
Abstract
Background
Patients harboring activating mutations in epidermal growth factor receptors (EGFR) are particularly sensitive to EGFR tyrosine kinase inhibitors (TKIs). However, most patients develop an acquired resistance after a period of about 10 months. This study focuses on the therapeutic effect of NVP-BEZ235, a dual inhibitor of phosphatidylinositol-3-kinase/mammalian target of rapamycin (PI3K/mTOR), in gefitinib-resistant non-small cell lung cancer.
Methods
H1975 cell line was validated as a gefitinib-resistant cell model by the nucleotide-sequence analysis. We used the 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay to detect the growth of H1975 cell line in vitro. H1975 cells’ migration was detected by the migration assay. Xenograft models were used to investigate the growth of gefitinib-resistant non-small cell lung cancer in vivo. Western blot and immunohistochemical analysis were used to investigate the level of PI3K/protein kinase B(AKT)/mTOR signaling pathway proteins.
Results
We show that NVP-BEZ235 effectively inhibited the growth of H1975 cells in vivo as well as in vitro. Similarly, H1975 cell migration was reduced by NVP-BEZ235. Further experiments revealed that NVP-BEZ235 attenuated the phosphorylation of PI3K/AKT/mTOR signaling pathway proteins.
Conclusion
Taken together, we suggest that NVP-BEZ235 inhibits gefitinib-resistant tumor growth by downregulating PI3K/AKT/mTOR phosphorylation.
Introduction
Advanced non-small cell lung cancer (NSCLC) is the leading cause of cancer-related death worldwide.Citation1 The epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs), gefitinib (Iressa®; AstraZeneca, London, UK) and erlotinib, competitively bind with the active site of the EGFR kinase. A subgroup of patients with activating mutations in EGFR are particularly sensitive to EGFR TKIs.Citation2,Citation3 Exon 19 deletion mutations and the single-point substitution mutation L858R in exon 21 are the most prevalent among these activating mutations.Citation4 However, after a period of about 10 months of progression-free survival, most patients relapse because of the development of an acquired resistance to EGFR TKIs.Citation5 Further research exhibited that the secondary threonine-to-methionine substitution at codon 790(T790M) accounts for approximately half of the cases of the acquired resistance.Citation6,Citation7 A bulkier amino acid is introduced by the methionine substitution to the acetylene side chain and this alteration makes a steric hindrance that may interfere with the binding of EGFR TKIs, eventually leading to the development of EGFR TKIs resistance.Citation7 Approximately another 20%–25% of EGFR TKIs resistance arises due to the amplification of mesenchymal-epidermal transition (MET), another tyrosine kinase receptor.Citation8,Citation9 The MET amplification results in continuous activation of the phosphatidylinositol-3-kinase (PI3K)/protein kinase B (AKT) and MEK-ERK signaling pathway despite EGFR inhibition. Moreover, MET inhibitors re-sensitize these cancers to EGFR TKIs.Citation9,Citation10 The T790M mutation and proto-oncogene MET amplification, both of which are independent resistance mechanisms, as suggested by Bean et al,Citation9 tend to cause sustained activation of the PI3K-AKT-mammalian target of rapamycin (mTOR) signaling pathway.
It has been recognized that abnormal activation of the PI3K/AKT/mTOR signaling pathway is implicated with human cancer.Citation11 PI3K is a heterodimer comprised of a p85 regulatory and a p110 catalytic subunit. Once PI3K is activated, p110 phosphorylates the phosphatidylinositol-4,5-diphosphate(PIP2) to phosphatidylinositol-3,4,5-triphosphate(PIP3), which facilitates the phosphorylation of AKT at Thr308 by PDK1.Citation12 A second phosphorylation event at Ser473 by the mTOR-rictor complex is required for maximal AKT activity.Citation13 The downstream target of PI3K/AKT pathway, mTOR, a serine/threonine-specific protein kinase, phosphorylates S6 ribosomal protein, and eukaryotic initiation factor 4E binding protein 1, thereby regulating tumor cell growth and proliferation. NVP-BEZ235, which directly targets the PI3K-AKT-mTOR pathway, has been found to have potential application in clinical practice.Citation14 BEZ235 is an imidazo[4,5-c]quinoline derivative that inhibits PI3K and the downstream mTOR kinase activity by binding to the ATP-binding cleft of these enzymes. Moreover, BEZ235 inhibits the activation of the downstream effectors AKT, S6 ribosomal protein, and 4E binding protein 1 in breast cancer cells.Citation14–Citation16 Therefore, we hypothesized that blocking the common downstream PI3K-AKT-mTOR signaling pathway by BEZ235 may be an effective approach to overcome the EGFR TKIs resistance.
In this study, NCI-H1975 cell line, which harbors both T790M and L858R mutations in EGFR, was used as a model of gefitinib-acquired resistance to examine the inhibitory effect of NVP-BEZ235 on gefitinib-resistant tumors in vitro as well as in vivo.
Materials and methods
Cell line and culture conditions
H1975 cell line was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in Dulbecco’s Modified Eagle’s Medium (HyClone) supplemented with 10% fetal bovine serum (HyClone) at 37°C in 5% CO2.
Reagents
NVP-BEZ235 was purchased from Selleck Chemicals (Houston, TX, USA), dissolved in dimethyl sulfoxide (DMSO) (Amresco LLC, Solon, OH, USA) to 10 mmol/L. Gefitinib was kindly supplied by AstraZeneca and dissolved in DMSO to the concentration of 25 mmol/L. Both compounds were stored at −20°C, and further diluted to the needed concentration in Dulbecco’s Modified Eagle’s Medium. DMSO was added to the culture medium of the control cells and was constantly kept below 0.1%. Antibodies against Ser473-phospho-AKT(9271), Ser235/236-phospho-S6(2211), and ribosomal protein S6(2217) were procured from Cell Signaling Technology (Danvers, MA, USA). AKT was bought from Proteintech Group, Inc. (Chicago, IL, USA) and β-actin was from Santa Cruz Biotechnology Inc. (Dallas, TX, USA).
Cell growth assay
The 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method was employed to estimate the number of viable cells by following the manufacturer’s instructions (Sigma-Aldrich Co, St Louis, MO, USA). Briefly, H1975 cells were grown in 200 μL medium at 37°C in 5% CO2 for 72 hours in 96-well plates, and then incubated with 20 μL MTT (5 g/L) for 4 hours. Afterwards, 150 μL DMSO was added to each well, and the absorbance value at 490 nm was read on a Microplate reader (Eppendorf, Hamburg, Germany).
Nucleotide sequencing of EGFR DNA of H1975 cells
For detection of the given mutation in the EGFR coding sequence, DNA was extracted from H1975 cells, and all 18–21 exons were amplified by polymerase chain reaction with the primers being as follows: forward, 5′-TGAAGGCTGTCCAACGA-3′; reverse, 5′-TTCCAATGCCATCCACT-3′. The products were directly sequenced in both sense and anti-sense directions by Invitrogen (Thermo Fisher Scientific, Waltham, MA, USA).
Migration assay
H1975 cells migration upon treatment with BEZ235 was assessed by the migration assay. It was performed on a double chamber transwell as previously described.Citation17 Briefly, H1975 cells were seeded into 24-well plates and incubated with different concentrations of drugs. Dispersed by trypsin, 2×105 cells were seeded into the upper chamber in triplicate. Then, fresh medium containing 30% fetal bovine serum was added to the lower chamber. After incubation at 37°C for 24 hours, migrated cells were stained and counted in five randomly-selected fields.
Western blot analysis
To detect the levels of proteins involved in the PI3K/AKT/mTOR signaling pathway, Western blotting was performed. H1975 cells were treated with BEZ235 or gefitinib for 16 hours and whole cell extracts were prepared as described previously.Citation18 Equal amounts of proteins were separated on SDS-PAGE 8%–15% Bis-Tris gel and transferred to a polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA, USA). The membrane was then probed with the aforementioned primary antibodies respectively, and protein expression was visualized by using the ECL and the results were transferred to the photographic film.
Xenograft models of H1975 cell lines in BALB/c nude mice
Thirty-two male 6-week old BALB/c nude mice, weighing from 18 to 20 g, were obtained from Hubei Experimental Animal Research Center, People’s Republic of China. The animals were raised under the specific pathogen-free conditions in the Experimental Animal Center of Huazhong University of Science and Technology, Wuhan, People’s Republic of China. They were divided into four groups (n=8) and were subcutaneously implanted with H1975 cells (1×107/0.2 mL in each nude mouse). The length and width of the tumors were measured every other day following the first day of H1975 cells’ implantation. When the tumor volume reached 280 mm3 (tumor volume was calculated as follows: tumor volume = length × widthCitation2 × π/6), the mice were divided into four groups and drugs were administered: a blank control group, a GEF group (gefitinib at 60 mg/kg orally every other day), a BEZ group (BEZ235 at 40 mg/kg subcutaneous injection every other day), and a group with a combination of gefitinib and BEZ235. Two weeks after the drug administration, the mice were sacrificed and tumors were excised. The weight and volume of the tumors were measured, and cluster of differentiation (CD)31 and vascular endothelial growth factor (VEGF) were immunohistochemically determined. All animal experiments were performed with the approval of the institutional animal use and care committee.
Statistical analysis
The data were expressed as mean ± standard deviation from independent experiments. Statistical significance was evaluated by using the Student’s t-test. The level of significance was set at P<0.05. Origin 7.5 was used for plotting the data.
Results
Resistance of H1975 cell line to gefitinib and presence of secondary mutation of EGFR
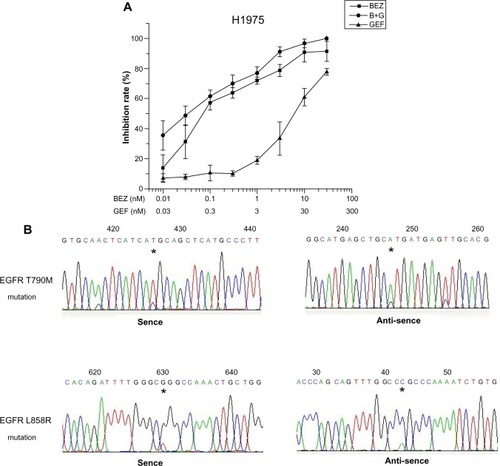
To understand whether the H1975 cell line is an ideal model of gefitinib resistance, we assessed the cellular proliferation by MTT assay, and amplification of the EGFR exons 18–21 in the H1975 cell line. After incubation with gefitinib for 72 hours, as shown in , H1975 cell growth was not inhibited until the concentration of gefitinib was over 3 μmol/L (P<0.05). The IC50 was 22.61 μmol/L, which was consistent with the result of a previous report.Citation8 It has been reported previously that the H1975 cell line carries L858R-T790M EGFR mutation and is resistant to gefitinib and erlotinib.Citation6,Citation19 To confirm the secondary T790M mutation in the H1975 cell line, polymerase chain reaction was performed to amplify the exons 18–21 of EGFR. The nucleotide sequencing exhibited that there existed a C-to-T point mutation in exon 20, which was due to the threonine-to-methionine substitution at codon 790 (T790M). Another T-to-G point mutation in exon 18, which led to arginine being substituted by leucine at codon 858 (L858R) was also observed ().
Figure 1 H1975 cell line was resistant to gefitinib and inhibited by BEZ235.
Abbreviations: B+G, NVP-BEZ235 combination with gefitinib; BEZ, NVP-BEZ235; EGFR, epidermal growth factor receptor; GEF, gefitinib; L858R, leucine-to-arginine substitution at codon 858; MTT, 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; SD, standard deviation; T790M, threonine-to-methionine substitution at codon 790.
Inhibition of H1975 cell growth by NVP-BEZ235 alone or in combination with gefitinib
After H1975 cells were incubated for 72 hours with an increasing concentration of BEZ235 alone or in combination with gefitinib, H1975 cell growth was strongly inhibited when BEZ235 concentration was more than 0.1 μmol/L (P<0.05), as shown in . The IC50 of BEZ235 alone was 0.151 μmol/L. When BEZ235 was given with gefitinib, the cell growth was suppressed at a higher inhibition ratio compared to the treatment with BEZ235 alone. The IC50 of gefitinib and BEZ235 were 0.177 μmol/L and 0.056 μmol/L respectively. The calculated combination index was 0.378, which indicated a strong synergistic effect of these two compounds.
Inhibition of H1975 cell migration by NVP-BEZ235
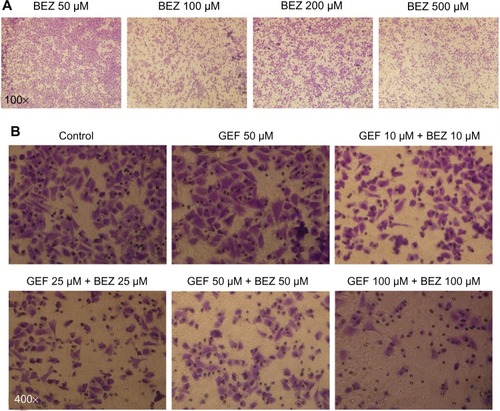
The inhibitory effect of BEZ235 on the migration of H1975 cells was examined by migration assay. shows cell migration was significantly inhibited by BEZ235 alone. The difference of the inhibitory effect between each group suggested that the inhibition acts in a dose-dependent manner. When administrated and combined with gefitinib, BEZ235 inhibited H1975 cell migration in the same manner, as seen in .
Figure 2 H1975 cell migration was inhibited by BEZ235.
Abbreviations: BEZ, NVP-BEZ235; GEF, gefitinib.
Inhibitory effect of NVP-BEZ235 on the growth of H1975 xenografts in nude mice
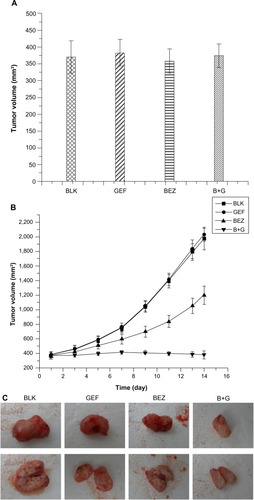
In view of the in vitro growth-inhibiting effects of BEZ235 on H1975 cells, we verified its in vivo inhibitory action in nude mice. The mice were randomized into four groups with similar average tumor volume (). As expected, the tumor volume was decreased two weeks after BEZ235 administration. At the end of treatment, the average tumor volume of the blank group was 1973.22±156.04 mm3, while the average tumor volume of BEZ235 group was 1198.76±123.28 mm3 and 379.92±54.74 mm3 for the combination group, respectively. The tumor volumes of BEZ235 alone and the combination groups were significantly different from that of the blank group (P<0.05). These in vivo findings further demonstrated that BEZ235 significantly inhibited the growth of H1975 xenografts ().
Figure 3 H1975 cells were implanted into nude mice. (A) The tumor volume did not show significant difference, before treatment with different components. (B) The tumor volumes started to show differences with different components’ treatment. (C) The observation of tumors after 2 weeks of different components’ treatment.
Inhibition of PI3K/AKT/mTOR signaling pathway by NVP-BEZ235 via reducing phosphorylation in vitro and in vivo
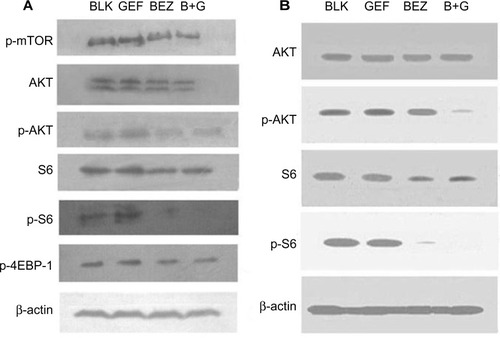
To understand the mechanisms by which BEZ235 inhibits gefitinib-resistant NSCLC, we examined the effects of BEZ235 on the expression of crucial proteins of PI3K/AKT/mTOR signaling pathway both in vitro and in vivo. The doses of drugs (BEZ235 at 200 nM and gefitinib at 0.1 μmol/L) in the four groups (DMSO control, 0.1 μmol/L gefitinib, 200 nmol/L BEZ235 and combination use of gefitinib and BEZ235) were similar to the concentrations used in previous studies.Citation14,Citation20,Citation21 With the in vitro study, H1975 cells were incubated in the presence of different components for 16 hours. We observed that the total protein AKT and S6 expression was not significantly altered by any treatment. However, the level of the phosphorylated proteins, Ser473-p-AKT and Ser235/236-p-S6, were decreased in the presence of BEZ235 and the combination of two drugs, while gefitinib did not cause any change in the expression of phosphorylated proteins (). Similar results were confirmed by the protein analysis of the tumor xenografts as shown in .
Figure 4 Analysis of PI3K/AKT/mTOR signaling pathway protein.
Abbreviations: AKT, protein kinase B; B+G, NVP-BEZ235 combination with gefitinib; BLK, blank control; BEZ, NVP-BEZ235; GEF, gefitinib; mTOR, mammalian target of rapamycin; 4EBP-1, 4E binding protein 1; PI3K, phosphoinositide 3-kinase; p, phosphorylated.
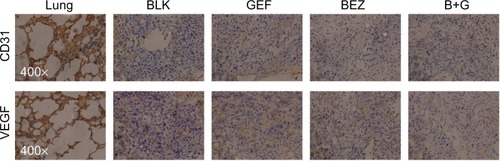
Inhibitory effect of NVP-BEZ235 in combination with gefitinib on VEGF and CD31 expression
Aberrant PI3K signaling impairs phosphatase and tensin homologue function, which plays an important part in tumor angiogenesis and in normal vascular formation.Citation22,Citation23 Therefore, we examined whether BEZ235 could interfere with tumor angiogenesis in nude mice xenografts. Immunohistochemical detection showed a significant reduction in VEGF and CD31 staining in BEZ235-treated tumors as compared with the controls (), suggesting that BEZ235 may be involved in the inhibition of tumor angiogenesis.
Figure 5 BEZ235 combination with gefitinib reduced VEGF and CD31 expression in vivo.
Abbreviations: B+G, NVP-BEZ235 combination with gefitinib; BEZ, NVP-BEZ235; BLK, blank control; CD, cluster of differentiation; GEF, gefitinib; VEGF, vascular endothelial growth factor.
Discussion
Development of acquired resistance is crucial for the treatment of NSCLC with EGFR TKI because of its high initial response rate.Citation24 Secondary mutation of EGFR, including T790M and proto-oncogene MET amplification, have been reported as the two most important mechanisms responsible for gefitinib acquired resistance.Citation7,Citation9 These two even share a common consequence: aberrant activation of the PI3K/AKT/mTOR signaling pathway.
PI3K is a pivotal kinase involved in the transmission of signals, and the correlation of aberrant PI3K signaling with cancer development has long been confirmed.Citation25–Citation28 In cancers, PI3K activation is frequently over-stimulated by amplificationCitation29 and/or mutationsCitation30 of the PI3KCA gene, as well as by functional deficiency of the phosphatase and tensin homologue, an important tumor suppressor gene.Citation31 These studies have identified PI3K as an attractive therapeutic target for cancer treatment. BEZ235 is a potent and reversible adenosine triphosphatase competitor that inhibits p110 of PI3K and mTOR and suppresses several downstream effectors of the PI3K pathway.Citation13
Isolated from a non-smoking female patient suffering from lung adenocarcinoma, H1975 cell line harbors EGFR T790M and L858R mutations, and has been used as a gefitinib-resistant cell model.Citation19
Our in vitro study demonstrated that BEZ235 effectively inhibited the growth of H1975 cells. In vivo BEZ235 treatment of nude mice xenografts showed that the tumors’ size decreased significantly. In addition, BEZ235 inhibited H1975 cells migration in a dose-dependent manner. Taken together, these results suggest that BEZ235 might be an effective alternative for the treatment of NSCLC resistant to gefitinib.
To understand the underlying mechanism of growth inhibition induced by BEZ235 in gefitinib-resistant NSCLC, we examined the protein levels of the PI3K/AKT/mTOR signaling pathway after drug treatment. Both in vitro and in vivo studies demonstrated that administration of the BEZ235 resulted in phosphorylated proteins’ reduction. Thus, we were led to speculate that down-regulation of pAKT and pS6 is one of the mechanisms by which BEZ235 inhibits the proliferation of H1975 cells. These findings were in agreement with previous studies which demonstrated that the inhibition of cell growth by BEZ235 is associated with PI3K/AKT/mTOR phosphorylation.Citation17,Citation21,Citation32,Citation33
Gefitinib has been reported to decrease VEGF expression in squamous cell carcinoma cells in either a hypoxia-inducible factor (HIF-1)-dependent or HIF-1-independent manner.Citation34 Furthermore, the HIF-1α has also been confirmed to be a downstream factor of the mTOR signaling pathway,Citation35 suggesting that the EGFR/PI3K/AKT/mTOR signal axis regulates VEGF expression and thereby tumor angiogenesis. Consistent with the finding of studies with different malignancies, BEZ235 induced a significantly reduced VEGF expression in xenograft animal models.Citation16,Citation36,Citation37 Parallel results were observed for CD31, a biomarker that has been used to measure microvessel density in tumor tissues.
In summary, the present study has demonstrated that BEZ235 specifically inhibited the growth of gefitinib-resistant tumors, both in cellular models and in xenografts. Further evidence showed that this growth inhibition was associated with the PI3K/AKT/mTOR phosphorylation. Our study provided a foundation for the further clinical use of BEZ235 in the treatment of gefitinib-resistant tumors.
Acknowledgments
This study was supported by Wu Jieping medical foundation, Beijing, People’s Republic of China (No 320.6700.09069). We are indebted to AstraZeneca for kindly supplying gefitinib, and to Meera Dassarath for her assistance in the preparation of the manuscript.
Disclosure
The authors have no conflicts of interest in this work.
References
- SiegelRNaishadhamDJemalACancer statistics, 2012CA Cancer J Clin2012621102922237781
- SharmaSVBellDWSettlemanJHaberDAEpidermal growth factor receptor mutations in lung cancerNat Rev Cancer20077316918117318210
- LynchTJBellDWSordellaRActivating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinibN Engl J Med2004350212129213915118073
- GazdarAFActivating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitorsOncogene200928Suppl 1S24S3119680293
- EngelmanJASettlemanJAcquired resistance to tyrosine kinase inhibitors during cancer therapyCurr Opin Genet Dev2008181737918325754
- PaoWMillerVAPolitiKAAcquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domainPLoS Med200523e7315737014
- KobayashiSBoggonTJDayaramTEGFR mutation and resistance of non-small-cell lung cancer to GefitinibN Engl J Med2005352878679215728811
- KuboTYamamotoHLockwoodWWMET gene amplification or EGFR mutation activate MET in lung cancers untreated with EGFR tyrosine kinase inhibitorsInt J Cancer200912481778178419117057
- BeanJBrennanCShihJYMET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinibProc Natl Acad Sci U S A200710452209322093718093943
- EngelmanJAZejnullahuKMitsudomiTMET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signalingScience200731658271039104317463250
- VivancolSawyersCLThe phosphatidylinositol 3-Kinase-AKT pathway in human cancerNat Rev Cancer20022748950112094235
- SarbassovDDGuertinDAAliSMSabatiniDMPhosphorylation and regulation of Akt/PKB by the rictor-mTOR complexScience200530757121098110115718470
- EngelmanJALuoJCantleyLCThe evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolismNat Rev Genet20067860661916847462
- MairaSMStaufferFBrueggenJIdentification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activityMol Cancer Ther2008771851186318606717
- BaumannPMandl-WeberSOduncuFSchmidmaierRThe novel orally bioavailable inhibitor of phosphoinositol-3-kinase and mammalian target of rapamycin, NVP-BEZ235, inhibits growth and proliferation in multiple myelomaExp Cell Res2009315348549719071109
- LiuTJKoulDLaFortuneTNVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomasMol Cancer Ther2009882204221019671762
- ZitoCRJilaveanuLBAnagnostouVMulti-level targeting of the phosphatidylinositol-3-kinase pathway in non-small cell lung cancer cellsPLoS One201272e3133122355357
- SlivaDLabarrereCSlivovaVSedlakMLloydFPJrHoNWGanoderma lucidum suppresses motility of highly invasive breast and prostate cancer cellsBiochem Biophys Res Commun2002298460361212408995
- KobayashiSJiHYuzaYAn alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptorCancer Res200565167096710116103058
- FaberACLiDSongYDifferential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibitionProc Natl Acad Sci U S A200910646195031950819850869
- SerraVMarkmanBScaltritiMNVP-BEZ235, a dual PI3K/m TOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutationsCancer Res200868198022803018829560
- DaidoSKanzawaTYamamotoATakeuchiHKondoYKondoSPivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cellsCancer Res200464124286429315205343
- HamadaKSasakiTKoniPAThe PTEN/PI3K pathway governs normal vascular development and tumor angiogenesisGenes Dev200519172054206516107612
- CostaDBKobayashiSTenenDGHubermanMSPooled analysis of the prospective trials of gefitinib monotherapy for EGFR-mutant non-small cell lung cancersLung Cancer20075819510317610986
- ErtmerAHuberVGilchSThe anticancer drug imatinib induces cellular autophagyLeukemia200721593694217330103
- VogtPKBaderAGKangSPhosphoinositide 3-kinase: from viral oncoprotein to drug targetVirology2006344113113816364744
- WorkmanPClarkePARaynaudFIvan MontfortRLDrugging the PI3 kinome: from chemical tools to drugs in the clinicCancer Res20107062146215720179189
- WorkmanPClarkePAGuillardSRaynaudFIDrugging the PI3 kinomeNat Biotechnol200624779479616841064
- SugawaNEkstrandAJJamesCDCollinsVPIdentical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomasProc Natl Acad Sci U S A19908721860286062236070
- GalliaGLRandVSiuIMPIK3CA gene mutations in pediatric and adult glioblastoma multiformeMol Cancer Res200641070971417050665
- LiJYenCLiawDPTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancerScience19972755308194319479072974
- XuCXLiYYuePThe combination of RAD001 and NVP-BEZ235 exerts synergistic anticancer activity against non-small cell lung cancer in vitro and in vivoPLoS One201166e2089921695126
- Brünner-KubathCShabbirWSaferdingVThe PI3 kinase/mTOR blocker NVP-BEZ235 overrides resistance against irreversible ErbB inhibitors in breast cancer cellsBreast Cancer Res Treat2011129238740021046231
- PoreNJiangZGuptaACernigliaGKaoGDMaityAEGFR tyrosine kinase inhibitors decrease VEGF expression by both hypoxia-inducible factor (HIF)-1-independent and HIF-1-dependent mechanismsCancer Res20066663197320416540671
- HudsonCCLiuMChiangGGRegulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycinMol Cell Biol200222207004701412242281
- RoperJRichardsonMPWangWVThe dual PI3K/mTOR inhibitor NVP-BEZ235 induces tumor regression in a genetically engineered mouse model of PIK3CA wild-type colorectal cancerPLoS One201169e2513221966435
- SchnellCRStaufferFAllegriniPREffects of the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 on the tumor vasculature: implications for clinical imagingCancer Res200868166598660718701483