ROS) to induce tumor cells death. Therefore, the role of antioxidant response as a mechanism of resistance to post-operative adjuvant cancer therapies is now becoming clearer. The nuclear factor (erythroid-derived 2)-like 2 (Nrf2) is a Cap“n”Collar (CNC), basic leucine zipper (bZIP) transcription factor recognized as the primary controller of redox homeostasis in cells. In carcinogenesis, Nrf2 is reputed to possess a good and a bad side, thereby playing a dual role. Evidence abounds indicating that Nrf2 activation protects cells by preventing chemical- and radiation-induced carcinogenesis.Citation13,Citation14 It is reported that Nrf2 inhibits carcinogenesis when its downstream targets are expressed. These downstream targets scavenge ROS, repair oxidative damage and ensure quick enzymatic modification and elimination of chemical carcinogens.Citation13 Recent evidence in the last decade has revealed the existence of a ‘dark side’ to Nrf2 activation in carcinogenesis, in contrast to its protective role in normal cells. It is well recognized now that Nrf2 is highly over-expressed in different cancer types, including lung, breast, colorectal, ovarian, pancreatic, liver and leukemia among others.Citation15–17 Cancers with aberrant activation of Nrf2 are characterized by tumor phenotypes including facilitated tumor progression, metastasis, chemoresistance and radio-resistance.Citation18
Photodynamic therapy (PDT) is an innovative and minimally invasive therapeutic approach employed for either curative or palliative treatment in early-stage and late-stage cancers, respectively.Citation19 During PDT, light exposure activates a non-toxic photosensitizing agent administered to the patient, leading to the production of cytotoxic ROS in the presence of molecular oxygen. These reactive species are utilized to eliminate tumor cells. PDT presents several advantages over traditional cancer treatments, such as minimal systemic toxicity, selective tumor destruction capability, absence of severe side effects, and the ability for repeat procedures. Therefore, it can be applied either alone or in combination with other treatments, such as surgery, radiotherapy, chemotherapy, and immunotherapy. Clinically, PDT has received approval and demonstrated successful application in treating conditions such as non-melanoma skin cancers, cutaneous T cell lymphoma, biliary tract cancer, esophageal cancer, as well as squamous cell carcinoma of the head, neck, and bladder.Citation20–23 Despite the overwhelming effectiveness of PDT in most solid tumors, there are indications that some cancers are relatively recalcitrant, and some initially responsive cancers may develop acquired resistance to PDT. Resistance to PDT has been attributed to sub-optimal accumulation of photosensitizers (PS) in the tumor, insufficient ROS production due to poor photophysical and physicochemical properties of PS, and PDT-induced ROS generation resulting in the activation of survival signal transduction pathways.Citation19,Citation24 Similar to what is obtained for other therapeutic modalities, activation of survival pathways has emerged as a major mechanism of resistance to PDT. It is now known that post-PDT, tumor cells activate signal transduction pathways that mediate PS extrusion, pro-survival autophagy, DNA damage repair, as well as up-regulated antioxidant response to mitigate PDT efficacy.Citation25 Although the mechanism underlying tumor resistance to PDT is not fully understood, we present in this review, the current knowledge regarding tumor resistance to PDT with particular focus on Nrf2 signaling pathway(s) and its role in mediating antioxidant response up-regulation and induction of drug efflux transporters in PDT treated tumors.
Cancers and Oxidative Stress-Based Therapies
Reactive species (ROS) are natural byproducts of cellular metabolism, possessing the potential for both beneficial and harmful effects. ROS generated during normal physiological and cellular reactions are usually neutralized by enzymatic antioxidants, such as superoxide dismutase (SOD), glutathione peroxidase (GPx), and catalase (CAT), as well as non-enzymatic antioxidants including glutathione (GSH), thioredoxin (Trx), peroxiredoxin (Pdx) and glutaredoxin (Gdx). When present in low to moderate concentrations, ROS function as second messengers in cell signaling, overseeing various cellular processes such as proliferation, differentiation, cell cycle progression, growth arrest, apoptosis, modulation of enzyme activity, mediation of inflammation, and the elimination of pathogens and foreign particles. An imbalanced defense mechanism of antioxidants, an overabundance of reactive species, or the influx of free radicals from the environment can lead to an excessive accumulation of reactive oxygen species (ROS). This condition, known as oxidative stress, poses a harmful threat, potentially causing damage to vital cell structures such as lipid membranes, proteins, and nucleic acids. The prolonged presence of elevated oxidative stress has been widely acknowledged as a contributing factor to the pathophysiology of various diseases, including cancers.Citation26 Elevated levels of reactive oxygen species (ROS) and the resulting oxidative stress can give rise to unfavorable outcomes, including the activation of oncogenes, genomic instability, de-differentiation, and increased mitogenesis. These events have all been linked to the initiation, development, progression, invasion, and metastasis of cancer.Citation26
While tumor cells are known to constitutively display a pro-oxidative shift in comparison with normal cells, the precise mechanism leading to this elevated oxidative stress in malignant cells and solid tumors remains unclear. However, malfunction of the mitochondrial respiratory chain, oncogenic activation and transformation, aberrant metabolism, inactivation of tumor suppressors, and a dysfunctional antioxidant response have been suggested as contributing to the elevated oxidative stress observable in malignant cells.Citation27 The heightened production of reactive oxygen species (ROS) serves as the origin of DNA-damaging agents in cancer cells, potentially fostering genetic instability. Simultaneously, ROS-induced mitochondrial dysfunction modifies the cellular apoptotic response to anticancer agents, contributing to the development of drug resistance in tumor cells.Citation27,Citation28 Despite the deleterious effects of elevated ROS in cancer cells, recent advances have exploited this feature to develop innovative therapeutic strategies for selectively targeting and killing cancer cells. Non-surgical cancer treatment approaches, such as chemotherapy, radiation therapy and photodynamic therapy, shared a common mechanism of ROS production. The approach involves subjecting cancer cells to intensified ROS attack induced by these ROS-generating procedures. The aim is to overwhelm the cellular antioxidant defense, pushing cancer cells beyond the tolerable ROS threshold that distinguishes cell survival from cell death. Cancer cells, owing to their elevated intrinsic oxidative state, require less additional ROS to surpass the threshold leading to cell death compared to normal cells. This is attributed to the higher oxidative status of cancer cells. However, it is suggested that ROS levels that can kill tumor cells have a less drastic impact on normal cells because normal cells operate at lower oxidative status and possess efficient ROS-induced injuries repair mechanisms.Citation26 Because ROS induced toxicity to both cancerous and non-cancerous tissues, a major concern in the employment of oxidative stress-based therapy in cancers is how to target and kill tumor cells selectively and effectively. Thus, normal tissues will be better protected from harmful effect of therapeutic ROS if oxidative stress-based therapies are precisely targeted to the diseased tumor cells, as is the case with photodynamic therapy.
Photodynamic Therapy as an Oxidative Stress Event
PDT is a minimally invasive treatment modality that has been approved for clinical use, with demonstrated effectiveness against various cancer types. The process entails the introduction of a photosensitizing drug via intravenous, systemic, or topical means, with a preference for localization within the targeted tumor. Subsequent exposure of the tumor-localized photosensitizer to visible light, at a wavelength corresponding to the absorption peak of the PS, triggers the production of cytotoxic reactive oxygen species in the presence of molecular oxygen. These ROS generated act to eliminate tumors through direct cell killing, damage to tumor-associated vasculature, or the activation of an anti-tumor immune response.Citation29–31 PDT can induce direct killing of tumor cells through the three primary mechanisms of apoptosis, necrosis, and autophagy-associated cell death. In vitro studies have identified apoptosis as the primary mode of cell death after PDT. However, evidence has shown that factors such as the subcellular localization of the photosensitizer (PS) in various organelles (eg, lysosome, mitochondria, melanosome, and endoplasmic reticulum), the dose of PDT (PS concentration × light fluence), drug-light interval (DLI), glucose deprivation, and cell genotype all play pivotal roles in determining the predominant mechanism of cell death.Citation32–36 The effectiveness of PDT is dependent on the simultaneous presence of three essential components: a photosensitizer, molecular oxygen, and a visible light source. Photosensitizers play a crucial role in the efficacy of PDT. For a PS to be ideal for photodynamic therapy, it must be chemically pure and stable, accumulate preferentially in and be retained by the target tumor, must not promote dark cytotoxicity, be rapidly cleared from normal tissue, and must possess considerable triplet quantum yield, contributing to the production of ROS after light exposure.Citation31,Citation32
Photochemical Mechanisms of ROS Production in PDT
The photosensitizer (PS) molecule initially exists in a singlet ground state (S0), with paired electrons exhibiting opposite spins in a low-energy molecular orbital. When irradiated with quantum light, the PS absorbs photon leading to the formation of an excited singlet state (S1). The S1 state, characterized by the excitation of one electron into a higher-energy molecular orbital, is inherently unstable and short-lived, with several potential fates. The S1 state PS may decay back to S0 undergoing molecular relaxation and internal conversion resulting in the emission of a photon (fluorescence) and the generation of heat, respectively. In most PS, the energy of the photon is redistributed over two unpaired electrons with the same spin orientation, causing the S1 to undergo intersystem crossing to a longer-lived triplet state (T1) characterized by lower energy. In the T1 state, the PS takes advantage of its longer lifetime to interact with surrounding molecules, and it is well established that generation of cytotoxic species produced during PDT occurs in this state. The PS in the T1 state can undergo two types of reactions. It can undergo a direct reaction with biomolecules in its vicinity, transferring a proton to form a radical anion or generate a radical cation as a result of the transfer of electron in what is referred to as a type I reaction. The radicals generated can interact with molecular oxygen to form superoxide anion (O2•-), the primary ROS produced in type I reaction. Type II reaction occurs when there is a direct transfer of energy from the T1 photosensitizer to molecular oxygen in its natural triplet state (3O2) to generate highly reactive singlet oxygen (1O2) (see ). While the type II reaction, which generates singlet oxygen, is considered the major cytotoxic pathway in PDT, evidence suggests that both types of reactions can occur at the same time during PDT. The ratio of these processes depends on factors such as the type of photosensitizer used, substrate concentration, and oxygen levels.Citation30,Citation37 Furthermore, reports from other studies also revealed that the high efficacy of PDT results from synergistic action of ROS (O2•-, HO• and 1O2) generated during type I and II reactions.Citation29,Citation38
Figure 1 Photochemical generation of ROS during PDT. Upon absorption of light of appropriate wavelength by the PS, an electron is elevated to an excited singlet state (S1) from the singlet ground state (S0). This excited singlet state is transient and can release energy through photon emission (fluorescence). Alternatively, it may undergo intersystem crossing, leading to the formation of a long-lived excited triplet state (T1). The T1 state is responsible for generating reactive species, including O2•-, H2O2, HO• and 1O2 through both type I and type II reactions.
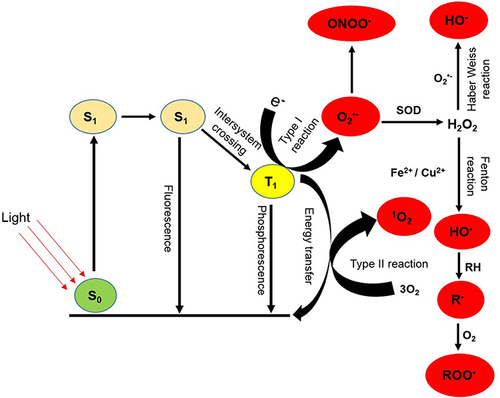
Superoxide (O2•-) radical produced in type I reaction is a relatively stable and not particularly reactive in biological systems. It does not by itself cause direct oxidative damage to nucleic acids, lipids or carbohydrates. It can undergo a dismutation reaction with itself, catalyzed by the enzyme superoxide dismutase (SOD) to generate H2O2, a non-radical molecule required for the functioning of many enzymes. Though not in itself highly cytotoxic, O2•- is able to generate highly cytotoxic secondary products by reacting with other biologically relevant radicals. O2•- can interact with H2O2 through a Haber-Weiss or an iron- or copper-mediated breakup of H2O2 (Fenton reaction), giving rise to the formation of the highly reactive and cytotoxic HO• radical.Citation39 Furthermore, O2•- can react with HO• to form 1O2 or with nitric oxide (NO•) to generate peroxynitrite (OONO•) another cytotoxic product (). HO• damage is diffusion rate-limited and due to its extremely high reactivity, will damage biomolecules in close proximity to its site of formation. It can add itself to and/or abstract an electron from an organic substrate such as fatty acid side chain of membranes forming a hydroxylated adduct or an oxidized substrate, respectively.Citation37 Both products are also radicals and initiate a chain reaction when they react with other molecules such as oxygen in the ground state to generate a peroxyl radical (ROO−) which further interacts with another organic substrate propagating a chain reaction.
1O2 and HO• are the two most important ROS generated during PDT because of their extreme reactivity towards biomolecules. Although both and other ROS are short-lived (1O2 < 40 ns and HO• < 1 ns), evidence has shown that PDT induced prolong oxidative stress in vitro.Citation30,Citation40–42 One of the outcomes of ROS-mediated damage in PDT-treated cells is the peroxidation of membranous lipids. Despite the fact that lipids are less abundant than proteins, many factors make membranous organelles and cell membrane prime target for PDT-derived ROS. The cell membrane is particularly vulnerable to damage from reactive oxygen species (ROS) due to the high susceptibility of polyunsaturated fatty acid (PUFA) residues in phospholipids to oxidation. In addition, molecular oxygen (O2) is highly soluble in the lipid environment, this increases the likelihood of an excited photosensitizer (PS) encountering O2 and generating singlet oxygen (1O2) and other ROS, especially compared to a hydrophilic environment.Citation29 ROS produced during photodynamic therapy (PDT) can initiate lipid peroxidation by abstracting hydrogen from lipids or, in the case of 1O2, by directly adding to mono- or polyunsaturated fatty acids, generating a lipid radical that stabilizes itself by reacting with oxygen to form a peroxyl radical. This peroxyl radical then attacks a neighboring fatty acid, leading to the creation of a stable lipid hydroperoxide and simultaneously generating another lipid radical. This chain reaction propagates, ultimately causing the destruction of lipid bilayers in membranes.Citation39,Citation43 Lipid hydroperoxides can undergo conversion into unstable conjugated dienes, which in turn decompose into various chemically reactive and biologically active products, including malondialdehyde (MDA), trans-4-hydroxy-2-nonenal, and 2-propenal (acrolein). These products resulting from lipid peroxidation have the capacity to modify macromolecules, disrupt metabolism, and influence cell signaling pathways, ultimately culminating in tumor cell destruction.
Proteins, being central to the majority of cellular functions, are abundant in biological systems, and consequently represent crucial targets for reactive oxygen species (ROS) generated through photodynamic therapy (PDT). HO• initiates the attack on the polypeptide backbone via an abstraction of the α-hydrogen atom from an amino acid residue leading to the formation of a carbon-centered radical. In the presence of O2, this radical evolves into a peroxyl radical, which, upon interaction with the protonated form of superoxide, is further transformed into alkyl peroxides.Citation39,Citation44 While PDT-derived ROS can target all amino acids in protein, cysteine, because of its thiol group exhibits the highest susceptibility. Nonetheless, though generally to a lesser degree than cysteine, methionine, tyrosine, and tryptophan are also prone to photo-oxidation.Citation45 ROS attack on proteins can lead to the oxidation of amino acid residue side chains, as well as the oxidation of the protein backbone, resulting in the formation of protein–protein cross-linkages. Although PDT-derived ROS attack on proteins primarily resulted in detrimental effects on protein function, disruption of normal redox homeostasis in photosensitized cells also induced cell death.
Reactive oxygen species-induced damage to DNA is an important cause of cell death during PDT. The HO• radical, produced via type I reaction during PDT is the most destructive reactive species. It has the capability to interact with and modify all components of the DNA molecule, including the purine and pyrimidine bases, as well as the deoxyribose sugar backbone. This interaction results in various forms of damage such as base or sugar lesions, single-strand breaks, double-strand breaks, abasic site formation, and the formation of DNA–DNA or DNA–protein cross-links. DNA damage has been reported in many cancer cells post-PDT. The formation of the double-strand breaks (DSBs) marker, γ-H2AX has been reported in HaCAT keratinocytes and WM35 melanoma cells.Citation46,Citation47 Singlet oxygen exhibits selectivity in its oxidative modification of DNA, with guanine being the most susceptible organic base to 1O2 oxidation. This susceptibility leads to the formation of 8-oxo-7,8-dehydro-2-deoxyguanosine,Citation48 a product that has been identified following photodynamic therapy.Citation49 Additionally, aldehydic products of lipid peroxidation, such as acrolein and 4-HNE, can also attack and modify DNA. This process results in the creation of bulky exocyclic adducts, which can contribute to DNA–DNA and DNA–protein crosslinking, ultimately impairing the binding of transcription factors. Although cells are equipped with the capability to repair oxidative DNA damage, however, unrepaired damage prior to DNA replication may induce cell death, DNA mutation, replication errors and genomic instability in photosensitized tumors leading to eventual tumor destruction.
ROS-Mediated Cytotoxicity to Tumor Cells After PDT
Singlet oxygen and other cytotoxic photoproducts generated upon irradiation during PDT start a cascade of biochemical events that may induce damage and kill tumor cells via three main biological mechanisms. These include (1) direct killing of tumor cells (2) destruction of tumor-associated vasculature and (3) stimulation of anti-tumor immune response. While all three processes have been implicated in the overall outcome of PDT effect, the relative contribution of each process is difficult to pinpoint. Therefore, the focus of this review will be on PDT-induced direct killing of tumor cells.
Direct killing of tumor cells in PDT is mediated via three different processes of apoptosis, necrosis and autophagy. This differential direct cell killing is known to depend on many factors including physicochemical characteristic and subcellular localization of PS, overall PDT dose (PS concentration vs light fluence), as well as drug-light interval (DLI). Many reports have shown that photosensitizers localize in different organelles (mitochondrion, lysosome, ER, melanosome and plasma membrane), and this is crucial in determining the predominant mechanism of cell death.Citation32,Citation35,Citation50 The subcellular localization of a PS may be determined by its ionic status, with cationic photosensitizers accumulating in the mitochondrion, while anionic ones accumulate in the lysosome.Citation37,Citation50 All other conditions being equal, it is generally accepted that photo-oxidative reaction with photosensitizers localizing to the mitochondria lead to apoptosis, while PDT with compounds targeting the cytoplasmic membrane, lysosomes, ER or Golgi membrane leads to necrosis. The cell death mechanism activated following PDT may also be a function of the quantity and type of ROS generated, as well as the degree of oxidative damage. Many reports have established that mild oxidative damage increases the likelihood of cells dying by undergoing apoptosis. In contrast, severe damage hinders ATP production and the activation of apoptotic pathways, compelling the cell to undergo necrosis.Citation29,Citation51 Whether PDT elicits an apoptotic, necrotic or autophagic cell death may also be cell type-specific. For example, Davids et alCitation52 demonstrated that hypericin-mediated PDT triggers distinct mechanisms of cell death in melanoma cells that are dependent on the presence or absence of melanin. The study revealed that in melanocytes and pigmented UCT-MEL 1 cells, a necrotic mode of cell death was induced by hypericin-mediated PDT, while apoptosis is the cell death mechanism triggered in keratinocytes and unpigmented A375 cells. In a study with two variants of mouse B16 melanoma cell line, Sparsa et alCitation53 reported that 5-ALA-mediated PDT induced a p53-dependent apoptotic cell death in pigmented B16F10 cells and an autophagic response leading to a caspase-independent cell death in unpigmented B16G4F cells. Apoptotic cell death mediated by PDT may be via the extrinsic (activation of death receptors) or intrinsic (mitochondrial outer membrane permeabilization and subsequent release of cytochrome C) pathway. Whichever pathway dominates may also be a reflection of many factors, including type of PS and cell type. The effectiveness of photo-activated hypericin in killing different melanoma cell types was reported by Kleemann et al.Citation35 In the study, photo-activation of hypericin resulted in an extrinsic (A375, unpigmented) and intrinsic (UCT-MEL 1, pigmented) caspase-dependent apoptotic mode of cell death, as well as a caspase-independent apoptotic mode not involving apoptotic-inducing factor in 501 Mel melanoma cells. While the exact mechanistic pathways responsible for the elimination of apoptotic tumor cells following PDT are not fully understood, there is a broad agreement that the irradiation of a PS triggers intrinsic (mitochondrial-dependent) apoptosis through the classical pathway. This process involves inducing photo-damage and deactivating the anti-apoptotic Bcl-2 protein family, consequently causing the pro-apoptotic Bax and Bak proteins to translocate into the outer mitochondrial membrane. The resulting permeabilization of the mitochondrial membrane stimulates the release of cytochrome c, initiating a cascade-like activation of effector caspases. These activated caspases then propel the cell through an irreversible process leading to apoptosis.
Many studies have shown that autophagy is triggered in tumor cells during PDT, however it is still unclear if the process is a pro-death or a cytoprotective mechanism. Buytaert et alCitation54 reported that PDT can stimulate an autophagic cell death mechanism, at least under conditions of apoptosis inhibition in Bax-/- and Bak-/- murine embryonal fibroblasts (MEFs) subjected to hypericin-mediated PDT. Davids et alCitation55 additionally documented the activation of a mechanism of cytoprotective autophagy in melanoma cells as a response to oxidative stress induced by hypericin-mediated PDT. Cancer cells may respond to oxidative stress-based therapy including PDT, chemotherapy, and radiotherapy by initially inducing autophagy. However, persistent oxidative stress can overwhelm the cells’ endogenous antioxidant defense capacity, resulting in a transition from autophagy to a potential senescent phenotype in an effort to prolong cellular survival. This transition may ultimately lead the cells to die either by apoptosis or necrosis.Citation56–58 Additional evidence revealed that autophagy may contribute to PDT-induced apoptosis, with the two processes occurring independently of one another,Citation59 or simultaneously.Citation60,Citation61 Overall, the emerging consensus is that apoptotic, necrotic, and autophagic cell death may be induced by PDT in tumor cells and that the predominating pathway is a function of many variables including cell type, the type of PS, its physico-chemical properties and intracellular localization, light dose (local PS concentration vs fluence), drug-light interval (DLI), as well as oxygen concentration in tissues.
KEAP1/Nrf2 Signaling Pathway
Nrf2 (encoded by the gene, NFE2L2), a basic region-leucine zipper (bZIP)-type factor belonging to the Cap ‘n’ Collar (CNC) transcription factor family, plays an important role in the regulation of redox, protein and metabolic homeostasis.Citation62 Having a molecular weight of 68 kDa, Nrf2 is a protein containing 605 amino acids. Structurally, Nrf2 is composed of seven (7) highly conserved Nrf2 erythroid-derived cap “n” collar homology (ECH; Neh) domains (Neh1-7; ). Neh1 is a CNC-bZIP domain, and it plays several important roles. It is the region that is necessary for DNA binding at antioxidant response element/electrophile response element (ARE/EpRE) of gene promoters, as well as interaction with small MAF proteins.Citation63,Citation64 The domain is also important as it regulates the stability of Nrf2 by its association with UbcM2, a ubiquitin-conjugating enzyme.Citation64,Citation65 Neh2 domain is located at the N-terminal, and it contains DLG and ETGE amino acid motifs that are critical for binding to KEAP1 (Kelch-like ECH-associated protein 1), and seven ubiquitin-lysine rich residues for targeting Nrf2 for ubiquitination and proteosomal degradation.Citation64,Citation66 Neh3 domain found at the C-terminal, and Neh4 and Neh5 domains are all independent transactivation domains. A region within Neh3 has been reported to be associated directly with chromodomain helicase DNA-binding protein 6 (CHD6).Citation67 Both Neh4 and Neh5 domains of Nrf2 contain acid-rich residues where the cooperative binding of co-activators (such as CREB-binding protein (CBP) and steroid receptor co-activator 3 (SRC3) and/or co-repressors (such as glucocorticoid receptor; GR, and 3-hydroxy-3-methylglutaryl reductase degradation; HRD1)) take place to mediate activation and repression of Nrf2 signaling, respectively.Citation64,Citation68–72 Neh6 is a serine rich domain, and it is composed of DSGIS and DSAPGS amino acid motifs where β-transducin repeat-containing protein (β-TrCP) binds to promote Nrf2 ubiquitination and proteosomal degradation.Citation64,Citation73,Citation74 Direct binding of retinoid X receptor-alpha (RXRα) takes place at the Neh7 domain leading to repression of the transcriptional activation of Nrf2.Citation75
Figure 2 Protein structure of (A) Nrf2 and (B) KEAP1 showing different domains.
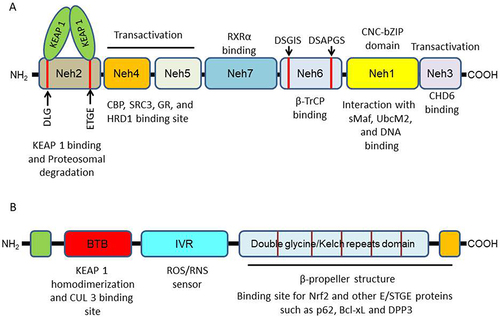
KEAP1, a dimeric protein primarily located in the cytoplasm, stands out as the most extensively studied intracellular regulator of Nrf2. Structurally, it consists of five conserved domains () namely, an N-terminal region (NTR), a conserved Broad-Complex/TramTrack/Bric-a-Brac (BTB) domain, an intervening region (IVR), a double glycine repeat (DGR) domain (comprising six subdomains, also known as the Kelch repeats domain), and a C-terminal region (CTR).Citation64,Citation76 The BTB domain is crucial for KEAP1 homodimerization and interaction with Cullin E3 ubiquitin ligase (CUL 3), a pivotal event for Nrf2 ubiquitination.Citation77,Citation78 The IVR domain, rich in cysteine, operates as a redox stress sensor.Citation79,Citation80 The DGR/Kelch repeats domain, in conjunction with the CTR, forms a six-bladed propeller structure necessary for binding with the Neh2 domain of Nrf2 and other proteins with ETGE and/or STGE conserved motifs, including p62, Bcl-xL, and dipeptidyl peptidase 3.Citation64,Citation81–83
Physiological Role of Nrf2
Nrf2 has historically functioned as the primary controller of the expression of cytoprotective genes, specifically those involved in antioxidant and detoxification processes. Nonetheless, emerging evidence suggests that Nrf2 possesses additional functions beyond its role in regulating redox homeostasis. In normoxic environments, Nrf2 typically remains confined to the cytoplasm, where it forms a complex with its inhibitory partner, KEAP1. KEAP1 functions as a substrate adaptor for the Cullin 3-based E3 ubiquitin ligase enzyme complex (Cul 3), facilitating the ubiquitination of Nrf2. This ubiquitination leads to the subsequent proteasomal degradation of Nrf2, thereby maintaining its basal levels at a minimum, and preventing the induction of target genes downstream of Nrf2. In the presence of a stressor such as ROS and small-molecule electrophiles, oxidation of the sulfhydryl groups in the Nrf2-binding domain of KEAP1 takes place. This led to a change in conformation which impairs the binding of Nrf2 to KEAP1. Consequently, the ubiquitination of Nrf2, and its subsequent proteosomal degradation is stopped. Thus, Nrf2 accumulates in the cytoplasm from where it is eventually translocated into the nucleus (). Once in the nucleus, Nrf2 heterodimerizes with the small musculo-aponeurotic fibrosarcoma (Smaf) protein. Subsequent binding of the Nrf2-sMAF complex to the antioxidant or electrophile response element (ARE/EpRE) sequence orchestrated the transcriptional regulation of numerous downstream target genes crucial for preserving cellular redox homeostasis.Citation84,Citation85 The downstream targets activated by Nrf2 include genes involved in (i) glutathione biosynthesis (glutamate cysteine ligase modifier subunit, GCLM; glutamate cysteine ligase catalytic subunit, GCLC; glutathione peroxidases, GPxs; glutathione reductase, GR), (ii) xenobiotic metabolism (glutathione-s-transferases, GSTs; microsomal epoxide hydroxylases, EH-1; NAD(P)H: quinone oxidoreductase 1, NQO1; and heme oxygenase-1, HO-1), (iii) scavenging of ROS (superoxide dismutase, SOD; catalase, CAT; thioredoxin reductase 1, Trxn 1; sulfiredoxin 1, Srxn 1; and peroxiredoxin 1, Prdx 1), (iv) drug efflux (multi-drug resistance protein (Mdrp) gene family), and (v) apoptosis and autophagy (p62, Bcl-2 and Bcl-xL).Citation86
Figure 3 Activation and response of Nrf2. Under normal condition, KEAP1 binds Nrf2 and package it for ubiquitination and proteosomal degradation with the help of Cullin 3-E3 ubiquitin ligase. In the presence of a stressor such as ROS and electrophiles, Nrf2 is activated and translocated into the nucleus where its binding to the antioxidant response element sequence leads to transcription of different cytoprotective genes.
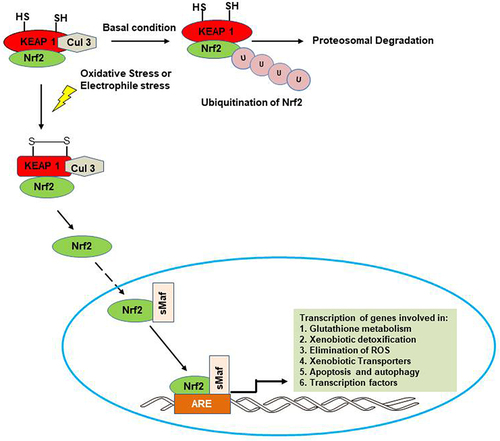
While the accepted cellular trigger for Nrf2 activation involves modifying the cysteine residues of KEAP1 through chemical adduction, oxidation, nitrosylation, or glutathionylation, emerging evidence suggests that Nrf2 activation can be regulated through alternative mechanisms. These include the phosphorylation of Nrf2 by upstream protein kinases such as PKC, PI3K/Akt, GSK-3β, and JNK, the binding of other proteins like caveolin and p21 to Nrf2, epigenetic modification of Nrf2 by microRNAs such as miR-144, miR-28, and miR-200a, as well as the direct interaction of heme with Bach1, facilitating the heterodimerization of Nrf2 with sMAF. These diverse mechanisms have all been reported to contribute to the repression and/or activation of Nrf2.Citation87,Citation88
Tumor Suppression Role of Nrf2 in Cancer
Because of the cytoprotective functions of target genes downstream of Nrf2, it is usually regarded as a vital controller of cellular defense against ROS- and electrophile-induced stress. The target genes expressed as a result of Nrf2 activation code for various enzymes that are involved in processes such as scavenging ROS and RNS, xenobiotic detoxification, xenobiotic efflux, apoptosis and autophagy among others. Normal functioning of the Nrf2/KEAP1 signaling pathway is vital to inhibiting oxidative stress and prevents damage to tissues and organs. Research findings have shown that mutations resulting in diminished Nrf2 activity can foster oxidative and inflammatory damage, consequently contributing to the promotion of various conditions like cardiovascular disorder, cancer, diabetes, neurodegenerative disease, and aging.Citation89 Reports showing that induction of Nrf2 can suppress tumorigenesis, especially in its early stages, are available in literature. Many studies using chemical agents, as well as genetic manipulations that promote Nrf2 activity, have demonstrated the suppression of the tumorigenesis process.Citation90 The anti-cancer properties of synthetic and naturally-occurring chemopreventive substances, which are known inducers of Nrf2, are well reported. Sulforaphane, a natural product obtained from broccoli, has been shown to inhibit the promotion and progression stage of tumorigenesis in different types of cancers via the activation of Nrf2/ARE-mediated genes.Citation14,Citation91–93 Using a LSL-KrasG12D/+;LSL-Trp53R172H/+;Pdx-1-Cre triple mutant (KPC) transgenic mouse model of pancreatic cancer, Liby et al,Citation94 reported that synthetic oleanane triterpenoids (potent chemopreventive and Nrf2 activity inducer) delay tumorigenesis and prolong survival by inhibiting the transcription of oncogenes including K-Ras and TP53 in the pancreas. Similarly, in a BRCA1 mutated mouse model of breast cancer, the same synthetic chemopreventive agent was reported to delay tumor development in the breast and extend survival BRCA1 mutated mice.Citation95
Further evidence of the tumor suppression role of Nrf2 was obtained from studies involving the use of Nrf2 knockout mouse models. Carcinogens-treated Nrf2 deficient mice were reported to show increase susceptibility to carcinogenesis and develop large number of tumors in the liver, fore-stomach and urinary bladder compared with wild-type mice.Citation96–99 Furthermore, Nrf2 knockout mice were reported to show increased susceptibility to colitis-associated colorectal cancer in contrast to their genetically unaltered counterparts.Citation100 Similarly, evidence from studies employing Nrf2 knockout mouse model revealed that Nrf2 is essential for the chemoprevention efficacy of oltipraz, 1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl] imidazole, and sulforaphane in different carcinogen-induced carcinogenesis models.Citation101–104 Another piece of evidence by Satoh et alCitation105 indicated that Nrf2 deficiency produces a conducive microenvironment for cancer cells metastasis to the lung, while its overexpression as a result of KEAP1 knockout increased resistance to metastasis of cancer cells to the lung. A reduced expression of Nrf2 has been reported in individuals with single nucleotide polymorphism (SNP) in the Nrf2 promoter region, making such individuals susceptible to increased risk of lung cancer.Citation106,Citation107 All these studies showed that Nrf2 may play an important role as an inhibitor of the tumorigenesis process and form the basis of the use of Nrf2 activators in clinical trials for the prevention of cancer and other diseases.
Oncogenic Role of Nrf2 in Cancer
Many studies, including those discussed above, have shown that Nrf2 is essential for chemopreventive agents to block carcinogenesis and protect normal cells from stressors such as endogenously generated ROS, radiation, environmental toxin and xenobiotics.Citation85,Citation101,Citation102 Recent evidence, however, has indicated that the role of Nrf2 is more complicated with its reported role in promoting cancer cell survival and resistance in many types of cancer therapy. Many reports have suggested that Nrf2 and its downstream targets are constitutively up-regulated in many tumors and this confers a survival advantage to mammalian cells under adverse conditions.Citation108–111 Similarly, overexpression of Nrf2 is associated with poor prognosis in some tumors.Citation112–114 This constitutive overexpression has been adduced primarily to somatic gain of function mutations in Nrf2 or loss of function mutations in KEAP1 which has been reported in many human cancers, including lung adenocarcinoma,Citation115,Citation116 hepatocellular carcinoma,Citation117,Citation118 breast,Citation119 colorectal,Citation120 gall bladder,Citation118 ovarian carcinomas,Citation112 skin squamous cell carcinoma,Citation121 endometrial,Citation122 and papillary adenocarcinoma.Citation123 Other mechanisms that lead to the constitutive activation of the Nrf2-KEAP1 pathway have also been reported. They include epigenetic modifications in the KEAP1 promoter, negative regulation of Nrf2-KEAP1 pathway by micro RNAs, aberrant accumulation of proteins disrupting Nrf2-KEAP1 interactions, as well as metabolism-induced KEAP1 modification.Citation63
Nrf2 activation might promote tumorigenesis in different ways, including mediating tumor cell proliferation. It is reported that constitutive activation of Nrf2 in tumor cells leads to increased expression of metabolic enzymes such as glucose-6-phosphate dehydrogenase, phosphogluconate dehydrogenase and transketolase. These enzymes promote glucose and glutamine metabolism in the pentose phosphate pathway and contribute to purine and amino acid synthesis, all key events driving metabolic reprogramming for tumor cell proliferation.Citation14,Citation124 There is evidence of a correlation between Nrf2 and tumor cell proliferation, with studies indicating that KEAP1−/− cell lines proliferate faster than their wild-type counterpart, while Nrf2−/− cells showed slower proliferation.Citation13,Citation109,Citation125–127 Furthermore, reduction in tumor cell proliferation associated with down-regulated Ki67 expression and p53-induced senescence has been reported with Nrf2 knockdown.Citation13,Citation128,Citation129 A study by Rojo et alCitation130 reported that loss of PTEN and subsequent inhibition of GSK-3, results in upregulation of PI3K-AKT and Nrf2 signaling, leading to increased proliferation rates and tumorigenicity in human endometrioid tumors. Functional crosstalk between oncogenic proteins (such as BRAF, KRAS and MYC) and Nrf2 signaling is another mechanism by which Nrf2 can promote cancer cell proliferation. Mutation in these oncogenic proteins is known to increase constitutive activation of Nrf2 in different tumors, including human pancreatic cancer.Citation18 Silencing Nrf2 in KRAS mutant cells inhibits oncogene-mediated proliferation and tumorigenesis in primary cells and tissues of mouse expressing KRASG12D and BRAFV619E and in human pancreatic cancer, indicating that Nrf2 plays proliferative role in tumorigenesis.Citation128
An important hallmark of cancers is their ability to evade apoptosis. Constitutive activation of Nrf2 promotes tumor development and progression by inhibiting apoptosis of cancer cells. Overexpression of Nrf2 in cancer cells results in transcription of gene coding for various cytoprotective enzymes which scavenge ROS and promote resistance to ROS-driven apoptosis. Interaction between Nrf2 and p53 signaling may also contribute to the ability of cancer cells to evade apoptosis. Over-activation of Nrf2 leads to transcription of downstream targets including x-CT, NQO1 and GST, all of which counteract the activity of p53, a tumor suppressor known for induction of cell growth arrest and apoptosis.Citation14,Citation131 Similarly, Nrf2 downstream targets, glutathione-s-transferase pi 1 (GSTP1) and p62 prevent apoptosis by blocking the activation of c-Jun N-terminal kinase (JNK), a pro-apoptosis protein,Citation132 and initiating selective autophagy of KEAP1.Citation14,Citation133 Further evidence from a study in mouse hepatoma (Hep1) and human hepatoblastoma (HepG2) cells revealed that Nrf2 induced an overexpression of Bcl-2 (B-cell lymphoma 2), leading to a repression in etoposide-induced apoptosis and increased cell survival.Citation134
Another mechanism through which Nrf2 contributes to tumorigenesis is via its role in metastasis. Constitutive Nrf2 activation in pancreatic adenosquamous carcinoma cells is reported to promote epithelial-to-mesenchymal (EMT) transition and invasion by lowering the expression of E-cadherin.Citation135 Silencing of Nrf2 suppresses radiation-induced invasion and migration by up-regulating E-cadherin expression and down-regulating N-cadherin expression in non-small cell lung cancer.Citation136 Further evidence revealed that constitutively activated Nrf2 suppressed the degradation of Bach 1, a pro-metastatic transcription factor, thereby promoting lung cancer metastasis.Citation18,Citation137 In addition, Nrf2 over-activation promotes proliferation and metastasis in MCF-7 and MDA-MB-231 human breast cancer cell lines by increasing RhoA gene expression.Citation127 It has also been reported that Nrf2 overexpression-induced metastasis is mediated via matrix metalloproteases, as evidence showed that deletion of Nrf2 mediated by shRNA repressed matrix metalloprotease 2 (MMP 2) expression and increased E-cadherin expression, leading to decreased invasion and migration in esophageal squamous cell carcinoma.Citation138 Surprisingly, a number of studies have shown that Nrf2 might as well play anti-metastatic role in cancers. During metastatic process in human prostate cancer, a reduction in the expression of Nrf2 and its target genes were reported.Citation139 Similarly, loss of Nrf2 increases migration, invasion and metastasis mediated by up-regulated SMAD signaling in hepatocellular carcinoma (HCC) cell lines.Citation140 In A549 lung cancer cell lines, Nrf2 inhibition facilitated TGF-β1-induced cell motility or migration via NOX4-ROS signaling.Citation141 These studies indicate that the pro- or anti-metastatic role of Nrf2 in cancer is not unequivocal, thus further studies are still warranted.
Development of intrinsic and acquired resistance to conventional treatments such as chemotherapy and radiation therapy is the most significant role of Nrf2 in tumorigenesis. Treatment of cancer cells with chemotherapeutic drugs including cisplatin, doxorubicin, etoposide, paclitaxel and others, as well as radiation therapy, induced higher ROS level to kill cancer cells. In response to high ROS level in cancer cells, Nrf2 mediates the transcription of several cytoprotective genes, including those coding for ROS scavenging enzymes, Phase II detoxifying enzymes, drug resistance proteins, drug efflux transporters, and other stress response molecules. The induction of these genes therefore provides avenue for survival to cancer cells. A lot of evidence has connected aberrant activation of Nrf2 to development of therapeutic resistance and recurrence in different cancers including breast, colorectal, lung, leukemia, liver, pancreatic and ovarian cancer.Citation15 Stable overexpression of Nrf2, as well as Nrf2 activation by tert-butylhydroquinone increases the resistance of human breast adenocarcinoma and neuroblastoma cells to doxorubicin, etoposide and cisplatin.Citation14,Citation142 Data from same study also revealed that genetic knockdown of Nrf2 sensitizes lung carcinoma cells to all three chemotherapeutic drugs by repressing the mRNA expression of NQO1 and glutathione.Citation142 Studies in human doxorubicin-resistant ovarian carcinoma cells indicated an elevated Nrf2 level compared with wild-type control cells.Citation143 Similarly, other studies reported that cisplatin- and carboplatin-resistant lung carcinoma cells were also characterized by upregulated Nrf2 level.Citation115,Citation144
A key mechanism through which Nrf2 drives resistance to chemotherapy and radiation therapy in cancer cells is via upregulation of endogenous antioxidants. Emerging evidence in most cancers points to the induction of antioxidant defense as having a role to play in resistance to most cancer therapies. Cancer cells are believed to endure persistent oxidative stress, a condition thought to contribute to tumor progression. Several studies have demonstrated that cancer cells display higher levels of ROS relative to normal cells.Citation145–148 The heightened ROS levels, and consequently, the consistently elevated oxidative stress in cancer cells, explain the presence of an elaborate and sophisticated intracellular antioxidant network. This characteristic has led many to hypothesize that it could enhance the resistance of cancer cells to chemotherapy and radiotherapy. Induction of Nrf2 and resultant expression of downstream antioxidant proteins has been associated with resistance to chemotherapy and radiotherapy. In human colorectal cancer cells, it is reported that activation of Nrf2 and subsequent stimulation of thioredoxin reductase 1 (TRX1) reduces ROS generation and drive resistance to 5 fluorouracil.Citation149 Studies on NSCLC and small cell lung carcinoma (SCLC) cells revealed that increasing resistance to cisplatin is due to decreased platinum-DNA binding and intracellular platinum accumulation driven by elevated levels of antioxidants.Citation150–152 In addition, Kim et alCitation153 reported that persistent activation of the Nrf2/ARE pathway is the driver for the enhanced expression of antioxidant proteins such as GCL, HO-1, thioredoxin and peroxiredoxin 1 observed in tamoxifen-resistant breast cancer cells. Furthermore, resistance to 5-FU and γ-irradiation in esophageal squamous carcinoma was attributed to an upregulation of GCLC and GSR caused by a gain of function mutation in Nrf2.Citation154 Other studies have shown that NQO1 an important downstream target of Nrf2 was upregulated between 2-3-fold in chemoresistant lung, colon, pancreas, and breast cancers.Citation15,Citation155–158 The role and contribution of another Nrf2 downstream target GSTP1 to chemoresistance was highlighted in breast cancer where its overexpression is reported to be an unfavorable marker for aggressiveness and poor prognosis.Citation159,Citation160 Further evidence revealed that the expression of GPx1 and GPx2, both members of the glutathione peroxidase (GPx) family were upregulated in cisplatin-resistant NSCLC and A549 cell lines, respectively.Citation161,Citation162 Evidence also showed that resistance to gemcitabine in pancreatic ductal adenocarcinoma (PDAC) can be attributed to a reduction in ROS generation due to antioxidant expression, especially increase in isocitrate dehydrogenase 1 (IDH1) expression.Citation163 Similar to these findings, resistance to gemcitabine in pancreatic cancer cells was reported to be due to inhibition of basal and gemcitabine-induced ROS formation by overexpression of SOD2 and CAT, both ROS-scavenging enzymes.Citation164 Conversely, several studies have shown that genetic or pharmacological inhibition of Nrf2 was able to repress Nrf2-mediated antioxidant gene expression, and thus increase the sensitivity of different tumors to chemotherapeutic drugs and radiotherapy.Citation66 Inhibition of Nrf2 by siRNA repressed the expression of GSH, thioredoxin 1, peroxiredoxin 1 and NQO1, thus increasing the sensitivity of chemoresistant ovarian cancer cells to cisplatin action.Citation165 Similarly in resistant prostate cancer cells, reduction in GSH content due to Nrf2 genetic inhibition enhances the vulnerability of the cells to chemotherapy and radiotherapy.Citation166 Also enhanced sensitization to doxorubicin was observed in human NSCLC A549 cells when GSH level was down-regulated as a result of pharmacological inhibition of Nrf2 by luteolin.Citation167
Another mechanism by which Nrf2 facilitates resistance to conventional therapies is via its crucial role in upregulating the expression of genes that encode drug efflux transporters. Constitutive activation of Nrf2 mediates an overexpression of various drug transport proteins such as multidrug resistance protein 1 (MDR1/ABCB1), multidrug resistance-associated proteins (MRPs/ABCCs), and breast cancer resistance protein (BCRP/ABCG2) across different tumor types.Citation14,Citation15,Citation18 These drug efflux pumps are members of the ATP-binding cassette (ABC) membrane transporter superfamily, and their upregulation decreases chemosynthetic drug accumulation in tumor cells, thus they are associated with chemoresistance in a number of tumors.Citation14,Citation15,Citation158 Constitutive induction of ABCC3, ABCG2 and P-glycoprotein are associated with poor prognosis characterized by poor or incomplete response, disease-free response and overall survival in adults with acute myeloid leukemia.Citation168,Citation169 Evidence has shown that ABCC1 was overexpressed in CUL3-silenced breast cancers and this confers resistance to paclitaxel and doxorubicin.Citation18,Citation170 Aberrant expression of ABCC1 resulting from Nrf2 activation promotes the efflux of doxorubicin leading to about 2-fold decrease in doxorubicin cytotoxicity in ovarian cancer, leukemia and NSCLC cells.Citation143,Citation171,Citation172 Resistance to sorafenib-induced apoptosis in hepatocellular carcinoma (HCC) was reported to be associated with Nrf2-induced upregulation of ABCC1, ABCC2 and MDR1.Citation173 A recent study showed that in tumor tissues from glioma patients, expression of Nrf2 correlates positively with ABCC1 (MRP1) expression and this is associated with tumor aggressiveness, chemoresistance and poor overall survival.Citation174 Similarly, Nrf2-driven induction of ABCC2 has been implicated in tamoxifen-resistant breast cancer cells and in different cisplatin-resistant human cancer cells.Citation175–177 Nrf2-mediated upregulation of other ABCCs, including ABCC3, ABCC4 and ABCC5 was reported to be the cause of intrinsic resistance to different anticancer drugs (including cisplatin, doxorubicin, etoposide and vincristine) in NSCLC, gastric cancer and hepatocarcinoma cells.Citation172,Citation177–179 ABCG2 (BCRP) is an ATP hydrolysis (energy)-dependent membrane transporter, and its expression is regulated by Nrf2 binding with ARE area of its promoter region.Citation15 Some studies revealed an upregulated expression of ABCG2 (between 3-4-fold) in cancer cells with elevated expression of Nrf2. This aberrant expression of ABCG2 was reported to contribute to chemoresistance in those cancer cells.Citation14,Citation180 Strong correlation between ABCG2 expression and resistance to 5-FU and irinotecan has been reported in breast and colorectal cancer, respectively.Citation181 In addition, ABCG2 was reported to be overexpressed in BRAF (V600E) mutant A375 melanoma cells and this confers acquired resistance to vemurafenib.Citation182 Other evidence for the role of ABCG2 in chemoresistance comes from genetic inhibition studies. Nrf2-silenced lung cancer cells showed a down-regulated ABCG2 expression which enhanced their sensitivity to mitoxantrone and topotecan.Citation183 Furthermore, a recent study revealed that the knockdown of Nrf2 repressed ABCG2 expression and reversed the resistance of PDAC cells to 5-fluorouracil.Citation184
Beyond adaptation due to upregulation of endogenous antioxidants and expression of drug efflux transporters, Nrf2 mediates resistance to chemotherapy and other conventional treatments in tumor cells via other mechanisms. The expression of many components of the autophagic system, a known pro-resistance process in tumor cells, is directly regulated by Nrf2. Evidence has shown that Nrf2 induces autophagy via upregulation of p62 with eventual reduction of ROS, and apoptosis inhibition in some tumor cells.Citation185,Citation186 In addition, promotion of angiogenesis and enhancing cancer stem cells (CSC) self-renewal are other mechanism by which Nrf2 promotes resistance to cancer therapies.Citation14,Citation18
Interestingly, the bi-directional mode of action of Nrf2 in tumorigenesis has brought into a clear perspective the importance of the crosstalk between its onco-suppressive and oncogenic roles. This is an indication that maintaining Nrf2 at its optimal level is vital for implementing effective anticancer strategies. The findings from the studies reviewed above have shown that this bidirectional mode of action may be influenced by the genetic makeup of negative regulators of Nrf2 such as KEAP1, as well as the functional crosstalk between Nrf2 and other signaling pathways including P13K/Akt/mTOR, GSK-3β, KRAS/BRAF/c-MYC, p53, NOTCH, STAT3 and NF-κB. The tumor-suppressor role of Nrf2 and its aberrant activation in promoting oncogenesis and mediating therapeutic resistance in different cancer types is complex, and it is a subject that has been addressed in detail in many excellent review articles available in literature.Citation13–18,Citation62
Nrf2, Cytoprotective Proteins and Resistance to Photodynamic Therapy in Cancers
Because of the multi-target mode of action of the PDT process, it is previously believed that the possibility of treated cells to develop protective strategy will be minimal. Thus, PDT is generally assumed to have a low probability of selecting for photo-resistant strain cells. However, more recent data are suggesting that PDT might also induce the cytoprotective mechanisms used by cancer cells to avoid the cytotoxic effect of traditional therapies. Generally, cancer cells are constitutively able to sustain ROS level in the non-toxic range due to the fact that the high ROS level co-evolves with a well-developed and efficient detoxifying antioxidant system. Consequently, this resilient antioxidant system endows tumor cells with high adaptability to oxidative stress-based therapies, including PDT. Although report on therapeutic resistance and the activated survival mechanisms in tumor cells after PDT is very limited, Broekgaarden et alCitation30 postulated that the survival of tumor cells post-PDT involves a number of interlinked survival pathways, of which antioxidant response mediated by the Nrf2 transcription factor is one. It is a well-known fact that ROS activates the Nrf2 pathway; however, reports on PDT activation of Nrf2 in tumors are limited. Some studies have reported on the activation of Nrf2 following PDT using different photosensitizer in certain tumors. Activation and translocation of Nrf2 into the nucleus was observed in T24 human bladder and HeLa cells following hypericin-mediated PDT,Citation187 as observed by us in pigmented MEL 1 and unpigmented A375 melanoma cells.Citation188 Additionally, Nrf2 downstream target genes including SOD, GCLc, GCLm, NQO 1 and HO-1 have been reported to be upregulated in many cancers after PDT.Citation51,Citation189,Citation190 We also observed similar upregulation in mRNA expression and activity of several Nrf2 downstream targets including SOD 1, SOD 2, GPx, TrX 1, CAT and GCLc after hypericin-mediated PDT (HYP-PDT) in melanoma cells.Citation188 The antagonistic nature of antioxidant defense mechanisms to PDT was further highlighted in other studies. Human breast carcinoma MCF-7 cells transfected with GPx gene were shielded from PDT-induced damage after exposure to singlet oxygen.Citation191 Furthermore, recent studies demonstrated the heterogeneity in the cellular response to 5-ALA-PDT and identified GPx 4 as a possible biomarker and an effector of PDT resistance in breast cancer and glioblastoma cell lines.Citation192,Citation193 In addition, HYP-PDT-induced cytotoxicity in human embryonic kidney (HEK)-293 cells over-expressing GSTP1 was reported to be diminished compared to wild-type HEK-293 cells.Citation194 These findings suggest that the upregulation of these genes is cytoprotective and may confer resistance to cancer cells after PDT.
The ability of many tumors to exploit the Nrf2-KEAP 1 pathway for survival and resistance to therapies, including PDT suggest that inhibition strategies for Nrf2 and its targets might attenuate these effects. Nrf2 knockdown increased ROS level and decreased HO-1 expression and thus increased the sensitivity of human ovarian cancer cells to methyl pyropheophorbide a (mPPa)-mediated PDT.Citation195 Also, Choi et alCitation196 reported that as a result of Nrf2 knockdown in multiple cancer cells, production of singlet oxygen and other ROS was increased leading to enhanced pheophorbide a (Pba)-PDT-mediated cytotoxicity and increased apoptotic/necrotic cell death of the cancer cells. Furthermore, data from a recent study revealed that pharmacological inhibition of Nrf2 by luteolin sensitizes human pancreatic and bladder T24 cancer cells to porphyrin TMPyP4-mediated PDT. Results from the study indicated that inhibition of Nrf2 by luteolin generates high ROS levels and favors apoptosis by upregulating RKIP expression, while SNAIL expression was downregulated.Citation197 The takeaway here is that when Nrf2 activity is inhibited, PDT efficacy is enhanced, suggesting a central role for Nrf2 in PDT resistance in cancers.
Specific inhibition of downstream targets of Nrf2 has also been used to shed light on the crucial role of Nrf2-mediated antioxidant response as a possible mechanism of resistance to PDT. Kimani et alCitation198 reported that different antioxidant inhibitors of the glutathione redox system and ROS scavenging enzymes potentiate the cytotoxicity of disulfonated aluminium-phthalocyanine mediated PDT in breast cancer cells. Similarly, inhibition of glutathione by buthionine sulfoximine (BSO) and carnosine enhanced the sensitivity of human breast adenocarcinoma cell lines, MDA-MB-231 and MCF7, respectively, to hypericin-mediated PDT.Citation199 A new approach to reduce GSH in tumor tissues relieves hypoxia and enhances the ROS killing effect of PDT by employing multifunctional nanosystem carriers that utilize meta-elements such as copper (Cu), manganese (Mn), iron (Fe) and gold (Au) has been reported to be effective in enhancing tumor cell destruction by PDT.Citation200–203 HO-1 is a downstream target of Nrf2 which has been closely associated with tumor cell survival post-PDT. The addition of an anti-HO-1 siRNA to WM541Lu human melanoma cells, 24 h prior to PDT increases the responsiveness of the melanoma cells to ALA-based PDT.Citation204 Zinc(II) protoporphyrin IX, an inhibitor of HO-1 significantly enhanced PDT-mediated cytotoxicity towards human ovarian carcinoma cells.Citation190 Another study employing siRNa knockdown of HO-1 showed an increased susceptibility of UM-UC-3 human urothelial cell line to ALA-mediated PDT.Citation205 Further evidence was provided by Kimakova et al,Citation206 when they demonstrated that resistance to HYP-PDT induced by SOD 2 over-expression in MCF-7 cells was abrogated when the cells were pre-treated with 2-methoxyestradiol (2-ME). Their data revealed that 2-ME increased the sensitivity of the resistant MCF-7 breast cancer cells by decreasing the clonogenic ability of the cells and promoting their apoptosis. Similar improved potentiation of antitumor effects of PDT was observed in other previous studies after genetic or pharmacological inhibition of SOD 2 by 2-ME in neurons, fibroblasts or Jurkat cells.Citation189,Citation207,Citation208 These reports demonstrate that inhibition of tumor antioxidant system represent a useful approach to enhance the efficacy of photodynamic therapy in different cancers.
Similar to what has been reported during the development of resistance to chemotherapy and other conventional treatments, the expression of drug efflux transporters has been identified as a vital mechanism that can drive tumor survival post-PDT. Efflux proteins such as P-gp, ABCG2 and ABCCs are overexpressed in majority of cancers. They are known to excrete therapeutic drugs and prevent their accumulation in tumor cells favoring resistance of tumors to different treatment protocols. In the context of PDT, ABCG2 is the most studied drug efflux transporter. It has been implicated in mediating excretion of most photosensitizers, and thus enhances the survival of tumor cells.Citation30,Citation209 Reports have shown that ABCG2 is involved in the efflux of a number of PS including protoporphyrin IX, hematoporphyrin IX, chlorin e6, pheophorbide a, pyropheophorbide a methyl ester, hypericin and aminoacridine.Citation209,Citation210
The importance of the Nrf2-ABCG2 axis in response to PDT-induced ROS generation, restoration of redox homeostasis and induction of tumor survival post-PDT has been highlighted in a number of studies. Overexpression of ABCG2 due to photoactivation of porphyrins in HepG2 cells was downregulated by Nrf2 siRNA knockdown suggesting that Nrf2 is a transcriptional inducer of ABCG2.Citation88,Citation211 Similar inhibition of ABCG2 expression coupled with increased intracellular Pba level and enhanced cytotoxicity was observed after Nrf2 knockdown in breast cancer MDA-MB-231 and HT-29 cells subjected to Pba-mediated PDT.Citation196 Further evidence revealed that increasing ROS generation by inhibiting the Nrf2-ABCG2 axis enhanced the sensitivity of human ovarian cancer cells to mPPa-mediated PDT.Citation195
Many photosensitizers are known substrates of ABCG2, with evidence showing that ABCG2 affects the pharmacokinetics of these photosensitizers and thus, mediates their efflux. Therefore, many studies have reported on inhibition of ABCG2 to block the efflux of photosensitizers and enhance the sensitivity of tumor cells to PDT. One of the earliest ABCG2 inhibitor used in PDT is the protein tyrosine kinase inhibitor (TKI), Imatinib mesylate. According to results from Liu et al,Citation212 Imatinib treatment increased the uptake of 2-(1-hexyloxyethyl)-2-devinyl pyropheophorbide a (HPPa, Photochlor), PpIX, and benzoporphyrin derivative monoacid ring A (BPD-MA, Verteporfin) and enhanced the efficacy of PDT in murine radiation induced fibrosarcoma 1 (RIF-1)-ABCG2+ cells and in mice bearing subcutaneous RIF-1 tumors. Gefitinib, another TKI, inhibited ABCG2-mediated efflux of protoporphyrin IX and thus enhanced the efficacy of ALA-PDT in four different malignant brain tumor cells.Citation213 Kim et alCitation214 reported an enhancement of pyropheophorbide a (PPa)-mediated PDT in colon cancer cells when ABCG2 was pharmacologically inhibited. Results from their study revealed that in HT-29 (ABCG2 high) and SW480 (ABCG2 low) colon cancer cell lines, expression of ABCG2 was negatively correlated with treatment efficacy. Inhibition of ABCG2 using Ko-143 enhanced the sensitivity of HT-29 cells by blocking PPa efflux and increasing singlet oxygen formation. These results were confirmed in vivo when they demonstrated that Ko-143 pre-treatment to HT-29 cells xenografted nude mice enhanced the sensitivity to PPa-PDT.Citation214 Similarly, doxorubicin-resistant A270 ovarian cancer cells overexpressing ABCG2 were reported to be more resistant to PPa-PDT compared to non-resistant wild-type ovarian cancer cells.Citation215 Further data from the study revealed that activation of the c-MET/PI3K signaling pathway is likely associated with doxorubicin resistance. Subsequently, repression of the c-MET and PI3K/Akt pathway, as well as Ko-143 pre-treatment result in down-regulated ABCG2 expression, increased PPa accumulation, increased ROS generation and enhanced cytotoxicity of PPa-PDT to doxorubicin-resistant cells.
Hypericin is a substrate for ABCG2, and the strong affinity of ABCG2 for hypericin was first confirmed by Jendzelovsky et al,Citation216 when they observed an over-expression of ABCG2 in HT29 cells treated with hypericin. Pre-treated of HT29 cells with an ABCG2 inhibitor fumitremorgin C (FTC), revealed an enhanced accumulation of hypericin in the cells. Subsequent treatment of the HT29 cells with proadifen represses ABCG2 activity and enhances HYP-PDT induced oxidative stress and apoptosis of the HT29 cells. A follow-up study by the same group showed that ABCG2 over-expressing promyelocytic leukemia (HL60) cells showed increased ability to efflux hypericin and are more resistant to HYP-PDT. Ko-143 treatment block ABCG2 over-expression increased hypericin accumulation and enhanced the sensitivity of HL60 cells to HYP-PDT-induced apoptosis.Citation217 Similarly, Khot et alCitation218 reported an overexpression of ABCG2 in HYP-mediated PDT-resistant 3D spheroidal modes of HT29 and HCT116 colorectal cancer cell lines compared to their respective 2D cell cultures. Furthermore, inhibiting ABCG2 activity as a result of co-treatment with Ko-143 enhanced the efficacy of HYP-PDT in the 3D spheroidal model of both cancer cell lines.Citation218 Combining HYP-PDT with inhibition of the 5-lipoxygenase (5-LOX) pathway (co-treatment with 5-LOX inhibitor, MK-886) was reported to decrease ABCG2 protein expression, increase intracellular accumulation of hypericin and improve the sensitivity of HT29 and MCF7 cancer cells to HYP-PDT.Citation219 Interestingly, a report from the laboratory of some of our collaborators indicated that HYP-PDT displayed differential ABCG2 expression in UCT Mel-1 (ABCG2 upregulation) and A375 (ABCG2 downregulation) melanoma cells. The observed increase in the expression of ABCG2 in A375 cells, however, did not translate to increased resistance, suggesting that ABCG2 expression may not influence HYP-PDT in those cell lines.Citation220 Overall, the studies reviewed here indicated that inhibition of ABCG2 to block the efflux of photosensitizers has emerged as a promising strategy to enhance the sensitivity of cancer cells to PDT. Inhibition of ABCG2 leads to increased intracellular accumulation of photosensitizers, higher ROS generation, and enhanced cytotoxicity, ultimately improving the effectiveness of PDT in different cancer types.
Conclusion and Future Perspectives
Photodynamic therapy (PDT) is a treatment modality for cancer that involves the use of photosensitizers, light, and oxygen to generate reactive oxygen species (ROS) that induce tumor cell death. However, the efficacy of PDT can be limited by the development of drug resistance. Nrf2 is a transcription factor that regulates the expression of genes involved in the cellular antioxidant response. Activation of Nrf2 in tumor cells can lead to the upregulation of expression of genes coding for antioxidant enzymes and drug efflux transporters. Antioxidant enzymes such as superoxide dismutase (SOD) and catalase, glutathione peroxidase, and heme oxygenase 1 can scavenge ROS generated by PDT, while drug efflux transporters can pump photosensitizer drugs out of tumor cells, thus limiting their accumulation and efficacy. Several studies reviewed here provide evidence showing that Nrf2 is aberrantly expressed in cancer cells resistant to PDT. Similarly, we reported evidence of overexpression of downstream targets of Nrf2, including various drug efflux transporters in cancer cells that are resistant to PDT. While the molecular mechanism(s) underlying the resistance of tumor cells to PDT may not be fully understood yet, overwhelming evidence has pointed to Nrf2-mediated antioxidant response and drug efflux transporter upregulation as possible mechanisms of resistance in PDT of cancers. To overcome this, strategy to downregulate the expression of Nrf2, including combining PDT with Nrf2 inhibitors to increase the cytotoxicity and efficacy of PDT needed to be vigorously pursued. Also, encapsulation of photosensitizers within nanocarriers in order to enhance their distribution to target tissues will help to protect PS from drug efflux transporters, and thus block a major limitation to PDT efficacy.
Consent for Publication
The details of images can be published and all authors provide consent for the article contents to be published.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
Acknowledgments
The authors acknowledge the assistance of Dr A.A. Oyagbemi with language editing of the manuscript.
Additional information
Funding
References
- Mattiuzzi C, Lippi G. Current cancer epidemiology. J Epidemiol Glob Health. 2019;9(4):217–222. doi:10.2991/jegh.k.191008.001
- Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
- World Health Organization. Global health estimates 2016: disease burden by cause, age, sex, by country and by region, 2000–2016. World Health Organization. 2018; Geneva. Available from: https://www.who.int/healthinfo/global_burden_disease/estimates/en/index1.html. Accessed August 30, 2021.
- Kalubula M, Shen H, Makasa M, Liu L. Epidemiology of cancers in Zambia: a significant variation in cancer incidence and prevalence across the nation. Malawi Med J. 2021;33(3):186–195. doi:10.4314/mmj.v33i3.6
- Kraybill WG, Harris J, Spiro IJ, et al. Long-term results of a Phase 2 study of neoadjuvant chemotherapy and radiotherapy in the management of high-risk, high-grade, soft tissue sarcomas of the extremities and body wall: radiation therapy oncology group trial 9514. Cancer. 2010;116(19):4613–4621. doi:10.1002/cncr.25350
- Uramoto H, Tanaka F. Recurrence after surgery in patients with NSCLC. Transl Lung Cancer Res. 2014;3(4):242–249. doi:10.3978/j.issn.2218-6751.2013.12.05
- Mallick S, Benson R, Hakim A, Rath GK. Management of glioblastoma after recurrence: a changing paradigm. J Egypt Natl Cancer Inst. 2016;28(4):199–210. doi:10.1016/j.jnci.2016.07.001
- Corrado G, Salutari V, Palluzzi E, Distefano MG, Scambia G, Ferrandina G. Optimizing treatment in recurrent epithelial ovarian cancer. Expert Rev Anticancer Ther. 2017;17(12):1147–1158. doi:10.1080/14737140.2017.1398088
- van der Merwe M, van Niekerk G, Fourie C, du Plessis M, Engelbrecht A-M. The impact of mitochondria on cancer treatment resistance. Cell Oncol. 2021;44(5):983–995. doi:10.1007/s13402-021-00623-y
- Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5(3):219–234. doi:10.1038/nrd1984
- Das CK, Mandal M, Kögel D. Pro-survival autophagy and cancer cell resistance to therapy. Cancer Metastasis Rev. 2018;37(4):749–766. doi:10.1007/s10555-018-9727-z
- Nikolaou M, Pavlopoulou A, Georgakilas AG, Kyrodimos E. The challenge of drug resistance in cancer treatment: a current overview. Clin Exp Metastasis. 2018;35(4):309–318. doi:10.1007/s10585-018-9903-0
- de la Vega MR, Chapman E, Zhang DD. NRF2 and the hallmarks of cancer. Cancer Cell. 2018;34(1):21–43. doi:10.1016/j.ccell.2018.03.022
- Wu S, Lu H, Bai Y. Nrf2 in cancers: a double‐edged sword. Cancer Med. 2019;8(5):2252–2267. doi:10.1002/cam4.2101
- Bai X, Chen Y, Hou X, Huang M, Jin J. Emerging role of NRF2 in chemoresistance by regulating drug-metabolizing enzymes and efflux transporters. Drug Metab Rev. 2016;48(4):541–567. doi:10.1080/03602532.2016.1197239
- Cykowiac M, Krajka-kuzniak V. Role of Nrf2 in pancreatic cancer. Antioxidants. 2022;11(1):98. doi:10.3390/antiox11010098
- Xue D, Zhou X, Qiu J. Emerging role of NRF2 in ROS-mediated tumor chemoresistance. Biomed Pharmacother. 2020;131:110676. doi:10.1016/j.biopha.2020.110676
- Choi BH, Kim JM, Kwak MK. The multifaceted role of NRF2 in cancer progression and cancer stem cells maintenance. Arch Pharm Res. 2021;44(3):263–280. doi:10.1007/s12272-021-01316-8
- Weijer R, Broekgaarden M, van Golen RF, et al. Low-power photodynamic therapy induces survival signaling in perihilar cholangiocarcinoma cells. BMC Cancer. 2015;15(1):1–17. doi:10.1186/s12885-015-1994-2
- Agostinis P, Berg K, Cengel KA, et al. Photodynamic therapy of cancer: an update. CA Cancer J Clin. 2011;61(4):250–281. doi:10.3322/caac.20114
- Baldea I, Filip AG. Photodynamic therapy in melanoma-An update. J Physiol Pharmacol. 2012;63(2):109–118.
- Van Straten D, Mashayekhi V, De Bruijn HS, Oliveira S, Robinson DJ. Oncologic photodynamic therapy: basic principles, current clinical status and future directions. Cancers. 2017;9(2):19. doi:10.3390/cancers9020019
- Manda G, Hinescu ME, Neagoe IV, et al. Emerging therapeutic targets in oncologic photodynamic therapy. Curr Pharm Des. 2018;24(44):5268–5295. doi:10.2174/1381612825666190122163832
- Rapozzi V, D’Este F, Xodo LE. Molecular pathways in cancer response to photodynamic therapy. J Porphyr Phthalocyanines. 2019;23(04n05):410–418. doi:10.1142/S1088424619300064
- Martins WK, Belotto R, Silva MN, et al. Autophagy regulation and photodynamic therapy: insights to improve outcomes of cancer treatment. Front Oncol. 2021;10:610472. doi:10.3389/fonc.2020.610472
- Manda G, Isvoranu G, Comanescu MV, Manea A, Butuner BD, Korkmaz KS. The redox biology network in cancer pathophysiology and therapeutics. Redox Biol. 2015;5:347–357. doi:10.1016/j.redox.2015.06.014
- Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8(7):579–591. doi:10.1038/nrd2803
- Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat. 2004;7(2):97–110. doi:10.1016/j.drup.2004.01.004
- Benov L. Photodynamic therapy: current status and future directions. Med Princ Pract. 2015;24(Suppl. 1):14–28.
- Broekgaarden M, Weijer R, van Gulik TM, Hamblin MR, Heger M. Tumor cell survival pathways activated by photodynamic therapy: a molecular basis for pharmacological inhibition strategies. Cancer Metastasis Rev. 2015;34(4):643–690. doi:10.1007/s10555-015-9588-7
- Davids LM, Kleemann B. Combating melanoma: the use of photodynamic therapy as a novel, adjuvant therapeutic tool. Cancer Treat Rev. 2011;37(6):465–475. doi:10.1016/j.ctrv.2010.11.007
- Abrahamse H, Hamblin MR. New photosensitizers for photodynamic therapy. Biochem J. 2018;473(4):347–364.
- Almeida RD, Manadas BJ, Carvalho AP. Duarte CB Intracellular signaling mechanisms in photodynamic therapy. Biochim Biophys Acta Rev Cancer. 2004;1704(2):59–86.
- Kiesslich T, Plaetzer K, Oberdanner CB, Berlanda J, Obermair FJ, Krammer B. Differential effects of glucose deprivation on the cellular sensitivity towards photodynamic treatment-based production of reactive oxygen species and apoptosis-induction. FEBS Lett. 2005;579(1):185–190. doi:10.1016/j.febslet.2004.11.073
- Kleemann B, Loos B, Scriba TJ, Lang D, Davids LM. St John’s Wort (Hypericum perforatum L.) photomedicine: hypericin-photodynamic therapy induces metastatic melanoma cell death. PLoS One. 2014;9(7):e103762. doi:10.1371/journal.pone.0103762
- Wyld L, Reed M, Brown N. Differential cell death response to photodynamic therapy is dependent on dose and cell type. Br J Cancer. 2001;84(10):1384. doi:10.1054/bjoc.2001.1795
- Castano AP, Demidova TN, Hamblin MR. Mechanisms in photodynamic therapy: part one-photosensitizers, photochemistry and cellular localization. Photodiagnosis Photodyn Ther. 2004;1(4):279–293. doi:10.1016/S1572-1000(05)00007-4
- Dąbrowski JM, Arnaut LG, Pereira MM, et al. Combined effects of singlet oxygen and hydroxyl radical in photodynamic therapy with photostable bacteriochlorins: evidence from intracellular fluorescence and increased photodynamic efficacy in vitro. Free Radic Biol Med. 2012;52(7):1188–1200. doi:10.1016/j.freeradbiomed.2011.12.027
- Ajuwon OR, Marnewick JL, Davids LM. Rooibos (Aspalathus linearis) and its major flavonoids - potential against oxidative stress-induced conditions. In: Gowder S, editor. Basic Principles and Clinical Significance of Oxidative Stress. Rijeka: Intech Europe; 2015:171–218.
- Mroz P, Pawlak A, Satti M, et al. Functionalized fullerenes mediate photodynamic killing of cancer cells: type I versus Type II photochemical mechanism. Free Radic Biol Med. 2007;43(5):711–719. doi:10.1016/j.freeradbiomed.2007.05.005
- Otake E, Sakuma S, Torii K, et al. Effect and mechanism of a new photodynamic therapy with glycoconjugated fullerene. Photochem Photobiol. 2010;86(6):1356–1363. doi:10.1111/j.1751-1097.2010.00790.x
- Sakharov D, Elstak E, Chernyak B, Wirtz K. Prolonged lipid oxidation after photodynamic treatment. Study with oxidation‐sensitive probe C11‐BODIPY581/591. FEBS Lett. 2005;579(5):1255–1260. doi:10.1016/j.febslet.2005.01.024
- Valko M, Rhodes C, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160(1):1–40. doi:10.1016/j.cbi.2005.12.009
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39(1):44–84. doi:10.1016/j.biocel.2006.07.001
- Butterfield DA, Dalle‐Donne I. Redox proteomics: from protein modifications to cellular dysfunction and disease. Mass Spectrom Rev. 2014;33(1):1–6. doi:10.1002/mas.21404
- Baldea I, Olteanu DE, Bolfa P, et al. Efficiency of photodynamic therapy on WM35 melanoma with synthetic porphyrins: role of chemical structure, intracellular targeting and antioxidant defense. J Photochem Photobiol B: Biol. 2015;151:142–152. doi:10.1016/j.jphotobiol.2015.07.019
- Baldea I, Olteanu DE, Bolfa P, Tabaran F, Ion R-M, Filip GA. Melanogenesis and DNA damage following photodynamic therapy in melanoma with two meso-substituted porphyrins. J Photochem Photobiol B: Biol. 2016;161:402–410. doi:10.1016/j.jphotobiol.2016.06.012
- Cooke MS, Loft S, Olinski R, et al. Recommendations for standardized description of and nomenclature concerning oxidatively damaged nucleobases in DNA. Chem Res Toxicol. 2010;23(4):705–707. doi:10.1021/tx1000706
- Al-Mutairi DA, Al-Mutairi DA, Craik JD, Batinic-Haberle I, Benov LT. Induction of oxidative cell damage by photo-treatment with zinc meta N-methylpyridylporphyrin. Free Radic Res. 2007;41(1):89–96. doi:10.1080/10715760600952869
- Fonda-Pascual P, Moreno-Arrones OM, Alegre-Sanchez A, et al. In situ production of ROS in the skin by photodynamic therapy as a powerful tool in clinical dermatology. Methods. 2016;109:190–202. doi:10.1016/j.ymeth.2016.07.008
- Buytaert E, Dewaele M, Agostinis P. Molecular effectors of multiple cell death pathways initiated by photodynamic therapy. Biochim Biophys Acta Rev Cancer. 2007;1776(1):86–107. doi:10.1016/j.bbcan.2007.07.001
- Davids LM, Kleemann B, Kacerovská D, Pizinger K, Kidson SH. Hypericin phototoxicity induces different modes of cell death in melanoma and human skin cells. J Photochem Photobiol B: Biol. 2008;91(2):67–76. doi:10.1016/j.jphotobiol.2008.01.011
- Sparsa A, Bellaton S, Naves T, et al. Photodynamic treatment induces cell death by apoptosis or autophagy depending on the melanin content in two B16 melanoma cell lines. Oncol Rep. 2013;29(3):1196–1200. doi:10.3892/or.2012.2190
- Buytaert E, Callewaert G, Hendrickx N, et al. Role of endoplasmic reticulum depletion and multidomain proapoptotic BAX and BAK proteins in shaping cell death after hypericin-mediated photodynamic therapy. FASEB J. 2006;20(6):756–758. doi:10.1096/fj.05-4305fje
- Davids LM, Kleemann B, Cooper S, Kidson SH. Melanomas display increased cytoprotection to hypericin-mediated cytotoxicity through the induction of autophagy. Cell Biol Int. 2009;33(10):1065–1072. doi:10.1016/j.cellbi.2009.06.026
- Gewirtz DA. Autophagy, senescence and tumor dormancy in cancer therapy. Autophagy. 2009;5(8):1232–1234. doi:10.4161/auto.5.8.9896
- Lu Z, Luo RZ, Lu Y, et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J Clin Invest. 2008;118(12):3917–3929. doi:10.1172/JCI35512
- Young AR, Narita M, Ferreira M, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23(7):798–803. doi:10.1101/gad.519709
- Kessel D, Oleinick NL. Initiation of autophagy by photodynamic therapy. Methods Enzymol. 2009;453:1–16.
- Kessel D, Arroyo AS. Apoptotic and autophagic responses to Bcl-2 inhibition and photodamage. Photochem Photobiol Sci. 2007;6(12):1290–1295. doi:10.1039/b707953b
- Kessel D, Vicente MGH, Reiners JJ. Initiation of apoptosis and autophagy by photodynamic therapy. Lasers Surg Med. 2006;38(5):482–488. doi:10.1002/lsm.20334
- Torrente L, DeNicola GM. Targeting NRF2 and its downstream processes: opportunities and challenges. Annu Rev Pharmacol Toxicol. 2022;62:279–300. doi:10.1146/annurev-pharmtox-052220-104025
- Menegon S, Columbano A, Giordano S. The dual roles of NRF2 in cancer. Trends Mol Med. 2016;22(7):578–593. doi:10.1016/j.molmed.2016.05.002
- Sánchez-Ortega M, Carrera AC, Garrido A. Role of NRF2 in lung cancer. Cells. 2021;10(8):1879. doi:10.3390/cells10081879
- Plafker KS, Nguyen L, Barneche M, Mirza S, Crawford D, Plafker SM. The ubiquitin-conjugating enzyme UbcM2 can regulate the stability and activity of the antioxidant transcription factor Nrf2. J Biol Chem. 2010;285(30):23064–23074. doi:10.1074/jbc.M110.121913
- Telkoparan-Akillilar P, Panieri E, Cevik D, Suzen S, Saso L. Therapeutic targeting of the NRF2 signaling pathway in cancer. Molecules. 2021;26(5):1417. doi:10.3390/molecules26051417
- Nioi P, Nguyen T, Sherratt PJ, Pickett CB. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol Cell Biol. 2005;25(24):10895–10906. doi:10.1128/MCB.25.24.10895-10906.2005
- Alam MM, Okazaki K, Nguyen LTT, et al. Glucocorticoid receptor signaling represses the antioxidant response by inhibiting histone acetylation mediated by the transcriptional activator NRF2. J Biol Chem. 2017;292(18):7519–7530. doi:10.1074/jbc.M116.773960
- Katoh Y, Itoh K, Yoshida E, Miyagishi M, Fukamizu A, Yamamoto M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells. 2001;6(10):857–868. doi:10.1046/j.1365-2443.2001.00469.x
- Kim JH, Yu S, Chen JD, Kong AN. The nuclear cofactor RAC3/AIB1/SRC-3 enhances Nrf2 signaling by interacting with transactivation domains. Oncogene. 2013;32(4):514–527. doi:10.1038/onc.2012.59
- Wu T, Zhao F, Gao B, et al. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014;28(7):708–722. doi:10.1101/gad.238246.114
- Zhang J, Hosoya T, Maruyama A, et al. Nrf2 Neh5 domain is differentially utilized in the transactivation of cytoprotective genes. Biochem J. 2007;404(3):459–466. doi:10.1042/BJ20061611
- Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, Hayes JD. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene. 2013;32(32):3765–3781. doi:10.1038/onc.2012.388
- Kang JS, Nam LB, Yoo OK, Keum YS. Molecular mechanisms and systemic targeting of NRF2 dysregulation in cancer. Biochem Pharmacol. 2020;177:114002.
- Wang H, Liu K, Geng M, et al. RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013;73(10):3097–3108. doi:10.1158/0008-5472.CAN-12-3386
- Tebay LE, Robertson H, Durant ST, et al. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med. 2015;88:108–146. doi:10.1016/j.freeradbiomed.2015.06.021
- Canning P, Cooper CD, Krojer T, et al. Structural basis for Cul3 protein assembly with the BTB-Kelch family of E3 ubiquitin ligases. J Biol Chem. 2013;288(11):7803–7814. doi:10.1074/jbc.M112.437996
- Zipper LM, Mulcahy RT. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J Biol Chem. 2002;277(39):36544–36552. doi:10.1074/jbc.M206530200
- Dinkova-Kostova AT, Holtzclaw WD, Cole RN, et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad of Sci. 2002;99(18):11908–11913. doi:10.1073/pnas.172398899
- Xie L, Gu Y, Wen M, et al. Hydrogen sulfide induces Keap1 S-sulfhydration and suppresses diabetes-accelerated atherosclerosis via Nrf2 activation. Diabetes. 2016;65(10):3171–3184. doi:10.2337/db16-0020
- Kopacz A, Kloska D, Forman HJ, Jozkowicz A, Grochot-Przeczek A. Beyond repression of Nrf2: an update on Keap1. Free Radic Biol Med. 2020;157:63–74. doi:10.1016/j.freeradbiomed.2020.03.023
- Li X, Zhang D, Hannink M, Beamer LJ. Crystal structure of the Kelch domain of human Keap1. J Biol Chem. 2004;279(52):54750–54758. doi:10.1074/jbc.M410073200
- Ogura T, Tong KI, Mio K, et al. Keap1 is a forked-stem dimer structure with two large spheres enclosing the intervening, double glycine repeat, and C-terminal domains. Proc Natl Acad Sci. 2010;107(7):2842–2847. doi:10.1073/pnas.0914036107
- Espinosa-Diez C, Miguel V, Mennerich D, et al. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015;6:183–197. doi:10.1016/j.redox.2015.07.008
- Furfaro A, Traverso N, Domenicotti C, et al. The Nrf2/HO-1 axis in cancer cell growth and chemoresistance. Oxid Med Cell Longev. 2016;2016:1958174. doi:10.1155/2016/1958174
- Milkovic L, Zarkovic N, Saso L. Controversy about pharmacological modulation of Nrf2 for cancer therapy. Redox Biol. 2017;12:727–732. doi:10.1016/j.redox.2017.04.013
- Bryan HK, Olayanju A, Goldring CE, Park BK. The Nrf2 cell defence pathway: keap1-dependent and-independent mechanisms of regulation. Biochem Pharmacol. 2013;85(6):705–717. doi:10.1016/j.bcp.2012.11.016
- Ishikawa T, Inoue Y, Ikegami Y, et al. A new strategy of ALA-photodynamic cancer therapy: inhibition of ABC transporter ABCG2. In: Efferth T, editor. Resistance to Targeted ABC Transporters in Cancer. Cham: Springer International Publishing; 2015:89–104.
- Egea J, González-Rodríguez Á, Gómez-Guerrero C, Moreno JA. Role of Nrf2 in disease: novel molecular mechanisms and therapeutic approaches. Front Pharmacol. 2019;10:1149. doi:10.3389/fphar.2019.01149
- Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12(8):564–571. doi:10.1038/nrc3278
- Dinkova-Kostova AT, Jenkins SN, Fahey JW, et al. Protection against UV-light-induced skin carcinogenesis in SKH-1 high-risk mice by sulforaphane-containing broccoli sprout extracts. Cancer Lett. 2006;240(2):243–252. doi:10.1016/j.canlet.2005.09.012
- Fahey JW, Haristoy X, Dolan PM, et al. Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo [a] pyrene-induced stomach tumors. Proc Natl Acad Sci. 2002;99(11):7610–7615. doi:10.1073/pnas.112203099
- Gills JJ, Jeffery EH, Matusheski NV, Moon RC, Lantvit DD, Pezzuto JM. Sulforaphane prevents mouse skin tumorigenesis during the stage of promotion. Cancer Lett. 2006;236(1):72–79. doi:10.1016/j.canlet.2005.05.007
- Liby KT, Royce DB, Risingsong R, et al. Synthetic Triterpenoids prolong survival in a transgenic mouse model of pancreatic cancer. Cancer Prev Res. 2010;3(11):1427–1434. doi:10.1158/1940-6207.CAPR-10-0197
- Kim EH, Deng C, Sporn MB, et al. CDDO-methyl ester delays breast cancer development in BRCA1-mutated mice. Cancer Prev Res. 2012;5(1):89–97. doi:10.1158/1940-6207.CAPR-11-0359
- Aoki Y, Sato H, Nishimura N, Takahashi S, Itoh K, Yamamoto M. Accelerated DNA adduct formation in the lung of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicol Appl Pharmacol. 2001;173(3):154–160. doi:10.1006/taap.2001.9176
- Iida K, Itoh K, Maher JM, et al. Nrf2 and p53 cooperatively protect against BBN-induced urinary bladder carcinogenesis. Carcinogenesis. 2007;28(11):2398–2403. doi:10.1093/carcin/bgm146
- Iizuka T, Ishii Y, Itoh K, et al. Nrf2‐deficient mice are highly susceptible to cigarette smoke‐induced emphysema. Genes Cells. 2005;10(12):1113–1125. doi:10.1111/j.1365-2443.2005.00905.x
- Kitamura Y, Umemura T, Kanki K, et al. Increased susceptibility to hepatocarcinogenicity of Nrf2‐deficient mice exposed to 2‐amino‐3‐methylimidazo [4, 5‐f] quinoline. Cancer Sci. 2007;98(1):19–24. doi:10.1111/j.1349-7006.2006.00352.x
- Khor TO, Huang MT, Prawan A, et al. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev Res. 2008;1(3):187–191. doi:10.1158/1940-6207.CAPR-08-0028
- Iida K, Itoh K, Kumagai Y, et al. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res. 2004;64(18):6424–6431. doi:10.1158/0008-5472.CAN-04-1906
- Ramos-Gomez M, Kwak M-K, Dolan PM, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in Nrf2 transcription factor-deficient mice. Proc Natl Acad Sci. 2001;98(6):3410–3415. doi:10.1073/pnas.051618798
- Xu C, Huang MT, Shen G, et al. Inhibition of 7, 12-dimethylbenz (a) anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2–related factor 2. Cancer Res. 2006;66(16):8293–8296. doi:10.1158/0008-5472.CAN-06-0300
- Yates MS, Kwak MK, Egner PA, et al. Potent protection against aflatoxin-induced tumorigenesis through induction of Nrf2-regulated pathways by the triterpenoid 1-[2-cyano-3-, 12-dioxooleana-1, 9 (11)-dien-28-oyl] imidazole. Cancer Res. 2006;66(4):2488–2494. doi:10.1158/0008-5472.CAN-05-3823
- Satoh H, Moriguchi T, Taguchi K, et al. Nrf2-deficiency creates a responsive microenvironment for metastasis to the lung. Carcinogenesis. 2010;31(10):1833–1843. doi:10.1093/carcin/bgq105
- Suzuki T, Shibata T, Takaya K, et al. Regulatory nexus of synthesis and degradation deciphers cellular Nrf2 expression levels. Mol Cell Biol. 2013;33(12):2402–2412. doi:10.1128/MCB.00065-13
- Yamamoto T, Yoh K, Kobayashi A, et al. Identification of polymorphisms in the promoter region of the human NRF2 gene. Biochem Biophys Res Commun. 2004;321(1):72–79. doi:10.1016/j.bbrc.2004.06.112
- Gañán-Gómez I, Wei Y, Yang H, Boyano-Adánez MC, García-Manero G. Oncogenic functions of the transcription factor Nrf2. Free Radic Biol Med. 2013;65:750–764. doi:10.1016/j.freeradbiomed.2013.06.041
- Homma S, Ishii Y, Morishima Y, et al. Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clin Cancer Res. 2009;15(10):3423–3432. doi:10.1158/1078-0432.CCR-08-2822
- H-K N, Surh Y-J. Oncogenic potential of Nrf2 and its principal target protein heme oxygenase-1. Free Radic Biol Med. 2014;67:353–365. doi:10.1016/j.freeradbiomed.2013.10.819
- Niture SK, Jaiswal AK. Nrf2-induced antiapoptotic Bcl-xL protein enhances cell survival and drug resistance. Free Radic Biol Med. 2013;57:119–131. doi:10.1016/j.freeradbiomed.2012.12.014
- Konstantinopoulos PA, Spentzos D, Fountzilas E, et al. Keap1 mutations and Nrf2 pathway activation in epithelial ovarian cancer. Cancer Res. 2011;71(15):5081–5089. doi:10.1158/0008-5472.CAN-10-4668
- Solis LM, Behrens C, Dong W, et al. Nrf2 and Keap1 abnormalities in non–small cell lung carcinoma and association with clinicopathologic features. Clin Cancer Res. 2010;16(14):3743–3753. doi:10.1158/1078-0432.CCR-09-3352
- Torrente L, Maan G, Oumkaltoum Rezig A, et al. High NRF2 levels correlate with poor prognosis in colorectal cancer patients and with sensitivity to the kinase inhibitor AT9283 in vitro. Biomolecules. 2020;10(10):1365. doi:10.3390/biom10101365
- Ohta T, Iijima K, Miyamoto M, et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008;68(5):1303–1309. doi:10.1158/0008-5472.CAN-07-5003
- Padmanabhan B, Tong KI, Ohta T, et al. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell. 2006;21(5):689–700. doi:10.1016/j.molcel.2006.01.013
- Fujimoto A, Furuta M, Totoki Y, et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet. 2016;48(5):500–509. doi:10.1038/ng.3547
- Shibata T, Kokubu A, Gotoh M, et al. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology. 2008;135(4):1358–1368. doi:10.1053/j.gastro.2008.06.082
- Nioi P, Nguyen T. A mutation of Keap1 found in breast cancer impairs its ability to repress Nrf2 activity. Biochem Biophys Res Commun. 2007;362(4):816–821. doi:10.1016/j.bbrc.2007.08.051
- Yoo NJ, Kim HR, Kim YR, An CH, Lee SH. Somatic mutations of the KEAP1 gene in common solid cancers. Histopathology. 2012;60(6):943–952. doi:10.1111/j.1365-2559.2012.04178.x
- Kim YR, Oh JE, Kim MS, et al. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J Pathol. 2010;220(4):446–451. doi:10.1002/path.2653
- Wong TF, Yoshinaga K, Monma Y, et al. Association of Keap1 and Nrf2 genetic mutations and polymorphisms with endometrioid endometrial adenocarcinoma survival. Int J Gynecol Cancer. 2011;21(8):1428–1435. doi:10.1097/IGC.0b013e31822d0eb2
- Li QK, Singh A, Biswal S, Askin F, Gabrielson E. KEAP1 gene mutations and NRF2 activation are common in pulmonary papillary adenocarcinoma. J Hum Genet. 2011;56(3):230–234. doi:10.1038/jhg.2010.172
- Mitsuishi Y, Taguchi K, Kawatani Y, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22(1):66–79. doi:10.1016/j.ccr.2012.05.016
- Lister A, Nedjadi T, Kitteringham NR, et al. Nrf2 is overexpressed in pancreatic cancer: implications for cell proliferation and therapy. Mol Cancer. 2011;10(1):1–13. doi:10.1186/1476-4598-10-37
- Zhang M, Zhang C, Zhang L, et al. Nrf2 is a potential prognostic marker and promotes proliferation and invasion in human hepatocellular carcinoma. BMC Cancer. 2015;15(1):1–12. doi:10.1186/s12885-015-1541-1
- Zhang C, Wang HJ, Bao QC, et al. NRF2 promotes breast cancer cell proliferation and metastasis by increasing RhoA/ROCK pathway signal transduction. Oncotarget. 2016;7(45):73593. doi:10.18632/oncotarget.12435
- DeNicola GM, Karreth FA, Humpton TJ, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475(7354):7354):106–109. doi:10.1038/nature10189
- Murakami S, Motohashi H. Roles of Nrf2 in cell proliferation and differentiation. Free Radic Biol Med. 2015;88:168–178. doi:10.1016/j.freeradbiomed.2015.06.030
- Rojo AI, Rada P, Mendiola M, et al. The PTEN/NRF2 axis promotes human carcinogenesis. Antioxid Redox Signal. 2014;21(18):2498–2514. doi:10.1089/ars.2014.5843
- Faraonio R, Vergara P, Di Marzo D, et al. p53 suppresses the Nrf2‐dependent transcription of antioxidant response genes. J Biol Chem. 2006;281(52):39776‐39784. doi:10.1074/jbc.M605707200
- Elsby R, Kitteringham NR, Goldring CE, et al. Increased constitutive c‐Jun N‐terminal kinase signaling in mice lacking glutathione S‐transferase Pi. J Biol Chem. 2003;278(25):22243‐22249. doi:10.1074/jbc.M301211200
- Jain A, Lamark T, Sjottem E, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element‐driven gene transcription. J Biol Chem. 2010;285(29):22576‐22591. doi:10.1074/jbc.M110.118976
- Niture SK, Jaiswal AK. Nrf2 protein up‐regulates antiapoptotic protein Bcl‐2 and prevents cellular apoptosis. J Biol Chem. 2012;287(13):9873‐9886. doi:10.1074/jbc.M111.312694
- Arfmann-Knübel S, Struck B, Genrich G, et al. The crosstalk between Nrf2 and TGF-β1 in the epithelial-mesenchymal transition of pancreatic duct epithelial cells. PLoS One. 2015;10(7):e0132978. doi:10.1371/journal.pone.0132978
- Zhao Q, Mao A, Guo R, et al. Suppression of radiation-induced migration of non-small cell lung cancer through inhibition of Nrf2-Notch axis. Oncotarget. 2017;8(22):36603. doi:10.18632/oncotarget.16622
- Lignitto L, LeBoeuf SE, Homer H, et al. Nrf2 activation promotes lung cancer metastasis by inhibiting the degradation of Bach1. Cell. 2019;178(2):316–329. doi:10.1016/j.cell.2019.06.003
- Shen H, Yang Y, Xia S, Rao B, Zhang J, Wang J. Blockage of Nrf2 suppresses the migration and invasion of esophageal squamous cell carcinoma cells in hypoxic microenvironment. Dis Esophagus. 2014;27(7):685–692. doi:10.1111/dote.12124
- Frohlich DA, McCabe MT, Arnold RS, Day ML. The role of Nrf2 in increased reactive oxygen species and DNA damage in prostate tumorigenesis. Oncogene. 2008;27(31):4353–4362. doi:10.1038/onc.2008.79
- Rachakonda G, Sekhar KR, Jowhar D, et al. Increased cell migration and plasticity in Nrf2-deficient cancer cell lines. Oncogene. 2010;29(25):3703–3714. doi:10.1038/onc.2010.118
- Ryu D, Lee JH, Kwak MK. NRF2 level is negatively correlated with TGF-β1-induced lung cancer motility and migration via NOX4-ROS signaling. Arch Pharm Res. 2020;43(12):1297–1310. doi:10.1007/s12272-020-01298-z
- Wang XJ, Sun Z, Villeneuve NF, et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29(6):1235–1243. doi:10.1093/carcin/bgn095
- Shim GS, Manandhar S, Shin DH, Kim TH, Kwak MK. Acquisition of doxorubicin resistance in ovarian carcinoma cells accompanies activation of the NRF2 pathway. Free Radic Biol Med. 2009;47(11):1619–1631. doi:10.1016/j.freeradbiomed.2009.09.006
- Singh A, Misra V, Thimmulappa RK, et al. Dysfunctional KEAP1–NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3(10):e420. doi:10.1371/journal.pmed.0030420
- Kalyanaraman B, Cheng G, Hardy M, Ouari O, Bennett B, Zielonka J. Teaching the basics of reactive oxygen species and their relevance to cancer biology: mitochondrial reactive oxygen species detection, redox signaling, and targeted therapies. Redox Biol. 2018;15:347–362. doi:10.1016/j.redox.2017.12.012
- Parascandolo A, Laukkanen MO. Carcinogenesis and reactive oxygen species signaling: interaction of the NADPH oxidase NOX1–5 and superoxide dismutase 1–3 signal transduction pathways. Antioxid Redox Signal. 2019;30(3):443–486. doi:10.1089/ars.2017.7268
- Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51(3):794–798.
- Toyokuni S, Okamoto K, Yodoi J, Hiai H. Persistent oxidative stress in cancer. FEBS Lett. 1995;358(1):1–3. doi:10.1016/0014-5793(94)01368-B
- Liu C, Zhao Y, Wang J, et al. FoxO3 reverses 5-fluorouracil resistance in human colorectal cancer cells by inhibiting the Nrf2/TR1 signaling pathway. Cancer Lett. 2020;470:29–42. doi:10.1016/j.canlet.2019.11.042
- Barrera G, Cucci MA, Grattarola M, Dianzani C, Muzio G, Pizzimenti S. Control of oxidative stress in cancer chemoresistance: spotlight on Nrf2 role. Antioxidants. 2021;10(4):510. doi:10.3390/antiox10040510
- Hospers GAP, Meuer C, De Leij L, Uges DRA, Mulder NH, De Vries EGE. A study of human small‐cell lung carcinoma (hSCLC) cell lines with different sensitivities to detect relevant mechanisms of cisplatin (CDDP) resistance. Int, J, Cancer. 1990;46(1):138–144. doi:10.1002/ijc.2910460125
- Jain N, Lam YM, Pym J, Campling BG. Mechanisms of resistance of human small cell lung cancer lines selected in VP‐16 and cisplatin. Cancer. 1996;77(9):1797–1808. doi:10.1002/(SICI)1097-0142(19960501)77:9<1797::AID-CNCR7>3.0.CO;2-9
- Kim SK, Yang JW, Kim MR, et al. Increased expression of Nrf2/ARE-dependent anti-oxidant proteins in tamoxifen-resistant breast cancer cells. Free Radic Biol Med. 2008;45(4):537–546. doi:10.1016/j.freeradbiomed.2008.05.011
- Shibata T, Kokubu A, Saito S, et al. NRF2 mutation confers malignant potential and resistance to chemoradiation therapy in advanced esophageal squamous cancer. Neoplasia. 2011;13(9):864–873. doi:10.1593/neo.11750
- Chian S, Li YY, Wang XJ, Tang XW. Luteolin sensitizes two oxaliplatin-resistant colorectal cancer cell lines to chemotherapeutic drugs via inhibition of the Nrf2 pathway. Asian Pac J Cancer Prev. 2014;15(6):2911–2916. doi:10.7314/APJCP.2014.15.6.2911
- Duong HQ, Yi YW, Kang HJ, et al. Inhibition of NRF2 by PIK-75 augments sensitivity of pancreatic cancer cells to gemcitabine. Int J Oncol. 2014;44(3):959–969. doi:10.3892/ijo.2013.2229
- Hou X, Bai X, Gou X, et al. 3′, 4′, 5′, 5, 7-pentamethoxyflavone sensitizes cisplatin-resistant A549 cells to cisplatin by inhibition of Nrf2 pathway. Mol Cells. 2015;38(5):396–401. doi:10.14348/molcells.2015.2183
- Zhong Y, Zhang F, Sun Z, et al. Drug resistance associates with activation of Nrf2 in MCF‐7/DOX cells, and wogonin reverses it by down‐regulating Nrf2‐mediated cellular defense response. Mol, Carcinog. 2013;52(10):824–834. doi:10.1002/mc.21921
- Huang J, Tan PH, Thiyagarajan J, Bay BH. Prognostic significance of glutathione-S-transferase-pi in invasive breast cancer. Mod Pathol. 2003;16(6):558–565. doi:10.1097/01.MP.0000071842.83169.5A
- Su F, Hu X, Jia W, Gong C, Song E, Hamar P. Glutathion-s-transferase π indicates chemotherapy resistance in breast cancer. J Surg Res. 2003;113(1):102–108. doi:10.1016/S0022-4804(03)00200-2
- Chen B, Shen Z, Wu D, et al. Glutathione peroxidase 1 promotes NSCLC resistance to cisplatin via ROS-induced activation of PI3K/AKT pathway. BioMed Res Int. 2019;2019:7640547. doi:10.1155/2019/7640547
- Du H, Chen B, Jiao NL, Liu YH, Sun SY, Zhang YW. Elevated glutathione peroxidase 2 expression promotes cisplatin resistance in lung adenocarcinoma. Oxid Med Cell Longev. 2020;2020:7370157. doi:10.1155/2020/7370157
- Zarei M, Lal S, Parker SJ, et al. Posttranscriptional upregulation of IDH1 by HuR establishes a powerful survival phenotype in pancreatic cancer cells. Cancer Res. 2017;77(16):4460–4471. doi:10.1158/0008-5472.CAN-17-0015
- Patel GK, Khan MA, Bhardwaj A, et al. Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK. Br J Cancer. 2017;116(5):609–619. doi:10.1038/bjc.2017.18
- Cho JM, Manandhar S, Lee HR, Park HM, Kwak MK. Role of the Nrf2-antioxidant system in cytotoxicity mediated by anticancer cisplatin: implication to cancer cell resistance. Cancer Lett. 2008;260(1–2):96–108. doi:10.1016/j.canlet.2007.10.022
- Zhang P, Singh A, Yegnasubramanian S, et al. Loss of Kelch-Like ECH-Associated Protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol Cancer Ther. 2010;9(2):336–346. doi:10.1158/1535-7163.MCT-09-0589
- Tang X, Wang H, Fan L, et al. Luteolin inhibits Nrf2 leading to negative regulation of the Nrf2/ARE pathway and sensitization of human lung carcinoma A549 cells to therapeutic drugs. Free Radic Biol Med. 2011;50(11):1599–1609. doi:10.1016/j.freeradbiomed.2011.03.008
- Benderra Z, Faussat AM, Sayada L, et al. MRP3, BCRP, and P-glycoprotein activities are prognostic factors in adult acute myeloid leukemia. Clin Cancer Res. 2005;11(21):7764–7772. doi:10.1158/1078-0432.CCR-04-1895
- Khot MI, Downey CL, Armstrong G, et al. The role of ABCG2 in modulating responses to anti-cancer photodynamic therapy. Photodiagnosis Photodyn Ther. 2020;29:101579. doi:10.1016/j.pdpdt.2019.10.014
- Loignon M, Miao W, Hu L, et al. Cul3 overexpression depletes Nrf2 in breast cancer and is associated with sensitivity to carcinogens, to oxidative stress, and to chemotherapy. Mol Cancer Ther. 2009;8(8):2432–2440. doi:10.1158/1535-7163.MCT-08-1186
- Xu X, Zhang Y, Li W, et al. Wogonin reverses multi-drug resistance of human myelogenous leukemia K562/A02 cells via downregulation of MRP1 expression by inhibiting Nrf2/ARE signaling pathway. Biochem Pharmacol. 2014;92(2):220–234. doi:10.1016/j.bcp.2014.09.008
- Young LC, Campling BG, Cole SP, Deeley RG, Gerlach JH. Multidrug resistance proteins MRP3, MRP1, and MRP2 in lung cancer: correlation of protein levels with drug response and messenger RNA levels. Clin Cancer Res. 2001;7(6):1798–1804.
- Gonzalez-Sanchez E, Marin JJ, Perez MJ. The expression of genes involved in hepatocellular carcinoma chemoresistance is affected by mitochondrial genome depletion. Mol Pharm. 2014;11(6):1856–1868. doi:10.1021/mp400732p
- de Souza I, Monteiro LKS, Guedes CB, et al. High levels of NRF2 sensitize temozolomide-resistant glioblastoma cells to ferroptosis via ABCC1/MRP1 upregulation. Cell Death Dis. 2022;13(7):1–13. doi:10.1038/s41419-022-05044-9
- Choi HK, Yang JW, Roh SH, Han CY, Kang KW. Induction of multidrug resistance associated protein 2 in tamoxifen-resistant breast cancer cells. Endocr Relat Cancer. 2007;14(2):293–303. doi:10.1677/ERC-06-0016
- Materna V, Liedert B, Thomale J, Lage H. Protection of platinum–DNA adduct formation and reversal of cisplatin resistance by anti‐MRP2 hammerhead ribozymes in human cancer cells. Int, J, Cancer. 2005;115(3):393–402. doi:10.1002/ijc.20899
- Paramasivan P, Kankia IH, Langdon SP, Deeni YY. Emerging role of nuclear factor erythroid 2-related factor 2 in the mechanism of action and resistance to anticancer therapies. Cancer Drug Resist. 2019;2(3):490–515. doi:10.20517/cdr.2019.57
- Zhang L, Fang CH, Fan YF. Detection of multidrug resistance-associated proteins MRP2, MRP3, and MRP5 mRNA expressions in hepatocarcinoma cells using SYBR real-time PCR. J South Med Univ. 2008;28(2):219–221.
- Zhang YH, Wu Q, Xiao XY, Li DW, Wang XP. Silencing MRP4 by small interfering RNA reverses acquired DDP resistance of gastric cancer cell. Cancer Lett. 2010;291(1):76–82. doi:10.1016/j.canlet.2009.10.003
- Doyle LA, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene. 2003;22(47):7340–7358. doi:10.1038/sj.onc.1206938
- Yuan J, Lv H, Peng B, Wang C, Yu Y, He Z. Role of BCRP as a biomarker for predicting resistance to 5-fluorouracil in breast cancer. Cancer Chemother Pharmacol. 2009;63(6):1103–1110. doi:10.1007/s00280-008-0838-z
- Wu CP, Sim HM, Huang YH, et al. Overexpression of ATP-binding cassette transporter ABCG2 as a potential mechanism of acquired resistance to vemurafenib in BRAF (V600E) mutant cancer cells. Biochem Pharmacol. 2013;85(3):325–334. doi:10.1016/j.bcp.2012.11.003
- Singh A, Wu H, Zhang P, Happel C, Ma J, Biswal S. Expression of ABCG2 (BCRP) is regulated by Nrf2 in cancer cells that confers side population and chemoresistance phenotype. Mol Cancer Ther. 2010;9(8):2365–2376. doi:10.1158/1535-7163.MCT-10-0108
- Kim EJ, Kim YJ, Lee HI, Jeong SH, Nam HJ, Cho JH. NRF2 knockdown resensitizes 5-fluorouracil-resistant pancreatic cancer cells by suppressing HO-1 and ABCG2 expression. Int J Mol Sci. 2020;21(13):4646. doi:10.3390/ijms21134646
- Liao W, Wang Z, Fu Z, et al. p62/SQSTM1 protects against cisplatin-induced oxidative stress in kidneys by mediating the cross talk between autophagy and the Keap1-Nrf2 signalling pathway. Free Radic Res. 2019;53(7):800–814. doi:10.1080/10715762.2019.1635251
- Mirzaei S, Mohammadi AT, Gholami MH, et al. Nrf2 signaling pathway in cisplatin chemotherapy: potential involvement in organ protection and chemoresistance. Pharmacol Res. 2021;167:105575. doi:10.1016/j.phrs.2021.105575
- Kocanova S, Buytaert E, Matroule J-Y, et al. Induction of heme-oxygenase 1 requires the p38MAPK and PI3K pathways and suppresses apoptotic cell death following hypericin-mediated photodynamic therapy. Apoptosis. 2007;12(4):731–741. doi:10.1007/s10495-006-0016-x
- Ajuwon OR, Lawal AO, Marnewick JL, Davids LM The transcription factor Nrf2 mediates endogenous antioxidants upregulation in response to hypericin-photodynamic therapy in melanoma cells. Poster presented at: Oxygen Club of California World Congress; May 4-6; 2016; University of California, Davis; CA.
- Dolgachev V, Oberley LW, Huang -T-T, et al. A role for manganese superoxide dismutase in apoptosis after photosensitization. Biochem Biophys Res Commun. 2005;332(2):411–417. doi:10.1016/j.bbrc.2005.04.141
- Nowis D, Legat M, Grzela T, et al. Heme oxygenase-1 protects tumor cells against photodynamic therapy-mediated cytotoxicity. Oncogene. 2006;25(24):3365–3374. doi:10.1038/sj.onc.1209378
- Wang HP, Qian SY, Schafer FQ, Domann FE, Oberley LW, Buettner GR. Phospholipid hydroperoxide glutathione peroxidase protects against singlet oxygen-induced cell damage of photodynamic therapy. Free Radic Biol Med. 2001;30(8):825–835. doi:10.1016/S0891-5849(01)00469-5
- Aniogo EC, George BP, Abrahamse H. Molecular effectors of photodynamic therapy-mediated resistance to cancer cells. Int J Mol Sci. 2021;22(24):13182. doi:10.3390/ijms222413182
- Mastrangelopoulou M, Grigalavicius M, Raabe TH, et al. Predictive biomarkers for 5‐ALA‐PDT can lead to personalized treatments and overcome tumor‐specific resistances. Cancer Rep. 2022;5(12):e1278.
- Dabrowski MJ, Maeda D, Zebala J, et al. Glutathione S-transferase P1-1 expression modulates sensitivity of human kidney 293 cells to photodynamic therapy with hypericin. Arch Biochem Biophys. 2006;449(1–2):94–103. doi:10.1016/j.abb.2006.02.009
- Tian S, Yong M, Zhu J, et al. Enhancement of the effect of methyl pyropheophorbide-a-mediated photodynamic therapy was achieved by increasing ROS through inhibition of Nrf2-HO-1 or Nrf2-ABCG2 signaling. Anticancer Agents Med Chem. 2017;17(13):1824–1836. doi:10.2174/1871520617666170327145857
- Choi BH, Ryoo IG, Kang HC, Kwak MK. The sensitivity of cancer cells to pheophorbide a-based photodynamic therapy is enhanced by Nrf2 silencing. PLoS One. 2014;9(9):e107158. doi:10.1371/journal.pone.0107158
- Ferino A, Rapozzi V, Xodo LE. The ROS-KRAS-Nrf2 axis in the control of the redox homeostasis and the intersection with survival-apoptosis pathways: implications for photodynamic therapy. J Photochem Photobiol B: Biol. 2020;202:111672. doi:10.1016/j.jphotobiol.2019.111672
- Kimani SG, Phillips JB, Bruce JI, MacRobert AJ, Golding JP. Antioxidant inhibitors potentiate the cytotoxicity of photodynamic therapy. Photochem Photobiol. 2012;88(1):175–187. doi:10.1111/j.1751-1097.2011.01022.x
- Theodossiou TA, Olsen CE, Jonsson M, Kubin A, Hothersall JS, Berg K. The diverse roles of glutathione-associated cell resistance against hypericin photodynamic therapy. Redox Biol. 2017;12:191–197. doi:10.1016/j.redox.2017.02.018
- Lin T, Zhao X, Zhao S, et al. O2-generating MnO2 nanoparticles for enhanced photodynamic therapy of bladder cancer by ameliorating hypoxia. Theranostics. 2018;8(4):990–1004. doi:10.7150/thno.22465
- Liu C, Wang D, Zhang S, et al. Biodegradable biomimic copper/manganese silicate nanospheres for chemodynamic/photodynamic synergistic therapy with simultaneous glutathione depletion and hypoxia relief. ACS Nano. 2019;13(4):4267–4277. doi:10.1021/acsnano.8b09387
- Wang C, Cao F, Ruan Y, Jia X, Zhen W, Jiang X. Specific generation of singlet oxygen through the Russell mechanism in hypoxic tumors and GSH depletion by Cu‐TCPP nanosheets for cancer therapy. Angew Chem. 2019;131(29):9951–9955. doi:10.1002/ange.201903981
- Zhang Y, Li S, Fang X, et al. Copper decorated Ti3C2 nanosystem with NIR-II-induced GSH-depletion and reactive oxygen species generation for efficient nanodynamic therapy. Nanophotonics. 2022;11(22):5189–5204. doi:10.1515/nanoph-2022-0599
- Frank J, Lornejad-Schafer M, Schoffl H, Flaccus A, Lambert C, Biesalski HK. Inhibition of heme oxygenase-1 increases responsiveness of melanoma cells to ALA-based photodynamic therapy. Int J Oncol. 2007;31(6):1539–1545.
- Miyake M, Ishii M, Kawashima K, et al. siRNA‐mediated knockdown of the heme synthesis and degradation pathways: modulation of treatment effect of 5‐aminolevulinic acid‐based photodynamic therapy in urothelial cancer cell lines. Photochem Photobiol. 2009;85(4):1020–1027. doi:10.1111/j.1751-1097.2009.00543.x
- Kimakova P, Solar P, Feckova B, et al. Photoactivated hypericin increases the expression of SOD-2 and makes MCF-7 cells resistant to photodynamic therapy. Biomed Pharmacother. 2017;85:749–755. doi:10.1016/j.biopha.2016.11.093
- Ğb JG, Nowis D, Skrzycki M, et al. Antitumor effects of photodynamic therapy are potentiated by 2-methoxyestradiol: a superoxide dismutase inhibitor. J Biol Chem. 2003;278(1):407–414. doi:10.1074/jbc.M209125200
- Wright KE, MacRobert AJ, Phillips JB. Inhibition of specific cellular antioxidant pathways increases the sensitivity of neurons to meta‐tetrahydroxyphenyl chlorin‐mediated photodynamic therapy in a 3D co‐culture model. Photochem Photobiol. 2012;88(6):1539–1545. doi:10.1111/j.1751-1097.2012.01185.x
- Hamblin MR. Drug efflux pumps in photodynamic therapy. In: Sosnik A, Bendayan R, editors. Drug Efflux Pumps in Cancer Resistance Pathways: From Molecular Recognition and Characterization to Possible Inhibition Strategies in Chemotherapy. Cambridge, Massachusetts: Academic Press; 2020:251–276.
- Robey RW, Steadman K, Polgar O, Bates SE. ABCG2-mediated transport of photosensitizers: potential impact on photodynamic therapy. Cancer Biol Ther. 2005;4(2):195–202. doi:10.4161/cbt.4.2.1440
- Hagiya Y, Adachi T, Ogura S-I, et al. Nrf2-dependent induction of human ABC transporter ABCG2 and heme oxygenase-1 in HepG2 cells by photoactivation of porphyrins: biochemical implications for cancer cell response to photodynamic therapy. J Exp Ther Oncol. 2008;7(2):153–167.
- Liu W, Baer MR, Bowman MJ, et al. The tyrosine kinase inhibitor imatinib mesylate enhances the efficacy of photodynamic therapy by inhibiting ABCG2. Clin Cancer Res. 2007;13(8):2463–2470. doi:10.1158/1078-0432.CCR-06-1599
- Sun W, Kajimoto Y, Inoue H, Miyatake SI, Ishikawa T, Kuroiwa T. Gefitinib enhances the efficacy of photodynamic therapy using 5-aminolevulinic acid in malignant brain tumor cells. Photodiagnosis Photodyn Ther. 2013;10(1):42–50. doi:10.1016/j.pdpdt.2012.06.003
- Kim JH, Park JM, Roh YJ, Kim IW, Hasan T, Choi MG. Enhanced efficacy of photodynamic therapy by inhibiting ABCG2 in colon cancers. BMC Cancer. 2015;15(1):1–9. doi:10.1186/s12885-015-1514-4
- Jung KA, Choi BH, Kwak MK. The c-MET/PI3K signaling is associated with cancer resistance to doxorubicin and photodynamic therapy by elevating BCRP/ABCG2 expression. Mol Pharmacol. 2015;87(3):465–476. doi:10.1124/mol.114.096065
- Jendželovský R, Mikeš J, Souček K, et al. Drug efflux transporters, MRP1 and BCRP, affect the outcome of hypericin-mediated photodynamic therapy in HT-29 adenocarcinoma cells. Photochem Photobiol Sci. 2009;8(12):1716–1723. doi:10.1039/b9pp00086k
- Jendželovský R, Jendželovská Z, Kucharova B, Fedoročko P. Breast cancer resistance protein is the enemy of hypericin accumulation and toxicity of hypericin-mediated photodynamic therapy. Biomed Pharmacother. 2019;109:2173–2181. doi:10.1016/j.biopha.2018.11.084
- Khot MI, Perry SL, Maisey T, et al. Inhibiting ABCG2 could potentially enhance the efficacy of hypericin-mediated photodynamic therapy in spheroidal cell models of colorectal cancer. Photodiagnosis Photodyn Ther. 2018;23:221–229. doi:10.1016/j.pdpdt.2018.06.027
- Kuchárová B, Mikeš J, Jendželovský R, et al. Potentiation of hypericin-mediated photodynamic therapy cytotoxicity by MK-886: focus on ABC transporters, GDF-15 and redox status. Photodiagnosis Photodyn Ther. 2015;12(3):490–503. doi:10.1016/j.pdpdt.2015.04.008
- Biteghe FN, Davids LM. A combination of photodynamic therapy and chemotherapy displays a differential cytotoxic effect on human metastatic melanoma cells. J Photochem Photobiol B: Biol. 2017;166:18–27. doi:10.1016/j.jphotobiol.2016.11.004