
Abstract
Lung cancer, mostly nonsmall cell lung cancer, continues to be the leading cause of cancer-related death worldwide. With the development of tyrosine kinase inhibitors that selectively target lung cancer-related epidermal growth factor receptor mutations, management of advanced nonsmall cell lung cancer has been greatly transformed. Improvements in progression-free survival and life quality of the patients were observed in numerous clinical studies. However, overall survival is not prolonged because of later-acquired drug resistance. Recent studies reveal a heterogeneous subclonal architecture of lung cancer, so it is speculated that the tumor may rapidly adapt to environmental changes via a Darwinian selection mechanism. In this review, we aim to provide an overview of both spatial and temporal tumor heterogeneity as potential mechanisms underlying epidermal growth factor receptor tyrosine kinase inhibitor resistance in nonsmall cell lung cancer and summarize the possible origins of tumor heterogeneity covering theories of cancer stem cells and clonal evolution, as well as genomic instability and epigenetic aberrations in lung cancer. Moreover, investigational measures that overcome heterogeneity-associated drug resistance and new assays to improve tumor assessment are also discussed.
Introduction
Despite many novel cancer treatments developed in the past decades, advanced lung cancer, mostly nonsmall cell lung cancer (NSCLC), is still the leading cause of cancer death and poses a great threat to public health. So far, early resection is still the only way to cure the disease. However, more than two-thirds of the patients are beyond the curable stage at diagnosis.Citation1 The estimated 5-year survival rate of advanced NSCLC is less than 10%, and the median life expectancy is only 4 months if left untreated.Citation1,Citation2 Chemotherapy may slightly prolong survival, but the adverse effects are sometimes unbearable, and the tumor eventually becomes resistant to the drug.Citation3
Based on the “oncogene addiction” theory,Citation4 the development of new compounds targeting tumor-driving pathways (eg, the epidermal growth factor receptor [EGFR] signaling pathway) brings new hope to cancer patients.Citation5 Identification of mutations in these pathways, followed by targeted therapy, gives rise to personalized therapy in several types of cancer.Citation6 In advanced NSCLC, tyrosine kinase inhibitors (TKIs; eg, gefitinib and erlotinib) competitively block EGFR and suppress tumor growth, conferring survival benefits with acceptable adverse effects on patients who have failed chemotherapy.Citation7 In patients with NSCLC and sensitive EGFR mutations, TKIs are recommended as first-line treatments because of their significantly increased response rates and prolonged progression-free survival in this group of patients.Citation8,Citation9
Although early results are encouraging, EGFR TKIs only show effects in a small number of patients (∼10%).Citation7,Citation8 Moreover, even tumors that initially respond to the TKIs eventually become refractory to the therapy,Citation9,Citation10 and the tumors rapidly regrow.Citation8,Citation11 Mechanisms underlying this later-acquired drug resistance are still unknown. However, recent reports reveal a critical role of tumor heterogeneity in the development of drug resistance.Citation12–Citation15 It is now believed that cancer is actually a process of clonal evolution and that every single tumor is a complex hierarchy of tumor subclones resulting from distinctive microenvironmental adaptation.Citation13 These heterogeneous tumor cells provide the material that Darwinian selection can work on; so the fittest subclones can survive through the therapeutic intervention and then dominate regrowth of the tumor.Citation13,Citation16 In this review, we provide an overview of currently recognized causes of tumor heterogeneity, including cancer stem cells (CSCs) and genomic instability, as well as epigenetic aberrations, and discuss their roles in NSCLC. Spatial and temporal tumor heterogeneity as a mechanism of the primary and acquired EGFR TKI resistance in advanced NSCLC is also elucidated. Finally, new approaches to tackling the challenge of tumor heterogeneity are summarized, which may bring new hope to future targeted therapy.
Origins of tumor heterogeneity in NSCLC
CSC and acquired drug resistance in NSCLC
According to the CSC theory, individual tumors are organized into a hierarchy composed of subsets of tumorigenic stems cells and their nontumorigenic progeny cells.Citation16–Citation19 Heterogeneous subclonal lineages in solid tumor are branched from distinctive CSCs and are dynamically maintained by these regenerating cells.Citation15,Citation16 Thus, identification of the CSCs followed by specific treatment targeting developmental signaling pathways (eg, Notch, hedgehog, and transforming growth factor-beta pathways) may be more effective in suppressing tumor growth and preventing drug resistance.Citation15,Citation20
The existence of CSCs in lung cancer is supported by the fact that only a small population of tumor cells (<1.5%) from adenocarcinoma samples possess clonal forming and tumorigenic ability.Citation21 These lung CSCs are thought to be derived from the self-renewing epithelial and bronchioalveolar cellsCitation19,Citation22–Citation24 as a result of oncogenic KRAS and EGFR activationCitation23,Citation25 and may exhibit increased aldehyde dehydrogenase activity.Citation20 It has been demonstrated that CSC-like cell population, characterized by elevated expression of cell cycle genes and increased aldehyde dehydrogenase activity, is increased in the EGFR mutant lung cancer cell line with acquired resistance to erlotinib,Citation26,Citation27 indicating a potential role of CSCs in EGFR TKI resistance. However, some nontumorigenic progeny subclones may regain tumorigenic capacity and transform back to stem-like cells.Citation16 Without specific biomarkers, it is very difficult to distinguish CSCs from nontumorigenic cells.Citation19,Citation20 Therefore, CSCs targeting therapy are still quite premature, and research in this field is complicated.Citation16,Citation18
Genomic and chromosomal instability of NSCLC
In addition to CSCs, genomic and chromosomal instability may also contribute to heterogeneity in solid tumors. In normal lung cells, the genome is replicated with high fidelity, and mutations are poorly tolerated, which is attributed to stringent intrinsic checking mechanisms such as base and nucleotide excision repair, mismatch repair, telomere maintenance, and double-strand break repair.Citation28 Malfunctioning checking mechanisms greatly increase the mutation rate and significantly accelerate the process of clonal evolution, leading to carcinogenesis and tumor heterogeneity.Citation28 Chromosomal instability has been shown to correlate with shorter survival in patients with NSCLC,Citation29 possibly because these unstable cancer cells display a higher multidrug-resistant capacity compared with stable cells.Citation30 In mice, lung adenocarcinoma with chromosome instability induced by overexpression of a mitotic checkpoint gene Mad2 is highly aneuploid, correlating with a higher tumor recurrent rate after anticancer treatment.Citation31 Moreover, next-generation sequencing techniques have identified a number of hallmark genomic mutations that are involved in DNA maintenance and mitotic progression, which may predict the prognosis of NSCLC.Citation32–Citation34 Some of these genomic mutations may be used for targets able to be drugged, highlighting the importance of maintaining genomic stability for tumor control.Citation15
Epigenetic aberrations of NSCLC
Apart from genomic mutations, gene expression and phenotypic changes of tumor cells can be affected by epigenetic aberrations including abnormal DNA methylation at CpG islands, dysregulated histone modification, and changes in pathways regulating these epigenetic mechanisms.Citation15,Citation35–Citation37 Compared with genomic mutations, epigenetic changes are reversible and more plastic under environmental pressures, adding further complexity to tumor heterogeneity and contributing to the development of drug resistance.Citation35,Citation38 In NSCLC, global DNA hypomethylation is considered a main cause of genomic instability,Citation39 as well as abnormal oncogene expression.Citation40 A recent analysis of cancer-specific differentially DNA-methylated regions by whole-genome bisulfite sequencing reveals drastic stochastic differences in the differentially methylated regions (DMRs) and a significant loss of sharply delimited methylation boundaries at CpG islands of the DNA in samples from several malignant tumors, including lung cancer.Citation41 Loss of the epigenetic marks in these cancer cells perhaps indicates the epigenome has been “reset”; thus, these cells have greater potential to reshape themselves under selective environmental pressures.Citation41 Consistently, status of global histone modification is strongly associated with prognosis of patients with NSCLC, and it can predict tumor recurrence in early-stage patients.Citation42,Citation43 Notably, epigenetic mechanisms may function in a different dimension from genomic heterogeneity during tumor evolution, through which the tumor cells are not only passively selected by the environments but also may actively change themselves to adapt external stress.Citation35,Citation41
Microenvironmental adaptation and tumor heterogeneity
The extrinsic compartments of tumor cells are composed of disorganized stromal cells including fibroblasts and vasculature and immune cells.Citation44 Distinct microenvironments characterized by varied degrees of selective pressures such as oxygen, acidity, and tumor growth factors may select for mutations that engender subclonal survival and expansion, thus exerting great influence on tumor heterogeneity.Citation45 Moreover, the extrinsic tumor microenvironment may also participate in the development of drug resistance by forming an adaptive, reciprocal signaling loop with tumor cells, thus providing a protective compartment in response to anticancer treatments.Citation44,Citation46 Hepatocyte growth factor (HGF), for instance, is secreted by stromal fibroblasts under the stimulation of tumor-derived factors and strongly propagates tumor expansion by activating MET signaling pathway in NSCLC.Citation47–Citation49 High levels of tumor HGF are associated with EGFR TKI resistance because the activation of MET induces the common downstream prosurvival signaling, bypassing EGFR inhibition.Citation49,Citation50 Moreover, the tumor vascular network featured by dysregulated angiogenesis and reorganization of existing vessels also contributes to tumor heterogeneity. Poorly developed tumor vasculature results in variations in oxygen and nutrient supply within the tumor, as well as affecting drug delivery.Citation44 Microvessel density as an index of angiogenesis may predict survival of the patients with lung cancer,Citation51 and elevated expression proangiogenic ligand vascular endothelial growth factor (VEGF) is associated with poorer prognosis in NSCLC.Citation52 It is therefore conceived that coinhibition of VEGF signaling may confer a benefit on tumor suppression. However, coadministration of VEGF targeted therapy has been shown to reduce the delivery of radiolabeled chemotherapy in patients with NSCLC, as determined by positron emission tomography.Citation53 Nevertheless, a combinational strategy targeting tumor stromal cells remains one of the most important research areas in personalized therapyCitation44 and will be further discussed later.
Spatial tumor heterogeneity and TKI resistance in NSCLC
Intertumor heterogeneity and TKI resistance
As previously mentioned, individual tumors are driven by distinct prosurvival signaling pathways as a result of heterogeneous genomic mutations.Citation5,Citation6 Optimal outcomes of personalized therapy can be achieved by matching the specific lesions with the corresponding treatments, whereas tumors without sensitive mutations are unsusceptible to treatment.Citation6 So far, a number of mutations with the tumor driving capacity have been identifiedCitation54–Citation57 (). Frequencies of these driver mutations in NSCLC are associated with histologic subtypes, sex, ethics, age, and past smoking history.Citation58–Citation60 The patients with wild-type EGFR are insensitive to gefitinibCitation9 or erlotinib treatment.Citation8,Citation61 KRAS and BRAF mutants, being mutually exclusively expressed with EGFR in ∼25% and 3% of patients with NSCLC, respectively,Citation60,Citation62 predict poorer prognosis in patients after EGFR-targeted treatment.Citation63,Citation64 Similarly, NSCLC patients harboring EML4-ALK fusion oncogene (∼7%)Citation65 are sensitive to TKI crizotinib but resistant to EGFR TKIs.Citation66,Citation67 Hence, intertumor heterogeneity of different mutations may confer primary resistance to any targeted therapy.Citation68,Citation69
Table 1 Distribution of known tumor-driving mutations and chromosomal fusions in advanced NSCLC
The sensitivity of different EGFR mutations to TKIs also varied between individuals. The EGFR gene is located on chromosome 7p12–7p13,Citation74 whereas the NSCLC-relevant mutations occur in exons 18–21, encoding the kinase domain of the receptor.Citation54 Although more than 188 EGFR mutations have been identified,Citation75 85% of TKI-sensitive clinical cases harbor only two major mutations:Citation76 in-frame deletions of exon 19 (45%–50%) and a point mutation L858R in exon 21 (40%–45%; ). Other uncommon TKI-sensitive mutations include amino acid substitution mutations of G719X in exon 18 and L861X in exon 21.Citation76,Citation77 These mutations cause profound activation of both prosurvival and antiapoptotic signaling cascades but also enhance the affinity of the receptor to gefitinib and erlotinib.Citation78,Citation79 In contrast, an insertion mutation in exon 20 (∼4% of EGFR mutations) has never been shown to confer any preferential binding or clinical responses to TKIsCitation77,Citation80,Citation81 but, similarly, elevates the activity of EGFR kinase.Citation82 In addition, deletion mutation in exon 19 seems to have a better response to gefitinib and erlotinib than mutations at other sites.Citation79,Citation83
Figure 1 Tumor-driving mutations in the tyrosine kinase domain of EGFR (epidermal growth factor receptor).
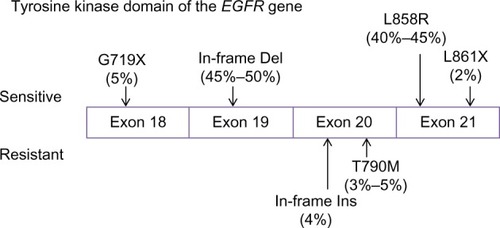
Variations in the expression of enzymes associated with EGFR signaling may also account for diverse responses to EGFR inhibition. BIM, a BCL2 proapoptotic family member regulated by ERK signaling, has recently been identified as a key mediator of TKI-induced apoptosis.Citation84–Citation86 Levels of pretreatment BIM expression in tumors specimens of NSCLC are different,Citation87 possibly as a result of an intronic deletion polymorphism in the coding region of BIM.Citation88 Low BIM RNA levels are associated with poor outcomes for EGFR inhibition.Citation87 In contrast, elevated tumoral expression of MCL1, an oncogenic member of the BCL2 family,Citation89,Citation90 may predispose NSCLC patients with EGFR-sensitizing mutants to native resistance.Citation91 In addition, mutations in PIK3CA, a p110α catalytic subunit of PI3K, may diminish gefitinib-induced apoptosis by activating bypass Akt signaling, thus conferring resistance to the TKIs.Citation92 The PIK3CA mutant is found in 4% of lung cancers and may cooccur with the EGFR mutation.Citation93
Intratumor heterogeneity and TKI resistance
Apart from differences between tumors, intratumor heterogeneity also presents at histologic, cellular, molecular, genetic, and epigenetic levels.Citation94 Early histological analysis of dissected NSCLC samples showed varied levels of extracellular receptors and stress-responsive genes in different regions of the tumor, indicating distinct subclonal interplay with external stress.Citation95,Citation96 Using a next-generation whole-genome sequencing technique, Gerlinger et al reveal that heterogeneous somatic mutations, divergent allelic profiles and ploidy heterogeneity vastly present in different regions of microdissected samples from the same renal tumor.Citation94 About 63%–69% of all somatic mutations are not detectable across every tumor region, and genes associated with good or poor prognosis can be detected at different regions of the same tumor, both of which reflect drastic subclonal diversity within the tumor.Citation94 The presence of intratumor heterogeneity largely diminishes the reliability of single-tumor biopsy prevalently used in hospital and poses a challenge to the current protocol of personalized therapy.Citation94
In lung cancer, whole-genome analysis of samples from different regions of the tumor is still lacking. By conducting deep digital sequencing analysis of 17 treatment-naïve lung adenocarcinomas, Govindan et alCitation15 found a multiclonal signature indicated by distinct variant allele frequency features in more than half of the samples. Some of these subclones were believed to be involved in progression and migration of the tumor.Citation15 It has been reported that some TKIs desensitizing mutations such as EGFR T790M mutationCitation97,Citation98 and amplification of METCitation98,Citation99 (both discussed later) may concomitantly present with TKIs sensitizing EGFR mutations in a minor group of patients before the TKI treatment. Considering heterogeneity within the tumor, it is not hard to imagine some less-prevalent subclones may be missed by the single-region biopsy, so that the actual coexistence rate of TKI-sensitive and TKI-resistant subclones may even be higher,Citation94 which may be further examined by genotyping microdissected samples from surgically resected lung tumors.
Notably, some researchers claim coexistence of both wild-type and mutant EGFR within the same tumor,Citation100,Citation101 as well as discordant EGFR mutational status between the primary tumor and the metastatic sites.Citation102,Citation103 These results are challenged by the reports showing highly homogeneous distribution of EGFR mutations across the primary tumorCitation104 and at the metastatic lesions.Citation104,Citation105 The high wild-type EGFR detection rate in the former studies was thought to be explained by normal tissue contamination, as well as technical variations (eg, the minor presence of EGFR mutations that may be neglected by less-sensitive methods).Citation104,Citation106,Citation107 Moreover, although some groups reported a conversion from EGFR mutant to wild-type after TKI treatment in occasional cases,Citation108 most of the studies show highly consistent EGFR mutations in tumors before and after the treatment.Citation57,Citation104,Citation109 It is hard to imagine why the conversion rate of mutant to wild-type after TKI treatment is so low if intratumor heterogeneity of EGFR mutation is not rare.Citation100,Citation110 In addition, Govindan et al applied computational analysis to the deep sequencing data of NSCLC samples and found that EGFR mutation exists in all subclones as a founder mutation of the tumor, suggesting this mutation may be acquired at the very initial phase of tumorigenesis.Citation15
Temporal tumor heterogeneity and acquired TKI resistance in NSCLC
TKI resistance after chemotherapy
Temporal heterogeneity reflects the dynamic tumor evolution in response to environmental changes including therapeutic interventions. Adaptive tumor subclones after this natural selection may then dominate the tumor clonal architecture, leading to disease progression.Citation14,Citation94,Citation111 Therefore, temporal tumor heterogeneity is perhaps more related to acquired resistance to chemotherapy or EGFR-targeted therapy in NSCLC.Citation108,Citation112
It has been noticed that the predictive value of pretreatment EGFR mutation status seen in the first-line TKI regimen is diminished in cohorts of TKI treatment after chemotherapy, and the tumor seems to be less sensitive to EGFR-targeted therapy after chemotherapy.Citation7,Citation9,Citation113 Bai et al compared EGFR mutation status before and after neoadjuvant chemotherapy in both lung tumor tissue and blood samples and found that EGFR mutant-positive NSCLC significantly decreased after chemotherapy.Citation114 However, these results may be misinterpreted because the reduction of EGFR mutant in the blood can also be affected by the amount of tumor cells being exposed to circulation, and chemotherapy may increase fibrotic content in the tumor, thus affecting the detection of mutant EGFR in the tumor.Citation115 De Pas et al compared EGFR expression on mediastinal lymph nodes at the primary biopsy with its expression on both the primary tumor and the residual mediastinal nodes after three cycles of chemotherapy in 47 patients with NSCLC and found reduced EGFR immunoreaction in 12% of the patients.Citation116 Platinum-based agents may reduce the sensitivity of lung cancer cells to EGFR TKIs as a result of PTEN loss and EGFR-independent AKT activation.Citation117 Although the precise mechanism of this chemotherapy-induced resistance to TKIs is still unclear, this issue merits further investigation because it may necessitate rebiopsy after chemotherapy for better guidance in targeted therapy.Citation109,Citation114
Secondary mutations and acquired TKIs resistance in NSCLC
In relapsed NSCLC after TKI treatment, secondary mutations that desensitize EGFR to its inhibitors are frequently detected (). T790M, which represents threonine-to-methionine substitution at position 790 in the gatekeeper residue of EGFR kinase domain because of a point mutation within exon 20, presents in about 50% of EGFR mutant NSCLCs that become resistant to TKIs after the treatment.Citation109,Citation112,Citation118,Citation119 EGFR of lung cancer cells with both TKI-sensitive mutations and T790M mutation regain affinity to ATP at the hydrophobic pocket, hence abrogating competitive inhibition of the receptor by gefitinib and erlotinib.Citation120 Other secondary mutations in the EGFR kinase domain that are associated with acquired resistance include L747S,Citation121 D761Y,Citation122 and T854A.Citation123 L747S resulting from deletion in exon 19 is located at the head of the loop between strand β3 and helix αC of the receptor and is believed to attenuate BIM upregulation and mitochondrial apoptosis induced by TKIs.Citation124 D761Y from exon 19 mutation occurs in the middle of the helix αC and forms a salt bridge that interacts with the α- and β-phosphates when ATP is present, reducing sensitivity of the EGFR to TKIs.Citation122 T854A mutation in exon 21 interferes the contact of erlotinib to the ATP pocket of the receptor, hence abrogating the inhibition of tyrosine phosphorylation by erlotinib.Citation123 These secondary mutations are rarer and cause less-potent resistance to TKIs than T790M.Citation54
Table 2 Temporal tumor heterogeneity and acquired TKI resistance in advanced NSCLC
Notably, it remains debatable whether T790M mutation truly developed after TKI exposure or whether the mutation preexists before the treatment and become more prevalent in the tumor architecture under selective pressure of the TKIs. There have been some reports showing T790M coexists with sensitive EGFR mutations before TKI treatments in a minor group of patients.Citation97,Citation125 Moreover, T790M mutation is significantly less prevalent in brain metastases, possibly because routine doses of TKIs hardly penetrate the blood–brain barrier and thus exert less selective pressure on the brain lesion.Citation54,Citation126,Citation127 In fact, the presence of secondary mutations in resistant NSCLC may not be an “all or none” phenomenon but, rather, a dynamic process in association with environmental pressure. Discontinuation of EGFR targeted therapy in resistant patients often leads to accelerated tumor growth and symptom progression,Citation128 suggesting at least some tumor cells are still under suppression by the EGFR inhibitors.Citation54,Citation128 Notably, in the absence of EGFR inhibition, the expression of mutations associated with acquired resistance may reversibly disappear, as revealed by serial biopsies.Citation112,Citation128 Consistently, reinitiation of EGFR-targeted therapy after a short period of TKI suspension may slow disease progression again in some cases.Citation112,Citation128,Citation129 Hence, larger cohorts are needed to evaluate whether dynamic mutation monitoring may benefit the patients who acquired resistance to targeted therapy.
Bypass signaling of EGFR inhibition
Compensatory activations of the “oncogene addiction” pathways via bypass signaling after EGFR inhibition represent another mechanism of acquired TKI resistance (). Focal amplification of MET oncogene, for instance, is observed in about 5%–20% of NSCLCs after treatment with EGFR inhibitors.Citation109,Citation112,Citation130 MET encodes a transmembrane tyrosine kinase receptor for HGF. Activation of MET phosphorylates the downstream ERBB3, activating Akt signaling. Amplification of MET oncogene causes hyperactivation of PI3K and Akt, bypassing the blockade of this pathway by EGFR inhibitors and promoting tumor survival.Citation130 In addition, amplification of HER2 and PI3KCA mutations, both of which activate the common downstream pathways, are detected in about 12% and 5% of EGFR mutant NSCLC patients, respectively.Citation57,Citation112,Citation131 Similar to T790M mutation, MET amplification may also exist before the treatment.Citation99 Turke et al reported MET amplification in rare tumor cells (<1%) from treatment-naïve NSCLC samples (5 of 27 patients).Citation99 Four of these five patients later developed TKI resistance as a result of MET amplification, suggesting acquired resistance may emerge as a result of the survival advantage of pre-existing resistant tumor subclones.Citation12,Citation99,Citation112
Phenotypic changes and epigenetic alterations in acquired TKI resistance
Apart from genetic aberrations, phenotypic changes of the tumor cells such as epithelial to mesenchymal transitionCitation132–Citation134 and transformation to a small-cell-like carcinomaCitation77,Citation81 are frequently observed in NSCLC after TKI treatments, possibly as a result of altered epigenetic modifications.Citation135 For instance, gefitinib-induced DNA hypermethylation is associated with decreased expression of micro-RNA (miRNA)-200c, resulting in overexpression of aldehyde dehydrogenase isoform 1, which contributes to the epithelial-to-mesenchymal transition and the presence of stem cell-like properties.Citation26 Moreover, DNA hypermethylation at the promoter region of death-associated protein kinase may silence this gene in the NSCLC cell line,Citation136 leading to erlotinib resistance, whereas transient induction of death-associated protein kinase by gene transfection resensitizes the cells to erlotinib.Citation136 Other hypermethylated tumor DNA such as Wnt antagonist SFRP5Citation137 and mitotic stress checkpoint gene CHFRCitation138 have also been reported to affect TKI sensitivity. Although it is still unclear to what extent epigenetic mechanisms are involved in acquired TKI resistance, considering its importance in clonal evolution and tumor heterogeneity, more efforts should be invested on research and new drug development in this area.Citation38
Advances in molecular diagnosis and tumor monitoring in NSCLC
Next-generation sequencing for the search of new molecular targets
Next-generation high-throughput sequencing techniques now allow us to map the genomic landscape of lung cancer at lower cost, but at orders of magnitude higher speed, compared with the traditional Sanger technique.Citation146,Citation147 These new platforms exponentially enhance our capacity to search for new oncogenes that can be made into drugs.Citation15,Citation148 Imielinski et alCitation149 conducted whole-exome and whole-genome sequencing on 183 paired samples of lung adenocarcinoma and normal lung tissues from treatment-naïve patients, from which 25 hallmark carcinogenic genes including 19 previously reported genes and 6 novel mutants (NKX2-1, TERT, PTEN, MDM2, CCND1, and MYC) were identified. They also detected frequent somatic mutations in epigenetic and splicing factor genes including U2AF1, ARID1A, RBM10, SETD2, and BRD3, suggesting epigenetic regulation as a new hallmark of lung carcinogenesis.Citation149 In 2012, a comprehensive genomic analysis of squamous cell lung cancer was reported by the Cancer Genome Atlas Research Network, showing statistically recurrent mutations in 11 genes, including TP53 mutation in almost all specimens and significantly altered signaling pathways such as NRF2/KEAP1, PI3K/AKT, and CKKN2A/RB1. By matching these genomic aberrations with the currently available US Food and Drug Administration-approved targeted therapeutic agent library, the researchers found 64% of patients in this study possess at least one gene that can potentially be a drug.Citation150 Moreover, the emerging single-cell profiling technique based on whole-genome amplification enables us to process sparse clinical biopsy samples from fine-needle aspirates or core biopsy specimens in which only hundreds of tumor cells are available.Citation151,Citation152 With the development of these advanced techniques, we should be able to obtain a better view of tumor pathology, and new drug development may be greatly accelerated in the foreseeable future.Citation146
Dynamic tumor monitoring by circulating biomarkers
Because cancer cells continue to evolve, dynamically monitoring molecular changes in the tumor should be involved in the personalized therapy. Rebiopsy of the tumor is a straightforward measure but usually confronts ethical issues.Citation153 Instead, dynamic monitoring circulating tumor biomarkers, also called liquid biopsy, may become a more realistic option.Citation154
Circulating tumor cells and DNA
Malignant tumor cells are known to present in the blood of patients.Citation155 However, isolation of these cancer cells from blood samples is rather difficult.Citation156 Maheswaran and coworkers developed a microfluidic-based device in which blood flows through highly condensed micropores coated with epithelial-cell adhesion molecule antibody. The device is able to capture and quantify circulating tumor cells (Ct-cells) at relatively higher efficiency.Citation156 By combining this isolating technique with the ultrasensitive Scorpion Amplification Refractory Mutation System, the researchers claimed 100% specificity and 92% sensitivity in detecting EGFR mutations and secondary T790M mutation from Ct-cells compared with direct tumor biopsy. Using this technique, they also found an increase of T790M mutant allele in Ct-cells after TKI treatment as a sign of drug resistance.Citation156 Moreover, using the whole-genome amplification technique, Ni et al recently reported whole-exome sequencing profiles of single Ct-cells obtained from patients with lung adenocarcinoma.Citation157 These cells exhibit distinct patterns of single-nucleotide variation, but highly reproducible copy number variations. Genes relevant to drug resistance and phenotypic transitions were found to be enriched after chemotherapy.Citation157 In addition, the detection of mutated tumor DNA in cell-free plasma has also been reported, and the cost is relatively lower, as cell isolation is not required.Citation158 However, the sensitivity of direct genotyping for DNA mutation in the plasma is markedly lower (43%–73% compared with for direct tumor biopsy),Citation110,Citation114,Citation159–Citation161 possibly because circulating tumor DNA (Ct-DNA) is more fragmented and it is difficult to extract fragmented tumor DNA from the blood for subsequent amplification.Citation160 Nevertheless, it has been reported that sensitive EGFR mutants are reduced in the Ct-DNA from the patients with NSCLC after TKI treatment, accompanied by the emergence of resistant DNA mutants.Citation114,Citation161 Although larger cohort studies are required before the spread of plasma genotyping, preliminary results have shown a promising future of both Ct-Cells and Ct-DNA in tumor monitoring.
Other circulating nucleotide biomarkers
In addition to Ct-Cells and Ct-DNA, specific miRNAs and methylated tumor DNA in the plasma may also be used as biomarkers of tumor evolution in patients with NSCLC.Citation162–Citation164 Compared with Ct-DNA and Ct-Cells, detection of methylated DNA in the plasma is less expensive and more time-saving.Citation35,Citation165 It also has been reported that methylated RARB2 and RASSF1A are increased in the plasma of patients with NSCLC.Citation166 Plasma levels of these methylated genes decrease after tumor resection and chemotherapy, whereas a rebound rise of these plasma biomarkers manifests tumor relapse.Citation166 So far, very few methylated biomarkers have been identified in the plasma with a predictive value of TKI resistance. Salazar et al showed that unmethylated CHFR may predict prolonged survival in patients receiving EGFR TKIs.Citation138 Other hypermethylated tumor DNA associated with TKI resistance, such as death-associated protein kinase,Citation136 SFRP5,Citation137 may also be worth further validation as plasma biomarkers for tumor monitoring. Moreover, by introducing microelectronic technology, multiple methylated tumor DNAs can be detected and quantified simultaneously, which may further improve efficiency of the detection.Citation167
miRNA is a class of short (18–25 nucleotides), noncoding RNA that binds to complementary mRNA for selective degradation, thus causing a cascade of effects.Citation168 Circulating miRNAs, being more stable than other plasma nucleotides, may emerge as another group of biomarkers for cancer monitoring.Citation168–Citation170 In female nonsmokers with lung adenocarcinoma, levels of plasma miR-195 and miR-122 can differentiate the patients who may benefit from EGFR TKI treatment.Citation171 Circulating miR-21, an important regulator of cell apoptosis and proliferation,Citation172,Citation173 was found to be elevated in the patients with NSCLC after TKI treatment, indicating drug resistance.Citation172 With the development of nanotechnology, highly sensitive probes in plasma are able to directly detect miRNA without labeling or amplification, which may significantly reduce the time and cost of cancer monitoring.Citation174
Proteomic biomarkers and VeriStrat
In addition to nucleotide biomarkers, proteins in the blood may also provide valuable information for tumor evaluation before and after EGFR-targeted therapy. VeriStrat, a mass spectrometry-based high-throughput platform, can analyze proteomic profiles using only 5 μL serum from the patients and subsequently classify the patients for “good” or “poor” response to TKIs.Citation175 To our knowledge, VeriStrat is the only liquid biopsy tool for NSCLC patient stratification before TKI treatment that has been validated in a Phase III clinical trial,Citation176,Citation177 which revealed its prognostic value in predicting overall survival and time to progression.Citation177–Citation180 However, the VeriStrat test seems to be more powerful in selecting patients who may have worse outcomes on TKIs than on chemotherapy, rather than finding patients who may benefit more from TKIs because the overall survival of “good” patients on either treatment is similar.Citation177 In addition, results of VeriStrat may also indicate temporal changes of the tumor. Lazzari et al processed sequentially collected serum samples from 111 NSCLC patients receiving gefitinib and found that one third of these patients converted from good to poor after the treatment, in relevance to drug resistance.Citation181 Further proteomic analysis identified overexpressed serum amyloid A protein 1 (SAA1) in plasma from patients with poor VeriStrat results, which is responsible for the generation of four mass signals in the test.Citation182 Dynamic change of this protein in the plasma during the course of TKI treatment may merit further investigations.Citation182
Advances in the next generation of targeted therapy
Next-generation irreversible TKIs
Apart from the advances in tumor diagnostics, new inhibitors of EGFR have also been developed and examined in clinical trials (). First-generation TKIs such as erlotinib and gefitinib inhibit EGF receptors by reversibly competing with ATP, but the inhibition can be abrogated by secondary EGFR mutations (eg, T790M) because of the increased affinity of EGFR to ATP.Citation5,Citation183 Second-generation TKIs (eg, afatinib [BIBW2992], dacomitinib [PF00299804], and neratinib [HKI-272]) form irreversible covalent bonds with the EGF receptor and also inhibit other EGFR family members (ERBB2 and ERBB4), and thus may be more potent in tumor suppression.Citation183–Citation185 Afatinib, an irreversible EGFR/ ERBB2 dual inhibitor, has been extensively studied in a serial of clinical trials of NSCLC.Citation186–Citation190 In patients who had failed both chemotherapy and TKI treatment, afatinib significantly prolonged progression-free survival and increased tumor response rate compared with placebo, although no benefit was seen in overall survival.Citation187 Dacomitinib, another pan-ERB inhibitor, also exhibits superiority over erlotinib as the second-line therapy after chemotherapy in unselected patients with NSCLC.Citation191 The main adverse events of the second-generation TKIs include diarrhea, dysphagia, and sore mouth,Citation192 possibly as a result of pan-ERB inhibition.Citation193 Severe gastrointestinal reactions of the second-generation TKIs may reduce bioavailability of the drugs.Citation193 To specifically target T790M mutation, several third-generation TKIs have been developed and are currently undergoing clinical or preclinical investigations. Of note, these highly specific inhibitors may cause much less damage to EGFR in the gastrointestinal tract and skin hair bulbs compared with their predecessors,Citation194,Citation195 and hence intestinal absorption of these drugs may be increased. It is hoped these new drugs may resolve the current clinical dilemma and improve outcomes of targeted therapy.Citation12
Table 3 Clinical trials of next-generation TKIs in advanced NSCLC
Combinational therapy
As previously discussed, activation of pathways bypassing EGFR is another adaptive mechanism against TKIs. In murine lung tumors cotransfected with mutant EGFR and inducible MET oncogenes, treatment with EGFR inhibitor alone failed to inhibit the tumor growth, but addition of the MET inhibitor crizotinib significantly shrinks the tumor.Citation195,Citation199 In HGF overexpressing lung cancer cell lines, crizotinib and gefitinib together suppress TKI-resistant tumor cell growth.Citation200 Clinical evaluation of the effectiveness of dual MET/EGFR inhibition is currently undergoing.Citation201,Citation202 Moreover, HSP90, a protein-folding chaperone participating in EGFR stabilization in the tumor, may also emerge as a target for tumor control.Citation203–Citation205 Inhibition of HSP90 in lung cancer cell lines and animal models increases the sensitivity to TKIs and enhances tumor suppression Citation203,Citation206,Citation207 In an early clinical trial, HSP90 inhibitor ganetespib showed a notable disease control rate in patients refractory to chemotherapy and EGFR TKIs, and the adverse effects were acceptable.Citation208 Further clinical cohorts are needed to evaluate the efficacy and safety of HSP90 inhibitors alone or in combination with other targeted treatments.Citation209
Abnormal activation of angiogenesis manifested by overexpression of VEGF is another hallmark of progressing NSCLC.Citation210,Citation211 In patients with advanced NSCLC, chemotherapy plus VEGF targeting monoclonal antibody bevacizumab has been shown to prolong progression-free survival (6.2 versus 4.5 months) and overall survival (12.3 versus 10.3 months) compared with chemotherapy alone.Citation212 Close crosstalk between VEGF and EGFR signaling cascades provides a rationale to concurrently inhibit both pathways. The benefit of combining bevacizumab with erlotinib for the treatment of recurrent NSCLC after chemotherapy has been observed in Phase I/II studies, and the adverse effects are tolerable.Citation213,Citation214 However, subsequent Phase III trials comparing combined treatment of erlotinib and bevacizumab with erlotinib alone as the second-line treatment showed no superiority of the combination in terms of progression-free survival and overall survival.Citation215,Citation216 Notably, these trials are all conducted in unselected patients.Citation210,Citation211 VeriStrat-based proteomic analysis may effectively distinguish NSCLC patients who are likely to benefit from the dual inhibition. Patients with a good VeriStrat result had significantly longer progression-free and overall survival (18.9 versus 6.3 weeks and 71.4 versus 19.9 weeks, respectively) than the patients with a poor result.Citation217 It is hoped that outcomes of the dual EGFR/VEGF inhibition may be improved in patients with activating EGFR mutations.Citation218
Summary and perspectives
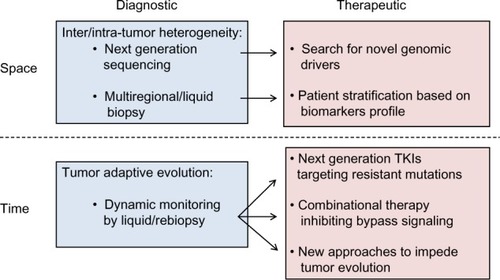
Despite tremendous efforts made to improve outcomes of cancer care in the past decades, the death rate of patients with advanced NSCLC is still very high. Personalized cancer treatments targeting specific tumor-driving mutations provide new options for patients who failed chemotherapy, greatly transforming cancer care.Citation219 However, despite an initial response, the patients eventually die from disease progression as a result of drug resistance. With a deeper understanding of tumor biology, we now realize this later-developed drug resistance may turn up as a result of tumor heterogeneity and clonal adaptation. In fact, complexity of the adaptive process is probably much beyond our imagination because CSCs, abnormalities in mitotic-maintaining genes, and epigenetic mechanisms may all contribute to the process. In terms of EGFR-targeted therapy, resistant mechanisms (eg, EGFR T790M mutation and MET amplification) emerge as a result of the tumor evolution after EGFR inhibition by gefitinib or erlotinib. Therefore, biomarkers that are sensitive to the resistant tumor clones and a new compound with specific effects on these resistant mechanisms have become the focus of research in this field (). Moreover, instead of biopsy at a single time, next-generation personalization may involve multiple rounds of tumor biopsies, followed by changes of the treatment to overcome the dynamic tumor evolution and drug resistance.Citation6,Citation220,Citation221 At present, laboratory findings are rapidly translated to clinical applications. Novel molecular targeted compounds and less invasive diagnostic tests are currently undergoing clinical evaluation, with some showing encouraging results. Cost/effectiveness evaluation is needed before these new tests and drugs can be spread for clinical use. Future studies should further elucidate the mechanism underlying tumor evolution, and measures to retard its process merit more investigation. With the advances seen in this highly active field, improvements in the clinical outcome of the patients with advanced NSCLC can be expected in the foreseeable future.
Figure 2 Schematic summary of measures to overcome tumor heterogeneity and drug resistance in lung cancer.
Abbreviation: TKI, tyrosine kinase inhibitors.
Disclosure
The authors report no conflicts of interest in this work.
References
- JemalASiegelRXuJWardECancer statistics, 2010CA Cancer J Clin201060527730020610543
- RappEPaterJLWillanAChemotherapy can prolong survival in patients with advanced non-small-cell lung cancer – report of a Canadian multicenter randomized trialJ Clin Oncol1988646336412833577
- SchillerJHHarringtonDBelaniCPEastern Cooperative Oncology GroupComparison of four chemotherapy regimens for advanced non-small-cell lung cancerN Engl J Med20023462929811784875
- WeinsteinIBJoeAKMechanisms of disease: Oncogene addiction – a rationale for molecular targeting in cancer therapyNat Clin Pract Oncol20063844845716894390
- GoffinJRZbukKEpidermal growth factor receptor: pathway, therapies, and pipelineClin Ther20133591282130324054705
- McDermottUSettlemanJPersonalized cancer therapy with selective kinase inhibitors: an emerging paradigm in medical oncologyJ Clin Oncol200927335650565919858389
- ShepherdFARodrigues PereiraJCiuleanuTNational Cancer Institute of Canada Clinical Trials GroupErlotinib in previously treated non-small-cell lung cancerN Engl J Med2005353212313216014882
- RosellRCarcerenyEGervaisRSpanish Lung Cancer Group in collaboration with Groupe Français de Pneumo-Cancérologie and Associazione Italiana Oncologia ToracicaErlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trialLancet Oncol201213323924622285168
- MokTSWuYLThongprasertSGefitinib or carboplatin- paclitaxel in pulmonary adenocarcinomaN Engl J Med20093611094795719692680
- HanJYParkKKimSWFirst-SIGNAL: first-line single-agent iressa versus gemcitabine and cisplatin trial in never-smokers with adenocarcinoma of the lungJ Clin Oncol201230101122112822370314
- ZhouCWuYLChenGErlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 studyLancet Oncol201112873574221783417
- GainorJFShawATEmerging paradigms in the development of resistance to tyrosine kinase inhibitors in lung cancerJ Clin Oncol201331313987399624101047
- MarteBTumour heterogeneityNature2013501746732724048064
- BurrellRAMcGranahanNBartekJSwantonCThe causes and consequences of genetic heterogeneity in cancer evolutionNature2013501746733834524048066
- GovindanRDingLGriffithMGenomic landscape of non-small cell lung cancer in smokers and never-smokersCell201215061121113422980976
- MeachamCEMorrisonSJTumour heterogeneity and cancer cell plasticityNature2013501746732833724048065
- ReyaTMorrisonSJClarkeMFWeissmanILStem cells, cancer, and cancer stem cellsNature2001414685910511111689955
- ShackletonMQuintanaEFearonERMorrisonSJHeterogeneity in cancer: cancer stem cells versus clonal evolutionCell2009138582282919737509
- SinghSChellappanSLung cancer stem cells: Molecular features and therapeutic targetsMol Aspects Med2013 http://dx.doi.org/10.1016/j.mam.2013.08.003
- PeacockCDWatkinsDNCancer stem cells and the ontogeny of lung cancerJ Clin Oncol200826172883288918539968
- CarneyDNGazdarAFBunnPAJrGuccionJGDemonstration of the stem cell nature of clonogenic tumor cells from lung cancer patientsStem Cells1982131491646294885
- RawlinsELHoganBLEpithelial stem cells of the lung: privileged few or opportunities for many?Development2006133132455246516735479
- KimCFJacksonELWoolfendenAEIdentification of bronchioalveolar stem cells in normal lung and lung cancerCell2005121682383515960971
- GiangrecoAArwertENRosewellIRSnyderJWattFMStrippBRStem cells are dispensable for lung homeostasis but restore airways after injuryProc Natl Acad Sci USA2009106239286929119478060
- LinCSongHHuangCAlveolar type II cells possess the capability of initiating lung tumor developmentPLoS ONE2012712e5381723285300
- ShienKToyookaSYamamotoHAcquired resistance to EGFR inhibitors is associated with a manifestation of stem cell-like properties in cancer cellsCancer Res201373103051306123542356
- Corominas-FajaBOliveras-FerrarosCCuyàsEStem cell-like ALDH(bright) cellular states in EGFR-mutant non-small cell lung cancer: a novel mechanism of acquired resistance to erlotinib targetable with the natural polyphenol silibininCell Cycle201312213390340424047698
- HoeijmakersJHGenome maintenance mechanisms for preventing cancerNature2001411683536637411357144
- BirkbakNJEklundACLiQParadoxical relationship between chromosomal instability and survival outcome in cancerCancer Res201171103447345221270108
- LeeAJEndesfelderDRowanAJChromosomal instability confers intrinsic multidrug resistanceCancer Res20117151858187021363922
- SotilloRSchvartzmanJMSocciNDBenezraRMad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawalNature2010464728743644020173739
- AlexandrovLBNik-ZainalSWedgeDCAustralian Pancreatic Cancer Genome InitiativeICGC Breast Cancer Consortium; ICGC MMML-Seq ConsortiumICGC PedBrainSignatures of mutational processes in human cancerNature2013500746341542123945592
- KimSCJungYParkJA high-dimensional, deep-sequencing study of lung adenocarcinoma in female never-smokersPLoS ONE201382e5559623405175
- SteadLFEganPDeveryAAn integrated inspection of the somatic mutations in a lung squamous cell carcinoma using next-generation sequencingPLoS ONE2013811e7882324244370
- BalgkouranidouILiloglouTLianidouESLung cancer epigenetics: emerging biomarkersBiomarkers Med2013714958
- LiuSVFabbriMGitlitzBJLaird-OffringaIAEpigenetic therapy in lung cancerFront Oncol2013313523755372
- RothschildSIEpigenetic Therapy in Lung Cancer – Role of microRNAsFront Oncol2013315823802096
- LundAHvan LohuizenMEpigenetics and cancerGenes Dev200418192315233515466484
- DaskalosANikolaidisGXinarianosGHypomethylation of retrotransposable elements correlates with genomic instability in non-small cell lung cancerInt J Cancer20091241818718823011
- DaskalosALogothetiSMarkopoulouSGlobal DNA hypomethylation-induced ΔNp73 transcriptional activation in non-small cell lung cancerCancer Lett20113001798620926182
- HansenKDTimpWBravoHCIncreased methylation variation in epigenetic domains across cancer typesNat Genet201143876877521706001
- BarlésiFGiacconeGGallegos-RuizMIGlobal histone modifications predict prognosis of resected non small-cell lung cancerJ Clin Oncol200725284358436417906200
- SongJSKimYSKimDKParkSIJangSJGlobal histone modification pattern associated with recurrence and disease-free survival in non-small cell lung cancer patientsPathol Int201262318219022360506
- JunttilaMRde SauvageFJInfluence of tumour micro-environment heterogeneity on therapeutic responseNature2013501746734635424048067
- TrédanOGalmariniCMPatelKTannockIFDrug resistance and the solid tumor microenvironmentJ Natl Cancer Inst200799191441145417895480
- MeadsMBGatenbyRADaltonWSEnvironment-mediated drug resistance: a major contributor to minimal residual diseaseNat Rev Cancer20099966567419693095
- NakamuraTMatsumotoKKiritoshiATanoYNakamuraTInduction of hepatocyte growth factor in fibroblasts by tumor-derived factors affects invasive growth of tumor cells: in vitro analysis of tumor-stromal interactionsCancer Res19975715330533139242465
- YamadaTMatsumotoKWangWHepatocyte growth factor reduces susceptibility to an irreversible epidermal growth factor receptor inhibitor in EGFR-T790M mutant lung cancerClin Cancer Res201016117418320008840
- YamadaTTakeuchiSKitaKHepatocyte growth factor induces resistance to anti-epidermal growth factor receptor antibody in lung cancerJ Thorac Oncol20127227228022089117
- SiegfriedJMWeissfeldLASingh-KawPWeyantRJTestaJRLandreneauRJAssociation of immunoreactive hepatocyte growth factor with poor survival in resectable non-small cell lung cancerCancer Res19975734334399012470
- MeertAPPaesmansMMartinBThe role of microvessel density on the survival of patients with lung cancer: a systematic review of the literature with meta-analysisBr J Cancer200287769470112232748
- HegdePSJubbAMChenDPredictive impact of circulating vascular endothelial growth factor in four phase III trials evaluating bevacizumabClin Cancer Res201319492993723169435
- Van der VeldtAALubberinkMBahceIRapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: implications for scheduling of anti-angiogenic drugsCancer Cell2012211829122264790
- PaoWChmieleckiJRational, biologically based treatment of EGFR-mutant non-small-cell lung cancerNat Rev Cancer2010101176077420966921
- PaoWGirardNNew driver mutations in non-small-cell lung cancerLancet Oncol201112217518021277552
- StellaGMLuisettiMPozziEComoglioPMOncogenes in non-small-cell lung cancer: emerging connections and novel therapeutic dynamicsThe lancet. Respir Med201313251261
- YuHAArcilaMERekhtmanNAnalysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancersClin Cancer Res20131982240224723470965
- JännePAEngelmanJAJohnsonBEEpidermal growth factor receptor mutations in non-small-cell lung cancer: implications for treatment and tumor biologyJ Clin Oncol200523143227323415886310
- MarchettiAMartellaCFelicioniLEGFR mutations in non-small-cell lung cancer: analysis of a large series of cases and development of a rapid and sensitive method for diagnostic screening with potential implications on pharmacologic treatmentJ Clin Oncol200523485786515681531
- KosakaTYatabeYEndohHKuwanoHTakahashiTMitsudomiTMutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implicationsCancer Res200464248919892315604253
- GarassinoMCMartelliOBrogginiMTAILOR trialistsErlotinib versus docetaxel as second-line treatment of patients with advanced non-small-cell lung cancer and wild-type EGFR tumours (TAILOR): a randomised controlled trialLancet Oncol2013141098198823883922
- BroseMSVolpePFeldmanMBRAF and RAS mutations in human lung cancer and melanomaCancer Res200262236997700012460918
- EberhardDAJohnsonBEAmlerLCMutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinibJ Clin Oncol200523255900590916043828
- PratilasCAHanrahanAJHalilovicEGenetic predictors of MEK dependence in non-small cell lung cancerCancer Res200868229375938319010912
- SodaMChoiYLEnomotoMIdentification of the transforming EML4-ALK fusion gene in non-small-cell lung cancerNature2007448715356156617625570
- ShawATYeapBYSolomonBJEffect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysisLancet Oncol201112111004101221933749
- ShawATYeapBYMino-KenudsonMClinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALKJ Clin Oncol200927264247425319667264
- JankuFStewartDJKurzrockRTargeted therapy in non-small-cell lung cancer – is it becoming a reality?Nat Rev Clin Oncol20107740141420551945
- LorussoVSilvestrisNMarechITORCH study: how much longer should we continue to use erlotinib in unselected patients with non-small-cell lung cancer?J Clin Oncol201331228828923233717
- LiTKungHJMackPCGandaraDRGenotyping and genomic profiling of non-small-cell lung cancer: implications for current and future therapiesJ Clin Oncol20133181039104923401433
- MogiAKuwanoHTP53 mutations in nonsmall cell lung cancerJ Biomed Biotechnol2011201158392921331359
- PanYZhangYLiYALK, ROS1 and RET fusions in 1139 lung adenocarcinomas: a comprehensive study of common and fusion pattern-specific clinicopathologic, histologic and cytologic featuresLung Cancer201484212112624629636
- SequistLVHeistRSShawATImplementing multiplexed genotyping of non-small-cell lung cancers into routine clinical practiceAnn Oncol201122122616262422071650
- MitsudomiTYatabeYEpidermal growth factor receptor in relation to tumor development: EGFR gene and cancerFEBS J2010277230130819922469
- YehPChenHAndrewsJNaserRPaoWHornLDNA-Mutation Inventory to Refine and Enhance Cancer Treatment (DIRECT): a catalog of clinically relevant cancer mutations to enable genome-directed anticancer therapyClin Cancer Res20131971894190123344264
- SharmaSVBellDWSettlemanJHaberDAEpidermal growth factor receptor mutations in lung cancerNat Rev Cancer20077316918117318210
- WuJYYuCJChangYCYangCHShihJYYangPCEffectiveness of tyrosine kinase inhibitors on “uncommon” epidermal growth factor receptor mutations of unknown clinical significance in non-small cell lung cancerClin Cancer Res201117113812382121531810
- MulloyRFerrandAKimYEpidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinibCancer Res20076752325233017332364
- CareyKDGartonAJRomeroMSKinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinibCancer Res200666168163817116912195
- YasudaHKobayashiSCostaDBEGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implicationsLancet Oncol2012131e23e3121764376
- WuJYWuSGYangCHLung cancer with epidermal growth factor receptor exon 20 mutations is associated with poor gefitinib treatment responseClin Cancer Res200814154877488218676761
- GreulichHChenTHFengWOncogenic transformation by inhibitor-sensitive and -resistant EGFR mutantsPLoS Med2005211e31316187797
- RielyGJPaoWPhamDClinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinibClin Cancer Res2006123 Pt 183984416467097
- DengJShimamuraTPereraSProapoptotic BH3-only BCL-2 family protein BIM connects death signaling from epidermal growth factor receptor inhibition to the mitochondrionCancer Res20076724118671187518089817
- GongYSomwarRPolitiKInduction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomasPLoS Med2007410e29417927446
- CraggMSKurodaJPuthalakathHHuangDCStrasserAGefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimeticsPLoS Med20074101681168917973573
- FaberACCorcoranRBEbiHBIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitorsCancer Discov20111435236522145099
- NgKPHillmerAMChuahCTA common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancerNat Med201218452152822426421
- FaberACLiDSongYDifferential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibitionProc Natl Acad Sci USA200910646195031950819850869
- SongLCoppolaDLivingstonSCressDHauraEBMcl-1 regulates survival and sensitivity to diverse apoptotic stimuli in human non-small cell lung cancer cellsCancer Biol Ther20054326727615753661
- CetinZOzbilimGErdoganALuleciGKarauzumSBEvaluation of PTEN and Mcl-1 expressions in NSCLC expressing wild-type or mutated EGFRMed Oncol201027385386019763916
- EngelmanJAMukoharaTZejnullahuKAllelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancerJ Clin Invest2006116102695270616906227
- KawanoOSasakiHEndoKPIK3CA mutation status in Japanese lung cancer patientsLung Cancer200654220921516930767
- GerlingerMRowanAJHorswellSIntratumor heterogeneity and branched evolution revealed by multiregion sequencingN Engl J Med20123661088389222397650
- CrouchECStoneKRBlochMMcDivittRWHeterogeneity in the production of collagens and fibronectin by morphologically distinct clones of a human tumor cell line: evidence for intratumoral diversity in matrix protein biosynthesisCancer Res19874722608660922822240
- BlackhallFHPintilieMWigleDAStability and heterogeneity of expression profiles in lung cancer specimens harvested following surgical resectionNeoplasia20046676176715720802
- InukaiMToyookaSItoSPresence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancerCancer Res200666167854785816912157
- SequistLVMartinsRGSpigelDFirst-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutationsJ Clin Oncol200826152442244918458038
- TurkeABZejnullahuKWuYLPreexistence and clonal selection of MET amplification in EGFR mutant NSCLCCancer Cell2010171778820129249
- BaiHWangZWangYDetection and clinical significance of intratumoral EGFR mutational heterogeneity in Chinese patients with advanced non-small cell lung cancerPLoS ONE201382e5417023418425
- TaniguchiKOkamiJKodamaKHigashiyamaMKatoKIntratumor heterogeneity of epidermal growth factor receptor mutations in lung cancer and its correlation to the response to gefitinibCancer Sci200899592993518325048
- GowCHChangYLHsuYCComparison of epidermal growth factor receptor mutations between primary and corresponding metastatic tumors in tyrosine kinase inhibitor-naive non-small-cell lung cancerAnn Oncol200920469670219088172
- KalikakiAKoutsopoulosATrypakiMComparison of EGFR and K-RAS gene status between primary tumours and corresponding metastases in NSCLCBr J Cancer200899692392919238633
- YatabeYMatsuoKMitsudomiTHeterogeneous distribution of EGFR mutations is extremely rare in lung adenocarcinomaJ Clin Oncol201129222972297721730270
- MatsumotoSTakahashiKIwakawaRFrequent EGFR mutations in brain metastases of lung adenocarcinomaInt J Cancer200611961491149416642476
- NagaiYMiyazawaHHuqunGenetic heterogeneity of the epidermal growth factor receptor in non-small cell lung cancer cell lines revealed by a rapid and sensitive detection system, the peptide nucleic acid-locked nucleic acid PCR clampCancer Res200565167276728216105816
- KamilaWKMichałSPawełKEGFR activating mutations detected by different PCR techniques in Caucasian NSCLC patients with CNS metastases: short reportClin Exp Metastasis20133081063107123892415
- JiangSXYamashitaKYamamotoMEGFR genetic heterogeneity of nonsmall cell lung cancers contributing to acquired gefitinib resistanceInt J Cancer2008123112480248618785203
- ArcilaMEOxnardGRNafaKRebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assayClin Cancer Res20111751169118021248300
- TaniguchiKUchidaJNishinoKQuantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomasClin Cancer Res201117247808781521976538
- KleinCASelection and adaptation during metastatic cancer progressionNature2013501746736537224048069
- SequistLVWaltmanBADias-SantagataDGenotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitorsSci Transl Med201137575ra26
- ThatcherNChangAParikhPGefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer)Lancet200536694961527153716257339
- BaiHWangZChenKInfluence of chemotherapy on EGFR mutation status among patients with non-small-cell lung cancerJ Clin Oncol201230253077308322826274
- Jair BarDURoni Borshtein, Hovav Nechushtan, Amir Onn. EGFR mutation in lung cancer: tumor heterogeneity and the impact of chemotherapyChin Clin Oncol20132113
- De PasTPelosiGde BraudFModulation of epidermal growth factor receptor status by chemotherapy in patients with locally advanced non-small-cell lung cancer is rareJ Clin Oncol200422244966497015611511
- ChinTMQuinlanMPSinghAReduced Erlotinib sensitivity of epidermal growth factor receptor-mutant non-small cell lung cancer following cisplatin exposure: a cell culture model of second-line erlotinib treatmentClin Cancer Res200814216867687618980981
- PaoWMillerVAPolitiKAAcquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domainPLoS Med200523e7315737014
- KobayashiSBoggonTJDayaramTEGFR mutation and resistance of non-small-cell lung cancer to gefitinibN Engl J Med2005352878679215728811
- YunCHMengwasserKETomsAVThe T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATPProc Natl Acad Sci USA200810562070207518227510
- CostaDBSchumerSTTenenDGKobayashiSDifferential responses to erlotinib in epidermal growth factor receptor (EGFR)-mutated lung cancers with acquired resistance to gefitinib carrying the L747S or T790M secondary mutationsJ Clin Oncol20082671182118418309959
- BalakMNGongYRielyGJNovel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitorsClin Cancer Res200612216494650117085664
- BeanJRielyGJBalakMAcquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinomaClin Cancer Res200814227519752519010870
- CostaDBHalmosBKumarABIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutationsPLoS Med20074101669167917973572
- TokumoMToyookaSIchiharaSDouble mutation and gene copy number of EGFR in gefitinib refractory non-small-cell lung cancerLung Cancer200653111712116730855
- JackmanDMHolmesAJLindemanNResponse and resistance in a non-small-cell lung cancer patient with an epidermal growth factor receptor mutation and leptomeningeal metastases treated with high-dose gefitinibJ Clin Oncol200624274517452016983123
- ClarkeJLPaoWWuNMillerVALassmanABHigh dose weekly erlotinib achieves therapeutic concentrations in CSF and is effective in leptomeningeal metastases from epidermal growth factor receptor mutant lung cancerJ Neurooncol201099228328620146086
- RielyGJKrisMGZhaoBProspective assessment of discontinuation and reinitiation of erlotinib or gefitinib in patients with acquired resistance to erlotinib or gefitinib followed by the addition of everolimusClin Cancer Res200713175150515517785570
- KurataTTamuraKKanedaHEffect of re-treatment with gefitinib (‘Iressa’, ZD1839) after acquisition of resistanceAnn Oncol200415117317414679138
- EngelmanJAZejnullahuKMitsudomiTMET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signalingScience200731658271039104317463250
- TakezawaKPirazzoliVArcilaMEHER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutationCancer Discov201221092293322956644
- BarrSThomsonSBuckEBypassing cellular EGF receptor dependence through epithelial-to-mesenchymal-like transitionsClin Exp Metastasis200825668569318236164
- UramotoHIwataTOnitsukaTShimokawaHHanagiriTOyamaTEpithelial-mesenchymal transition in EGFR-TKI acquired resistant lung adenocarcinomaAnticancer Res20103072513251720682976
- ChungJHRhoJKXuXClinical and molecular evidences of epithelial to mesenchymal transition in acquired resistance to EGFR-TKIsLung Cancer201173217618221168239
- WalterKHolcombTJanuarioTDNA methylation profiling defines clinically relevant biological subsets of non-small cell lung cancerClin Cancer Res20121882360237322261801
- OgawaTLiggettTEMelnikovAAMethylation of death-associated protein kinase is associated with cetuximab and erlotinib resistanceCell Cycle20121181656166322487682
- ZhuJWangYDuanJDNA Methylation status of Wnt antagonist SFRP5 can predict the response to the EGFR-tyrosine kinase inhibitor therapy in non-small cell lung cancerJ Exp Clin Cancer Res20123118023009178
- SalazarFMolinaMASanchez-RoncoMFirst-line therapy and methylation status of CHFR in serum influence outcome to chemotherapy versus EGFR tyrosine kinase inhibitors as second-line therapy in stage IV non-small-cell lung cancer patientsLung Cancer2011721849120705357
- LiHHuHWangRPrimary concomitant EGFR T790M mutation predicted worse prognosis in non-small cell lung cancer patientsOnco Targets Ther20143;7513524
- YanoSWangWLiQHepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutationsCancer Res200868229479948719010923
- YanoSYamadaTTakeuchiSHepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohortJ Thorac Oncol20116122011201722052230
- MazièresJPetersSLepageBLung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectivesJ Clin Oncol201331161997200323610105
- WangLHuHPanYPIK3CA mutations frequently coexist with EGFR/KRAS mutations in non-small cell lung cancer and suggest poor prognosis in EGFR/KRAS wildtype subgroupPLoS ONE201492e8829124533074
- CheungHWDuJBoehmJSAmplification of CRKL induces transformation and epidermal growth factor receptor inhibitor resistance in human non-small cell lung cancersCancer Discov20111760862522586683
- KimYHKweiKAGirardLGenomic and functional analysis identifies CRKL as an oncogene amplified in lung cancerOncogene201029101421143019966867
- DanielsMGBowmanRVYangIAGovindanRFongKMAn emerging place for lung cancer genomics in 2013J Thorac Dis20135Suppl 5S491S49724163742
- NavinNKendallJTrogeJTumour evolution inferred by single-cell sequencingNature20114727341909421399628
- KorfBRRehmHLNew approaches to molecular diagnosisJAMA2013309141511152123571590
- ImielinskiMBergerAHHammermanPSMapping the hallmarks of lung adenocarcinoma with massively parallel sequencingCell201215061107112022980975
- Network CGARCancer Genome Atlas Research NetworkComprehensive genomic characterization of squamous cell lung cancersNature2012489741751952522960745
- PotterNEErminiLPapaemmanuilESingle-cell mutational profiling and clonal phylogeny in cancerGenome Res201323122115212524056532
- NavinNHicksJFuture medical applications of single-cell sequencing in cancerGenome Med2011353121631906
- JakobsenJNSørensenJBIntratumor heterogeneity and chemotherapy-induced changes in EGFR status in non-small cell lung cancerCancer Chemother Pharmacol201269228929922130585
- MurtazaMDawsonSJTsuiDWNon-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNANature2013497744710811223563269
- AllardWJMateraJMillerMCTumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseasesClin Cancer Res200410206897690415501967
- MaheswaranSSequistLVNagrathSDetection of mutations in EGFR in circulating lung-cancer cellsN Engl J Med2008359436637718596266
- NiXZhuoMSuZReproducible copy number variation patterns among single circulating tumor cells of lung cancer patientsProc Natl Acad Sci USA201311052210832108824324171
- KimuraHKasaharaKKawaishiMDetection of epidermal growth factor receptor mutations in serum as a predictor of the response to gefitinib in patients with non-small-cell lung cancerClin Cancer Res200612133915392116818687
- GotoKIchinoseYOheYEpidermal growth factor receptor mutation status in circulating free DNA in serum: from IPASS, a phase III study of gefitinib or carboplatin/paclitaxel in non-small cell lung cancerJ Thorac Oncol20127111512121900837
- LiuXLuYZhuGThe diagnostic accuracy of pleural effusion and plasma samples versus tumour tissue for detection of EGFR mutation in patients with advanced non-small cell lung cancer: comparison of methodologiesJ Clin Pathol201366121065106923888061
- KuangYRogersAYeapBYNoninvasive detection of EGFR T790M in gefitinib or erlotinib resistant non-small cell lung cancerClin Cancer Res20091582630263619351754
- BearzattoAConteDFrattiniMp16(INK4A) Hypermethylation detected by fluorescent methylation-specific PCR in plasmas from non-small cell lung cancerClin Cancer Res20028123782378712473590
- HsuHSChenTPHungCHCharacterization of a multiple epigenetic marker panel for lung cancer detection and risk assessment in plasmaCancer200711092019202617876837
- AnQLiuYGaoYDetection of p16 hypermethylation in circulating plasma DNA of non-small cell lung cancer patientsCancer Lett20021881–210911412406555
- PorterJRStainsCISegalDJGhoshISplit beta-lactamase sensor for the sequence-specific detection of DNA methylationAnal Chem200779176702670817685552
- PonomaryovaAARykovaEYCherdyntsevaNVPotentialities of aberrantly methylated circulating DNA for diagnostics and post-treatment follow-up of lung cancer patientsLung Cancer201381339740323806794
- ShinYLabel-free methylation specific sensor based on silicon microring resonators for detection and quantification of DNA methylation biomarkers in bladder cancerSensors Actuators B: Chem2013177404411
- RedovaMSanaJSlabyOCirculating miRNAs as new blood-based biomarkers for solid cancersFuture Oncol20139338740223469974
- BoeriMVerriCConteDMicroRNA signatures in tissues and plasma predict development and prognosis of computed tomography detected lung cancerProc Natl Acad Sci USA201110893713371821300873
- HuZChenXZhaoYSerum microRNA signatures identified in a genome-wide serum microRNA expression profiling predict survival of non-small-cell lung cancerJ Clin Oncol201028101721172620194856
- ZhangHSuYXuFKongJYuHQianBCirculating microRNAs in relation to EGFR status and survival of lung adenocarcinoma in female non-smokersPLoS ONE2013811e8140824282590
- LiBRenSLiXMiR-21 overexpression is associated with acquired resistance of EGFR-TKI in non-small cell lung cancerLung Cancer2013
- VoliniaSCalinGALiuCGA microRNA expression signature of human solid tumors defines cancer gene targetsProc Natl Acad Sci USA200610372257226116461460
- WangYZhengDTanQWangMXGuLQNanopore-based detection of circulating microRNAs in lung cancer patientsNat Nanotechnol201161066867421892163
- TaguchiFSolomonBGregorcVMass spectrometry to classify non-small-cell lung cancer patients for clinical outcome after treatment with epidermal growth factor receptor tyrosine kinase inhibitors: a multicohort cross-institutional studyJ Natl Cancer Inst2007991183884617551144
- ButtsCAVeriStrat validated in patients with non-small-cell lung cancerLancet Oncol201415767167224831978
- GregorcVNovelloSLazzariCPredictive value of a proteomic signature in patients with non-small-cell lung cancer treated with second-line erlotinib or chemotherapy (PROSE): a biomarker-stratified, randomised phase 3 trialLancet Oncol201415771372124831979
- AmannJMLeeJWRoderHGenetic and proteomic features associated with survival after treatment with erlotinib in first-line therapy of non-small cell lung cancer in Eastern Cooperative Oncology Group 3503J Thorac Oncol20105216917820035238
- CarboneDPDingKRoderHPrognostic and predictive role of the VeriStrat plasma test in patients with advanced non-small-cell lung cancer treated with erlotinib or placebo in the NCIC Clinical Trials Group BR.21 trialJ Thorac Oncol20127111653166023059783
- StinchcombeTERoderJPetermanAHA retrospective analysis of VeriStrat status on outcome of a randomized phase II trial of first-line therapy with gemcitabine, erlotinib, or the combination in elderly patients (age 70 years or older) with stage IIIB/IV non-small-cell lung cancerJ Thorac Oncol20138444345123370367
- LazzariCSpreaficoABachiAChanges in plasma mass-spectral profile in course of treatment of non-small cell lung cancer patients with epidermal growth factor receptor tyrosine kinase inhibitorsJ Thorac Oncol201271404821964534
- MilanELazzariCAnandSSAA1 is over-expressed in plasma of non small cell lung cancer patients with poor outcome after treatment with epidermal growth factor receptor tyrosine-kinase inhibitorsJ Proteomics201276Spec9110122771314
- KwakELSordellaRBellDWIrreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinibProc Natl Acad Sci USA2005102217665767015897464
- JiHZhaoXYuzaYEpidermal growth factor receptor variant III mutations in lung tumorigenesis and sensitivity to tyrosine kinase inhibitorsProc Natl Acad Sci USA2006103207817782216672372
- LiDAmbrogioLShimamuraTBIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer modelsOncogene200827344702471118408761
- KatakamiNAtagiSGotoKLUX-Lung 4: a phase II trial of afatinib in patients with advanced non-small-cell lung cancer who progressed during prior treatment with erlotinib, gefitinib, or bothJ Clin Oncol201331273335334123816963
- MillerVAHirshVCadranelJAfatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trialLancet Oncol201213552853822452896
- SequistLVYangJCYamamotoNPhase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutationsJ Clin Oncol201331273327333423816960
- WuYLZhouCHuCPAfatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trialLancet Oncol201415221322224439929
- YangJCShihJYSuWCAfatinib for patients with lung adenocarcinoma and epidermal growth factor receptor mutations (LUX-Lung 2): a phase 2 trialLancet Oncol201213553954822452895
- RamalingamSSBlackhallFKrzakowskiMRandomized phase II study of dacomitinib (PF-00299804), an irreversible pan-human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non-small-cell lung cancerJ Clin Oncol201230273337334422753918
- YangJCHirshVSchulerMSymptom control and quality of life in LUX-Lung 3: a phase III study of afatinib or cisplatin/pemetrexed in patients with advanced lung adenocarcinoma with EGFR mutationsJ Clin Oncol201331273342335023816967
- SequistLVBesseBLynchTJNeratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: results of a phase II trial in patients with advanced non-small-cell lung cancerJ Clin Oncol201028183076308320479403
- ZhouWErcanDChenLNovel mutant-selective EGFR kinase inhibitors against EGFR T790MNature200946272761070107420033049
- NanjoSYamadaTNishiharaHAbility of the Met kinase inhibitor crizotinib and new generation EGFR inhibitors to overcome resistance to EGFR inhibitorsPLoS ONE2013812e8470024386407
- ReckampKLGiacconeGCamidgeDRA phase 2 trial of dacomitinib (PF-00299804), an oral, irreversible pan-HER (human epidermal growth factor receptor) inhibitor, in patients with advanced non-small cell lung cancer after failure of prior chemotherapy and erlotinibCancer201412081145115424501009
- JännePAvon PawelJCohenRBMulticenter, randomized, phase II trial of CI-1033, an irreversible pan-ERBB inhibitor, for previously treated advanced non small-cell lung cancerJ Clin Oncol200725253936394417761977
- ChiapporiAAEllisPMHammJTA phase I evaluation of oral CI-1033 in combination with paclitaxel and carboplatin as first-line chemotherapy in patients with advanced non-small cell lung cancerJ Thorac Oncol2006191010101917409987
- XuLKikuchiEXuCCombined EGFR/MET or EGFR/HSP90 inhibition is effective in the treatment of lung cancers codriven by mutant EGFR containing T790M and METCancer Res201272133302331122552292
- ChenXZhouJYZhaoJChenJJMaSNZhouJYCrizotinib overcomes hepatocyte growth factor-mediated resistance to gefitinib in EGFR-mutant non-small-cell lung cancer cellsAnticancer Drugs201324101039104623962905
- NakagawaTTakeuchiSYamadaTCombined therapy with mutant-selective EGFR inhibitor and Met kinase inhibitor for overcoming erlotinib resistance in EGFR-mutant lung cancerMol Cancer Ther201211102149215722844075
- GoldmanJWLauxIChaiFPhase 1 dose-escalation trial evaluating the combination of the selective MET (mesenchymal-epithelial transition factor) inhibitor tivantinib (ARQ 197) plus erlotinibCancer2012118235903591122605616
- RiceJWVealJMBarabaszATargeting of multiple signaling pathways by the Hsp90 inhibitor SNX-2112 in EGFR resistance models as a single agent or in combination with erlotinibOncol Res2009185–622924220225761
- WangXSongXZhuoWThe regulatory mechanism of Hsp90alpha secretion and its function in tumor malignancyProc Natl Acad Sci USA200910650212882129319965370
- ShimamuraTLowellAMEngelmanJAShapiroGIEpidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycinsCancer Res200565146401640816024644
- UenoTTsukudaKToyookaSStrong anti-tumor effect of NVP-AUY922, a novel Hsp90 inhibitor, on non-small cell lung cancerLung Cancer2012761263121996088
- ShimamuraTLiDJiHHsp90 inhibition suppresses mutant EGFR-T790M signaling and overcomes kinase inhibitor resistanceCancer Res200868145827583818632637
- SocinskiMAGoldmanJEl-HariryIA multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancerClin Cancer Res201319113068307723553849
- GaronEBFinnRSHamidiHThe HSP90 inhibitor NVP-AUY922 potently inhibits non-small cell lung cancer growthMol Cancer Ther201312689090023493311
- FerraraNThe role of vascular endothelial growth factor in pathological angiogenesisBreast Cancer Res Treat19953621271378534862
- SetoTHigashiyamaMFunaiHPrognostic value of expression of vascular endothelial growth factor and its flt-1 and KDR receptors in stage I non-small-cell lung cancerLung Cancer2006531919616697074
- SandlerAGrayRPerryMCPaclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancerN Engl J Med2006355242542255017167137
- RSO’NeillVJFehrenbacherLPhase II study of efficacy and safety of bevacizumab in combination with chemotherapy or erlotinib compared with chemotherapy alone for treatment of recurrent or refractory non small-cell lung cancerJ Clin Oncol200725304743475017909199
- HerbstRSJohnsonDHMininbergEPhase I/II trial evaluating the anti-vascular endothelial growth factor monoclonal antibody bevacizumab in combination with the HER-1/epidermal growth factor receptor tyrosine kinase inhibitor erlotinib for patients with recurrent non-small-cell lung cancerJ Clin Oncol200523112544255515753462
- HerbstRSAnsariRBustinFEfficacy of bevacizumab plus erlotinib versus erlotinib alone in advanced non-small-cell lung cancer after failure of standard first-line chemotherapy (BeTa): a double-blind, placebo-controlled, phase 3 trialLancet201137797801846185421621716
- JohnsonBEKabbinavarFFehrenbacherLATLAS: randomized, double-blind, placebo-controlled, phase IIIB trial comparing bevacizumab therapy with or without erlotinib, after completion of chemotherapy, with bevacizumab for first-line treatment of advanced non-small-cell lung cancerJ Clin Oncol201331313926393424101054
- AkerleyWBoucherKRichNA phase II study of bevacizumab and erlotinib as initial treatment for metastatic non-squamous, non-small cell lung cancer with serum proteomic evaluationLung Cancer201379330731123273522
- SpigelDRBurrisHAIIIGrecoFARandomized, double-blind, placebo-controlled, phase II trial of sorafenib and erlotinib or erlotinib alone in previously treated advanced non-small-cell lung cancerJ Clin Oncol201129182582258921576636
- RosellRBivonaTGKarachaliouNGenetics and biomarkers in personalisation of lung cancer treatmentLancet2013382989372073123972815
- GaspariniGLongoRThe paradigm of personalized therapy in oncologyExpert Opin Ther Targets201216Suppl 1S7S1622073968
- MaPCPersonalized targeted therapy in advanced non-small cell lung cancerCleve Clin J Med201279Electronic Suppl 1eS56eS6022614968