
Abstract
Myelofibrosis (MF) is a myeloid malignancy associated with a heavy symptomatic burden that decreases quality of life and presents a risk for leukemic transformation. While there are limited curative treatments, the recent discovery of the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway dysregulation has led to many clinical investigations for new treatment approaches. This review provides practical knowledge on the disease state, an overview of treatment options, and specifically focuses on the efficacy and safety of pacritinib in the management of MF. Pacritinib is a novel selective inhibitor of JAK2 and FMS-related tyrosine kinase 3 (FLT3) currently in Phase III trials for the treatment of MF. Thus far, studies have demonstrated clinical efficacy in reducing splenomegaly and constitutional symptoms. Common adverse events were gastrointestinal in nature, while hematologic toxicity was limited. However, it was announced that all ongoing clinical trials on pacritinib have been placed on hold by the US Food and Drug Administration in February 2016, due to concerns for increased intracranial hemorrhage and cardiac events. With comprehensive risk-benefit analysis of clinical trial data, the utility of pacritinib in the management of MF may be more clearly defined.
Introduction
Myelofibrosis (MF) is a clonal hematopoietic stem cell disorder that is characterized by ineffective erythropoiesis, abnormal proinflammatory cytokine expression, bone marrow fibrosis, and extramedullary hematopoiesis in organs such as the spleen and the liver. MF, along with polycythemia vera (PV) and essential thrombocythemia (ET), are myeloid malignancies that comprise a subgroup of Philadelphia chromosome-negative myeloproliferative neoplasms. MF can arise de novo as primary myelofibrosis (PMF) or evolve from preexisting PV or ET (post-PV/ET MF). Its pathogenesis is driven by multiple somatic mutations in the Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway. Understanding the various genetic mutations linked to MF is an area of active research that may provide prognostic value to guide treatment decisions as well as lead to novel therapeutic targets.
In the initial phase of MF, dysregulation of granulocyte and megakaryocyte proliferation results in abnormal cytokine and growth factor production. As the disease evolves, bone marrow fibrosis develops and is followed by progressive bone marrow failure and extramedullary hematopoiesis. Ultimately, the unchecked growth of differentiated myeloid cells may enable transformation into acute myeloid leukemia (AML).Citation1
The hallmark clinical manifestations of MF include abnormal blood counts, hepatosplenomegaly, and constitutional symptoms. Anemia is present in approximately 50% of patients at the time of diagnosis, which can be accompanied by either thrombocytopenia or thrombocytosis and either leukopenia or leukocytosis.Citation1 The majority of MF patients develop splenomegaly due to extramedullary hematopoiesis and suffer from abdominal distension, early satiety, and left upper quadrant pain. An increased production of selected proinflammatory cytokines and growth factors in MF leads to a hypercatabolic state, causing constitutional symptoms in patients, including fever, night sweats, weight loss, pruritus, fatigue, and bone pain. These debilitating symptoms negatively impact the quality of life, and along with the risk of AML transformation, contribute to the lowered life expectancy of patients with MF compared with the healthy population.Citation1–Citation3 The rate of leukemic transformation is estimated to be 3.9%–20%, and is associated with dismal median survival of 6 weeks for patients who received only supportive care and 7 months for treated patients.Citation2 The most common causes of death in patients with MF are leukemic transformation, thrombosis, hemorrhagic events, and infection.Citation4
Epidemiology and prognosis
Based on data from 2008 through 2010, the annual age-adjusted incidence rate for all MF in the USA is between 2 and 3 per 100,000, and 1 per 100,000 for primary MF. The median age at diagnosis ranges from 61 to 66 years, with more males than females (53% vs 47%) diagnosed each year.Citation5 There is a high degree of heterogeneity in clinical presentation, disease evolution, and overall prognosis. Median survival can range from more than 10 years in patients with indolent MF to less than 2 years in patients with aggressive disease.Citation5
Several models and scoring systems have been developed to incorporate patient- and disease-related prognostic factors to guide treatment decisions. The Dupriez system (also known as Lille score) uses only hemoglobin (Hgb) level and white blood cell count at time of diagnosis. As the understanding of MF pathogenesis evolves and data from large multicenter studies become available, the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT) proposes the use of the International Prognostic Scoring System (IPSS) at time of diagnosis and the Dynamic IPSS (DIPSS) as disease progresses ().Citation6–Citation8 The latest DIPSS-Plus algorithm incorporates three additional risk factors: red blood cell (RBC) transfusion requirement, platelet count, and unfavorable karyotypes ().Citation9 Although these prognostic scoring systems have been developed for patients with PMF, studies have not shown any prognostic or therapeutic differences between PMF or post-PV/ET MF.
Table 1 Primary myelofibrosis: Dupriez, IPSS, and DIPSS scoring algorithms
Table 2 Primary myelofibrosis: DIPSS-Plus scoring algorithm
Unfavorable cytogenetics are associated with increased risk for cytopenias, circulating blasts, and higher risk for leukemic transformation. On the other hand, patients who present with favorable cytogenetics (eg, normal karyotype, 13q−, 20q−, +9, chr 1 translocation/duplication) benefit from a significantly higher 5-year overall survival rate of 51% vs 8% in those with unfavorable karyotypes.Citation10 Specific genetic mutations may also have prognostic values in MF. A large international study examined the mutation status of five genes (ASXL1, EZH2, SRSF2, IDH1, and IDH2) in 797 patients with PMF. These somatic mutations have been identified to exert an adverse prognosis independent of the IPSS and DIPSS-Plus scores. Patients with at least two mutated genes had the lowest median survival of 2.6 years, compared with 7 years for patients with one mutation and 12.3 years for patients with no mutations.Citation11 Conversely, patients who harbored mutations in calreticulin (CALR) had lower DIPSS-Plus scores, higher platelet counts, and lower risk of anemia and leukocytosis. These patients also had longer overall survival compared with patients with mutations in oncogenes MPL (hazards ratio [HR] =1.7) and JAK2 (HR =2.6). Additionally, “triple negative” patients who had wild-type JAK2, MPL, and CALR were deemed to have high risk of disease, with overall survival of 2.5 years (HR =3.6) compared with 8.2 years in patients with CALR mutation.Citation12 However, these patients were not stratified by CALR mutation variant: type 1 vs type 2. In a different study of 532 PMF patients, 131 patients were CALR-mutated cases: 110 patients were classified as type 1/type 1-like, and 21 patients were type 2/type 2-like. Survival was shorter in patients with type 2/type 2-like (median 3.5 years) vs type 1/type 1-like CALR mutations (median 13.7 years) (P=0.003; HR =2.5, 95% confidence interval [95% CI] =1.4–4.5).Citation13 Thus, specific types of CALR mutations have differing prognostic implications, with type 1 having a better prognosis. It is likely that genetic profiles along with other patient- and disease-related factors, such as circulating cytokines and ferritin levels, will be incorporated into prognostic scoring models for MF in the future.Citation14
Conventional treatment options
Allogeneic hematopoietic stem cell transplantation (alloHSCT) remains the only potentially curative treatment for MF to date. In an analysis of 289 PMF patients (median age 47 years) who received myeloablative alloHSCT between 1989 and 2002, the 5-year overall survival rates were 37%, 30%, and 40%; and 100-day mortality rates were 18%, 35%, and 19% for human leukocyte antigen-identical sibling, unrelated, and other related transplants, respectively.Citation15 Successful treatment with nonmyeloablative conditioning transplant has also been reported. In a group of 103 patients (median age 55 years) who received reduced-intensity busulfan/fludarabine conditioning, the 5-year overall survival rates were 74% and 38% for matched and mismatched transplants respectively, with nonrelapsed mortality rates of 12% and 38%.Citation16 Unfortunately, MF is a disease of the elderly, and few patients are eligible for transplant due to high risk for toxicities from conditioning chemotherapy and post transplant complications such as infection, graft failure, and graft-versus-host disease. An ad hoc analysis was performed in 190 PMF patients who received alloHSCT compared with 248 patients treated with conventional therapies. Patients with DIPSS intermediate-2 or high-risk disease had a lower relative risk (RR) of death (0.55 and 0.37, respectively) compared with patients with low-risk disease (RR of death 5.6).Citation17 Thus, with careful screening for adequate performance status and no significant comorbidities, alloHSCT may benefit patients who present with higher-risk MF.
Patients who are not stem cell transplant candidates often require various therapies to manage MF-related symptoms.Citation18–Citation20 Splenectomy may alleviate splenomegaly-related symptoms, but is associated with high morbidity and mortality rates of 31% and 9%, respectively, because of perioperative bleeding, thrombosis, and infections.Citation21 Approximately 40% of patients treated with hydroxyurea achieve clinical improvement in leukocytosis, thrombocytosis, and splenomegaly, with response lasting 13.2 months.Citation22 Symptomatic anemia may be managed with blood transfusion, erythropoietin-stimulating agents (ESAs), androgens, or immunomodulating drugs. The use of ESAs in this setting may be limited by the potential concern for splenomegaly exacerbation.Citation23 Danazol is a synthetic androgen that can help achieve RBC-transfusion-independence and increase Hgb, with the most frequent toxicity being moderate transaminitis reported in 27% of patients.Citation24 Immunomodulating agents such as thalidomide, lenalidomide, and pomalidomide, with or without prednisone, have been studied for management of splenomegaly, cytopenias, and constitutional symptoms. Anemia improved in 19%–30% of patients, but many required dose reduction or interruption due to sedation, constipation, and paresthesias, especially with thalidomide.Citation19 Furthermore, a Phase III study comparing pomalidomide to placebo failed to demonstrate a difference in RBC-transfusion-independence rate.Citation25 These conventional drug therapies offer only modest response rates in MF-related symptoms, and none have been shown to change the natural course of disease progression or provide survival benefits to patients with MF.
Molecular pathogenesis: JAK/STAT pathway
In 2005, a single activating point mutation in the tyrosine kinase JAK2 was found to correlate highly with dysregulation of the JAK/STAT signaling pathway in the majority of patients with PV, ET, and PMF (). The four members of the JAK family are JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2). They are intracellular tyrosine kinases that interact with various cytoplasmic receptors for cytokines, including erythropoietin, thrombopoietin, granulocyte-macrophage colony-stimulating factor, interleukins (ILs), and interferons (IFNs). The binding of ligands to the cytokine receptors phosphorylates JAKs, which in turn activate downstream signaling molecules such as STAT, RAS, mitogen-activated protein (MAP), and phosphoinositide 3-kinase-Akt (PI3K-Akt). The transduction of signals regulates the transcription of genes that play a role in normal hematopoietic cell differentiation (JAK2), inflammatory response (JAK1), and innate and adaptive immunity (JAK3).Citation26–Citation28
Figure 1 JAK/STAT pathway and potential therapeutic targets.
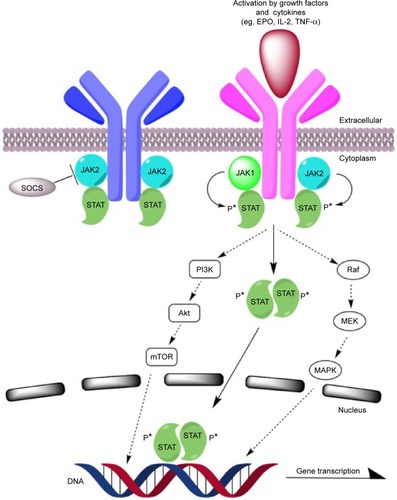
The upregulation of the JAK/STAT pathway has been observed in various solid tumors and hematologic malignancies such as breast, lung, Hodgkin’s lymphoma, and high-risk AML. It is also implicated in mediating resistance to BCR-ABL kinase inhibitors in chronic myelogenous leukemia (CML).Citation28 The point mutation JAK2V617F, in which valine is substituted with phenylalanine at codon 617, occurs in the autoinhibitory JH2 pseudokinase domain of the JAK2 protein, leading to a constitutively active JAK/STAT pathway without the need for cytokine or growth factor ligand binding. The mutation also confers resistance on negative regulation by suppressor of cytokine signaling (SOCS3) protein, which may lead to cytokine hypersensitivity and resistance to apoptosis.Citation26,Citation28
Plasma levels of various proinflammatory and profibrotic cytokines and growth factors are elevated in MF patients. These include IL-2R, IL-8, IL-10, tumor necrosis factor (TNF-α), hepatocyte growth factor (HGF), IFN-α, and IFN-γ inducible protein (IP-10). Overexpression of these cytokines is believed to be responsible for MF-related leukocytosis (IL-2R, HGF), thrombocytopenia (IP-10), transfusion-dependent anemia (IL-2R, IL-8, IL-10), splenomegaly (HGF), and constitutional symptoms (IL-8). Elevated levels of IL-2R or IL-8 may also have prognostic value for inferior overall and leukemia-free survival. The production of these cytokines in patients with MF has been shown to be downregulated through the inhibition of JAK1 and JAK2.Citation27,Citation29
It is unclear how cytokine expression, molecular mutations, and signaling pathways lead to the diverse clinical phenotypes observed in MF patients. Nonetheless, targeting of the JAK/STAT pathway, especially JAK2, becomes a major focus of attention in the development of novel targeted therapeutic agents for MF.
Approved JAK inhibitor
Ruxolitinib is a cyclopentylpropionitrile-derived small molecule that inhibits both JAK1 (half-maximal inhibitory concentration [IC50] =3.3 nmol/L) and JAK2 ([IC50] =2.8 nmol/L), with less specificity for JAK3 and TYK2.Citation26 The safety and efficacy of ruxolitinib in the treatment of MF was evaluated in two Phase III clinical trials: Controlled Myelofibrosis Study with Oral JAK Inhibitor Treatment (COMFORT-I) and COMFORT-II.Citation30,Citation31 Both studies enrolled patients with IPSS intermediate-2 or high-risk PMF or post-PV/ET MF, with platelet count ≥100×109/L, Eastern Cooperative Oncology Group performance status of ≤3, and palpable splenomegaly. Patients were included regardless of JAK2 mutation status. COMFORT-I was a double-blind, placebo-controlled trial in which 309 patients were randomized in a 1:1 ratio. In the ruxolitinib group, 41.9% of patients reached the primary endpoint of ≥35% reduction in spleen volume as assessed by magnetic resonance imaging (MRI) at 24 weeks, vs 0.9% in placebo group (P<0.001). Spleen volume response was maintained in 67% of patients at 48 weeks, and ruxolitinib was shown to improve total symptom burden at 24 weeks compared with placebo (45.9% vs 5.3%, P<0.001). A 2-year follow-up analysis reported durable spleen volume reduction, improved quality of life, and survival advantage at 102 weeks compared with placebo (HR =0.58, 95% CI =0.36–0.95; P=0.03), with 2-year survival probabilities of 82% and 73%, respectively.Citation32 COMFORT-II was an open-labeled trial in which 219 MF patients were randomized in a 2:1 ratio to receive ruxolitinib or best available therapy (BAT). The majority of patients in the BAT group received hydroxyurea (47%) and glucocorticoids (16%), while 33% received no active treatment. Only patients in the ruxolitinib group met the primary endpoint of ≥35% reduction in spleen volume at 48 weeks (28% vs 0% in BAT, P<0.001). Ruxolitinib group demonstrated reduced MF-associated symptoms and improved overall survival compared with the BAT group at 144 weeks (HR =0.48, 95% CI =0.28–0.85; P=0.009).Citation33 An ad hoc analysis comparing ruxolitinib-treated patients in the COMFORT-II trial with historical controls from a multicenter database who received solely conventional therapies confirmed the survival advantage of ruxolitinib (HR =0.61, 95% CI =0.41–0.91, P=0.0148).Citation34 The COMFORT-I and COMFORT-II data also showed a reduction in circulating inflammatory cytokine levels (eg, TNF-α and IL-6). Interestingly, clinical response rates were similar between patients with wild-type JAK2 and those with JAK2V617F. Furthermore, only a modest reduction in JAK2V617F allele burden was measured over time (7%–22% reduction at week 48).Citation33,Citation35
The rates of nonhematological toxicities were similar between ruxolitinib and placebo/BAT groups. Headache, dizziness, diarrhea, and ecchymosis occurred more frequently in the ruxolitinib group vs placebo group, but overall incidence of grade 3/4 toxicities was low. Incidence of infections was 50% during the first 6 months of ruxolitinib treatment. Recent in vitro and in vivo data demonstrated decreased maturation and activation of T-cells and natural killer (NK) cells in ruxolitinib-treated patients.Citation36,Citation37 The immunosuppressive property of ruxolitinib may contribute to increased viral reactivation and atypical infection. The majority of ruxolitinib patients experienced hematological toxicities, specifically anemia (all grades 96%; grade 3/4 42%–45%) and thrombocytopenia (all grades 68%–70%; grade 3/4 8%–13%), which were managed with dose modifications and/or blood product transfusion. The rate of ruxolitinib discontinuation due to hematological toxicities was low at <1%. Based on these two Phase III clinical trials, ruxolitinib was approved in November 2011 for the treatment of patients with IPSS intermediate- and high-risk MF. The starting dose of ruxolitinib is determined by the baseline platelet level of the patient: 20 mg orally twice daily (platelets >200×109/L), 15 mg orally twice daily (platelets 100–200×109/L), or 5 mg orally twice daily (platelets <100×109/L). Treatment is relatively well tolerated, but is often complicated by the development of cytopenias. Other JAK inhibitors with various pharmacokinetics and safety profiles have since been studied for the treatment of MF, including pacritinib, which will be the focus of this review.
Pacritinib
Pacritinib, SB1518, is a novel oral tyrosine kinase inhibitor with dual activity against JAK2 and FMS-related tyrosine kinase 3 (FLT3) under investigation for the treatment of various myeloid and lymphoid hematological malignancies. In August 2014, this small molecule received Fast Track designation by the US Food and Drug Administration (FDA) for the treatment of intermediate and high-risk MF. The drug candidate is currently in Phase III trials: PERSIST-1 and PERSIST-2 (ClinicalTrials.gov ID: NCT01773187 and NCT02055781).
Chemistry/molecular design
Pacritinib was designed using computational chemistry to be an adenosine triphosphate-site competitive kinase inhibitor of JAK2.Citation38 It has a low molecular weight of 472.58 g/mol; its chemical formula is C28H32N4O3 and chemical structure is 11-(2-pyrrolidin-1-ylethoxy)-14,19-dioxa-5,7,26-triazatetracyclo(19.3.1.1(2,6).1(8,12))heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene.Citation39 The compound is classified as a macrocycle, a ring structure of at least 12 atoms, and contains a pyrimidine core ().
Figure 2 Molecular structure of pacritinib.
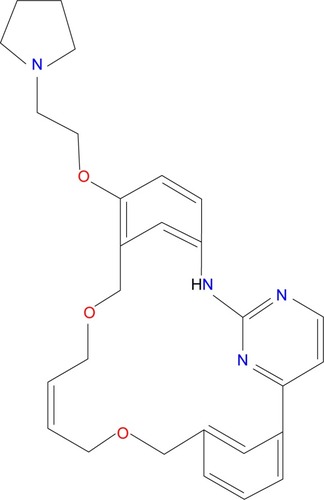
The molecular structure of pacritinib allows for hydrogen bonding with JAK2, which contributes to its selectivity and potency of the JAK2 tyrosine kinase.Citation39 It selectively inhibits wild-type JAK2 ([IC50] =23 nmol/L) and the mutant JAK2V617F ([IC50] =19 nmol/L); but not JAK1, JAK3, or TYK2 ([IC50] of 1,280, 520, and 50 nmol/L, respectively). Pacritinib also inhibits both wild-type FLT3 and the mutant form FLT3D835Y, with [IC50] of 22 and 6 nmol/L, respectively.Citation40,Citation41
Pharmacokinetics, pharmacodynamics, and metabolism
In vitro studies have provided information on pharmacodynamics and metabolism of pacritinib.Citation39 Results from metabolic studies suggest that pacritinib is metabolized mainly by cytochrome P450 CYP3A4 and does not inhibit the CYP isozymes (3A4, 1A2, 2C9, 2D6, 2C19). Findings from a bidirectional Caco-2 permeability assay established that pacritinib has high permeability and a low efflux ratio, and thus is unlikely to be a substrate for P-glycoprotein. Plasma protein binding assays have also demonstrated that pacritinib is more than 99% bound to plasma proteins. A study to assess the effect of food on drug bioavailability was performed using a single 200 mg dose of pacritinib in healthy volunteers. The mean plasma concentrations, peak plasma concentrations, and area under the curve were slightly higher, although not significantly higher, under fed conditions than fasted conditions. Therefore, it was concluded that there was no food effect on pacritinib.
Phase I studies of pacritinib in MF have provided pharmacokinetic data demonstrating that the drug is rapidly absorbed and does not accumulate with repeated cycles. In a Phase I study of pacritinib in myeloid diseases, including 31 patients with MF and five patients with AML, the mean time to peak concentration (Tmax) ranged from 3 to 5 hours, and the mean elimination half-life (t1/2) was 2–3 days.Citation42 The other Phase I study with 20 MF patients produced similar results: Tmax ranged from 3 to 6 hours, while t1/2 was 22–55 hours.Citation43 In both studies, pacritinib was administered orally once daily in 28-day cycles at doses ranging from 100 to 600 mg. Pharmacologically active levels were achieved at all doses. A dose-related linear increase in area under the concentration–time curve was observed with no drug accumulation.
Clinical efficacy in myelofibrosis
Phase II trials focusing on pacritinib for the treatment of MF demonstrated encouraging results. One study treated 33 patients with primary or secondary MF who had splenomegaly of at least 5 cm below the left costal margin and were not suitable for standard therapy.Citation44,Citation45 Pacritinib was administered orally as 400 mg daily in 28-day cycles. The primary endpoint was spleen response rate as measured by MRI from baseline up to week 24. Out of 30 patients, 29 (97%) patients had spleen volume reduction, and 17 (57%) patients had a reduction of ≥25%. Out of 31 patients assessed by physical exam, 12 (39%) patients had ≥50% spleen length reduction with seven (23%) patients having 100% reduction. In patients on treatment for 6 months, the intensity of disease-related symptoms decreased by 40%–65%. Another single-arm, open-label Phase II study of pacritinib in 34 patients with newly diagnosed intermediate and high-risk disease or previously treated MF was conducted.Citation46 According to DIPSS classification, 34% of patients were intermediate-1 risk, 37% were intermediate-2 risk, and 20% were high risk. This trial evaluated the primary endpoint of spleen response using pacritinib at the same dosing schedule. Out of 26 patients assessed by MRI, eight (31%) patients had ≥35% reduction in spleen volume. Out of 33 patients assessed by physical exam, 14 (42%) patients had ≥50% spleen length reduction. Fifteen (48%) patients had ≥50% reduction in total symptom score as assessed by the Myelofibrosis Symptom Assessment Form.
Two large randomized, controlled, open-label international Phase III trials of pacritinib for the treatment of MF are currently ongoing (). The primary endpoint for both studies is the proportion of patients achieving ≥35% reduction in spleen volume from baseline to week 24 by MRI or computed tomography. PERSIST-1 compares pacritinib 400 mg daily, randomized in a 2:1 ratio, vs BAT, which may include any physician-selected treatment agent except ruxolitinib, in 327 patients with primary and secondary MF regardless of platelet count.Citation47 Positive findings from PERSIST-1 were recently announced: pacritinib resulted in a significantly higher percentage of patients with spleen volume reduction and improvement in disease-related symptoms compared with BAT.Citation48 The spleen volume reduction rates at week 24 were 19.1% for pacritinib vs 4.7% for BAT in the intent-to-treat group and were 25% vs 5.9% in the evaluable population. It was observed that patients with the lowest platelet counts had the greatest efficacy with pacritinib. In patients with platelets <100×109/L and <50×109/L, spleen volume reduction rates were 23.5% and 33.3% in the pacritinib group, respectively. No patients in the BAT group with baseline platelets <100×109/L achieved the primary endpoint at week 24. PERSIST-2 has completed enrollment of approximately 300 patients with MF and baseline platelets ≤100×109/L.Citation49 Treatment-naïve or previously treated patients are randomized into three study arms, pacritinib 400 mg daily, pacritinib 200 mg twice daily, and BAT including ruxolitinib. This study could demonstrate the utility of pacritinib as first-line or second-line treatment in patients with MF and baseline cytopenias, either due to disease or prior therapy.
Table 3 Comparison of Phase III trials for pacritinib in myelofibrosis
On February 9, 2016 the US Food and Drug Administration placed all ongoing studies of pacritinib in myelofibrosis on a full clinical hold due to concerns about increased mortality due to intracranial hemorrhage and cardiac events among pacritinib-treated patients. Data are currently being collected from the PERSIST clinical trials and will be submitted to the FDA before a final decision will be rendered as to the potential for resumption of the trials.
Safety and tolerability
Prior to the recent FDA notification of concern over increased intracranial hemorrhage and cardiac events, pacritinib was considered well tolerated with manageable gastrointestinal (GI) symptoms and minimal myelosuppression and other hematologic adverse effects. In a Phase I dose-escalation trial of pacritinib, out of 36 patients with MF and AML, the main adverse events were grade 1/2 diarrhea (33%) and nausea (13%).Citation42 Grade 3 diarrhea (4%) and grade 3/4 thrombocytopenia (4%) were also noted. Dose limiting toxicities (DLTs) were only observed at the 600 mg dose, in which three of six patients developed GI toxicities requiring treatment interruption. In the other Phase I dose-escalation trial with 20 MF patients, the most common drug-related adverse events were diarrhea (89%), nausea and vomiting (39%), abdominal pain (22%), fatigue (22%), rash (17%), and dysgeusia (17%).Citation43 Overall, most events experienced were grade 1/2. Grade 3 toxicities were reported at the higher daily doses of 500 or 600 mg: diarrhea (11%), nausea (6%), abdominal pain (11%), and fatigue (11%). No DLTs were observed at the lower doses of 100, 200, or 400 mg. However, at the 600 mg daily dose, two of four patients experienced DLTs due to GI toxicities. Three patients in the 500 mg group also required treatment interruptions after cycle 1 due to diarrhea, dizziness, and elevated alanine aminotransferase, respectively. Thus, on the basis of tolerability profile and clinical efficacy, the suggested dose of pacritinib for Phase II trials was 400 mg daily.
The safety profile of pacritinib in MF was further examined in Phase II trials. Consistent with Phase I results, the most frequent adverse events were due to GI toxicity. One Phase II trial enrolled 33 MF patients, in which 32 were evaluable for safety.Citation44,Citation45 Diarrhea (81%), nausea (41%), vomiting (22%), fatigue (9%), rash (6%), and abdominal/extremity pain (6%) and insomnia (6%) were reported. These symptoms were readily manageable with supportive care, antidiarrheal, and antiemetic agents. There were minimal grade 3/4 toxicities, including grade 3 diarrhea (6%) and grade 3 rash (3%). Significant neutropenia and thrombocytopenia were uncommon, with no grade 3/4 events. Eighteen patients had adverse events requiring dose interruption or reduction, mostly due to diarrhea. There were three deaths reported; however, none were related to the study drug.
The other Phase II study enrolled 34 patients and observed similar nonhematological toxicities: diarrhea (77%), nausea (46%), fatigue (37%), and vomiting (31%).Citation46 Most adverse events were grade 1 or 2, though grade 3/4 events were also reported: fatigue (12%), diarrhea (9%), abdominal pain (6%), dehydration (6%), and aspartate aminotransferase increase (6%). Hematologic abnormalities such as anemia (all grades 34%; grade 3/4 26%) and thrombocytopenia (all grades 23%; grade 3/4 21%) were also noted. However, many patients had baseline cytopenias of Hgb <10 g/dL (40%) and platelets <100×109/L (43%). During the study period, mean Hgb levels remained steady, and platelet levels modestly decreased. Only three patients required dose interruption due to thrombocytopenia. Pacritinib was discontinued in nine patients (26%); however, only six cases were reasons related to the study drug including adverse reactions such as nausea, pruritus, hypersensitivity, thrombocytopenia, hyperbilirubinemia, and subdural hematoma. There were five deaths; two were due to disease progression, while three were due to severe adverse events, including one subdural hematoma possibly related to pacritinib and two sepsis cases considered unrelated to the study drug.
In the recently announced results of the Phase III PERSIST-1 trial comparing pacritinib and BAT, adverse events continued to be consistent with prior studies.Citation48 The most common adverse events were diarrhea, nausea, and vomiting (grade 3 were <5%, <1%, <1%, respectively). Hematologic adverse events were similar between the two treatment groups.
However, the concern that increased deaths among pacritinib-treated patients are being investigated and pacritinib treatment has been discontinued among all trial patients until further notice from the FDA. Subsequent to the initial announcement, compassionate use has been granted to certain patients who were felt to have been benefiting from the drug.
Discussion
The clinical application of pacritinib in the treatment of MF remains uncertain. Current data have demonstrated therapeutic benefits of decreasing splenomegaly and improving quality of life in patients with intermediate- or high-risk disease. In MF, it is believed that constitutional symptoms are linked to elevated plasma levels of proinflammatory cytokines; however, the effect of pacritinib on circulating cytokine levels via JAK inhibition is currently unknown. It also remains to be seen whether pacritinib confers survival benefits or modulates the natural course of disease progression in terms of mutation allele burden, bone marrow fibrosis, and AML transformation.
Although expected toxicities from pacritinib such as GI complaints are manageable, recent concerns over an excess of intracranial hemorrhage and cardiac events raise the possibility that unexpected adverse effects may complicate its use. Increased risk of infection and immunosuppression has not been described in currently available data. Significant drug-related hematological toxicities appear to be less common with pacritinib than with less selective JAK inhibitors, which is advantageous for MF patients who often present with baseline anemia and thrombocytopenia. The potential for pacritinib to have enhanced efficacy when used in combinatorial strategies, prior to alloHSCT or as an anti-leukemic therapy relies completely on analysis of the final toxicity and survival results from the PERSIST trials.
Besides pacritinib, other JAK2 inhibitors that target the dysregulated JAK/STAT pathway have been under investigation. However, the clinical development of many agents has been terminated because of significant adverse events. For example, XL019 was halted in a Phase I trial owing to peripheral neuropathy despite dose reductions.Citation50 Phase III trials of fedratinib (SAR302503) found significant neurotoxicity, specifically Wernicke’s encephalopathy,Citation51 while studies with lestaurtinib (CEP701) reported higher toxicities than therapeutic benefits.Citation52 Momelotinib (CYT387) is the only other JAK inhibitor still in development for the treatment of MF. This JAK1/2 inhibitor has also demonstrated clinical responses in splenomegaly, constitutional symptoms, and anemia in Phase I/II studies.Citation53 It is currently in a Phase III trial vs ruxolitinib for treatment of MF patients who are JAK inhibitor-naïve.Citation54
While the JAK/STAT pathway is the target of many investigational therapeutic agents for MF, it is just one of many components underlying the molecular pathogenesis of the disease. This is known in part because even though JAK2V617F mutation results in JAK/STAT dysregulation, it alone does not lead to the development of clinical MF and only 40%–60% of patients are JAK2V617F-positive. Furthermore, blast cells of transformed JAK2V617F-positive MF patients are often JAK2V617F-negative, suggesting that the mutation is not crucial to the leukemic transformation process.Citation55 Other members of the JAK family may play a key role in the manifestation of disease symptoms. For example, inhibition of JAK1 reduces levels of circulatory proinflammatory and proangiogenic cytokines, which may contribute to the improvement of symptoms reported by patients. Somatic mutations in other cell proliferation protein have been described in MF patients. Gain of function mutations in the thrombopoietin receptor myeloproliferative leukemia virus oncogene (MPL) is reported in approximately 5% of PMF patients who are JAK2V617F-negative.Citation56 Loss-of-function mutations in JAK/STAT pathway inhibitors SH2B3 or CBL genes have been identified as well.Citation26,Citation28 The interaction of these other signaling pathways with JAK/STAT provides additional therapeutic targets and treatment options for MF. Some current agents under investigation include mammalian target of rapamycin inhibitors (eg, everolimus), histone deacetylase inhibitors (eg, givinostat), pegylated IFN alpha (eg, PEG-IFN-α2a), proteasome inhibitors, PI3K inhibitors, and Hedgehog inhibitors. Various combinations of chemotherapy agents, hypomethylating agents, JAK inhibitors, and other pathway inhibitors are also being studied.Citation18
Conclusion
MF is a chronic hematological condition with a broad spectrum of disease symptoms that often lead to a poor quality of life with a risk of AML transformation. Although treatment options are currently limited, the novel class of JAK2 inhibitors has demonstrated disease response, as measured by improvement in splenomegaly and symptom burden. Current data on the clinical benefits and safety profile of pacritinib in the treatment of patients with MF are eagerly awaited, particularly in light of the unexpected toxicities seen with other JAK inhibitors resulting in cessation of further development. Full enrollment was achieved in PERSIST-1 and PERSIST-2 prior to the FDA clinical hold; once toxicities are clarified and resolved and it is determined whether the hold will be lifted, we may more clearly define the role of pacritinib in the management of MF.
Disclosure
The authors report no conflicts of interest in this work.
References
- Abdel-WahabOILevineRLPrimary myelofibrosis: update on definition, pathogenesis, and treatmentAnnu Rev Med20096023324518947294
- TamCSNussenzveigRMPopatUThe natural history and treatment outcome of blast phase BCR-ABL-myeloproliferative neoplasmsBlood200811251628163718566326
- GuptaVFoltzLSirhanSBusqueLTurnerAREmerging therapeutic options for myelofibrosis: a Canadian perspectiveAm J Blood Res20122317018623119228
- CervantesFPassamontiFBarosiGLife expectancy and prognostic factors in the classis BCR/ABL-negative myeloproliferative disordersLeukemia200822590591418385755
- MehtaJWangHIqbalSUMesaREpidemiology of myeloproliferative neoplasms in the United StatesLeuk Lymphoma201455359560023768070
- TefferiACME information: primary myelofibrosis: 2014 update on diagnostic, risk-stratification and managementAm J Hematol201489991592525124313
- CervantesFDupriezBPereiraANew prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and TreatmentBlood2009113132895290118988864
- PassamontiFCervantesFVannucchiAMA dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment)Blood201011591703170820008785
- GangatNCarmazzaDVaidyaRDIPSS plus: a refined dynamic International Prognositc Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion statusJ Clin Oncol201029439239721149668
- CaramazzaDBegnaKHGangatNRefined cytogenetic-risk categorization for overall and leukemia-free survival in primary myelofibrosis: a single center study of 433 patientsLeukemia2011251828820944670
- GuglielmelliPLashoTLRotunnoGThe number of prognostically detrimental mutations and prognosis in primary myelofibrosis: an international study of 797 patientsLeukemia20142891804181024549259
- TefferiALashoTLFinkeCMCALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: clinical, cytogenetic and molecular comparisonsLeukemia20142871472147724402162
- TefferiALashoTTischerAThe prognostic advantage of calreticulin mutations in myelofibrosis might be confined to type 1 or type 1-like CALR variantsBlood2014123152465246625301336
- ZhouAOhSTPrognostication in MF: from CBC to cytogenetics to molecular markersBest Pract Res Clin Haematol201427215516425189726
- BallenKKShresthaSSobocinskiKAOutcome of transplantation for myelofibrosisBiol Blood Marrow Transplant201016335836719879949
- KrögerNHollerEKobbeGAllogeneic stem cell transplantation after reduced-intensity conditioning in patients with myelofibrosis: a prospective, multicenter study of the Chronic Leukemia Working Party of the European Group for Blood and Marrow TransplantationBlood2009114265264527019812383
- KrogerNGioginoTScottBLImpact of allogeneic stem cell transplantation on survival of patients less than 65 years of age with primary myelofibrosisBlood2015125213347335025784679
- GowinKMesaREmerging therapies for the treatment of chronic Philadelphia chromosome-negative myeloproliferative neoplasm-associated myelofibrosisExpert Opin Investig Drugs2013221216031611
- CervantesFHow I treat myelofibrosisBlood2014124172635264225232060
- GeyerHLMesaRATherapy for myeloproliferative neoplasms: when, which agent, and how?Blood2014124243529353725472969
- TefferiAMesaRANagorneyDMSchroederGSilversteinMNSplenectomy in myelofibrosis with myeloid metaplasia: a single-institution experience with 223 patientsBlood20009572226223310733489
- Martinez-TrillosAGayaAMaffioliMEfficacy and tolerability of hydroxyurea in the treatment of the hyperproliferative manifestations of myelofibrosis: results in 40 patientsAnn Hematol201089121233123720567824
- HuangJTefferiAErythropoiesis stimulating agents have limited therapeutic activity in transfusion-dependent patients with primary myelofibrosis regardless of serum erythropoietin levelEur J Haematol200983215415519366369
- CervantesFAlvarez-LarránADomingoAArellano-RodrigoEMontserratEEfficacy and tolerability of danazol as a treatment for the anemia of myelofibrosis with myeloid metaplasia: long-term results in 30 patientsBr J Haematol2005129677177515953003
- TefferiAPassamontiFBarbuiTPhase 3 study of pomalidomide in myeloproliferative neoplasm (MPN)–associated myelofibrosis with RBC-transfusion dependence [abstract]Blood20131222139423687088
- TamCSVerstovsekSInvestigational Janus kinase inhibitorsExpert Opin Investig Drugs2013226687699
- GotlibJJAK inhibition in the myeloproliferative neoplasms: lessons learned from the bench and bedsideHematol Am Soc Hematol Educ Program20132013529537
- MascarenhasJOCrossNCMesaRAThe future of JAK inhibition in myelofibrosis and beyondBlood Rev201428518919625043171
- TefferiAVaidyaRCaramazzaDFinkeCLashoTPardananiACirculating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling studyJ Clin Oncol201129101356136321300928
- VerstovsekSMesaRAGotlibJA double-blind, placebo-controlled trial of ruxolitinib for myelofibrosisN Engl J Med2012366979980722375971
- HarrisonCKiladijianJJAl-AliHKJAK inhibition with ruxolitinib versus best available therapy for meylofibrosisN Engl J Med2012366978779822375970
- VerstovsekSMesaRAGotlibJEfficacy, safety and survival with ruxolitinib in patients with myelofibrosis: results of a median 2-year follow-up of COMFORT-IHaematologica201398121865187124038026
- CervantesFVannucchiMKildadjianJJThree-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosisBlood2013122254047405324174625
- PassamontiFMaffioliMCervantesFImpact of ruxolitinib on the natural history of primary myelofibrosis: a comparison of the DIPSS and the COMFORT-2 cohortsBlood2014123121833183524443442
- JungCWRuxolitinib changes the natural course of myelofibrosis and its transplant outcomeBlood Res2013481686923589801
- Parampalli YajnanarayanaSStübigTCornezIJAK1/2 inhibition impairs T cell function in vitro and in patients with myeloproliferative neoplasmsBr J Haematol2015169682483325824483
- SchönbergKRudolphJVonnahmeMJAK inhibition impairs NK cell function in myeloproliferative neoplasmsCancer Res201575112187219925832652
- PouslenAWilliamABlanchardSStructure-based design of oxygen-linked macrocyclic kinase inhibitors: discovery of SB1518 and SB1578, potent inhibitors of Janus kinase 2 (JAK2) and Fms-like tyrosine kinase-3 (FLT3)J Comput Aided Mol Des201226443745022527961
- WilliamADLeeACBlanchardSDiscovery of the macrocycle 11-(2-pyrrolidin-1-ylethoxy)-14,19-dioxa-5,7,26-triazatetracyclo(19.3.1.1(2,6).1(8,12))heptacosa-1(25),2(26),3,5,8,10,12(27), 16,21,23-decaene (SB1518), a potent Janus kinase 2/fms-like tyrosine kinase-3 (JAK2/FLT3) inhibitor for the treatment of myelofibrosis and lymphomaJ Med Chem201154134638465821604762
- HartSGohKCNovotny-DiermayrVSB1518, a novel macrocyclic pyrimidine-based JAK2 inhibitor for the treatment of myeloid and lymphoid malignanciesLeukemia201125111751175921691275
- VerstovsekSMachidaCDeanJPMylntHPacritinib. Inhibitor of tyrosine-protein kinase JAK2, inhibitor of FLT-3, treatment of myelofibrosisDrugs Future2013386375386
- VerstovsekSOdenikeOScottBPhase I dose-escalation trial of SB1518, a novel JAK2/FLT3 inhibitor, in acute and chronic myeloid diseases, including primary or post-essential thrombocytopenia/polycythemia vera myelofibrosis [abstract 3905, ASH]Blood20091141502
- SeymourJFToBGohAFirst report of the phase I study of the novel oral JAK2 inhibitor SB1518 in patients with myelofibrosis [abstract 1144]Haematologica201095Suppl 2472
- DeegHJOdenikeOScottBLPhase II study of SB1518, an orally available novel JAK2 inhibitor, in patients with myelofibrosis [abstract]J Clin Oncol201129Suppl 15 abstract 6515, ASCO
- VerstovsekMDDeegHGOdenikeOPhase 1/2 study of SB1518, a novel JAK2/FLT3 inhibitor, in the treatment of primary myelofibrosisBlood2010 abstract 3082, ASH
- KomrokjiRSSeymourJFRobertsAWResults of a Phase 2 study of pacritinib (SB1518), a JAK2/JAK2(V617F) inhibitor, in patients with myelofibrosisBlood2015125172649265525762180
- Cell TherapeuticsOral pacritinib versus best available therapy to treat myelofibrosis (PAC325) Available from: http://www.clinicaltrials.gov/ct2/show/NCT01773187. NLM identifier: NCT01773187Accessed June 5, 2015
- MesaRAEgyedMSzokeAResults of the PERSIST-1 Phase III study of pacritinib (PAC) versus best available therapy (BAT) in primary myelofibrosis (PMF), post-polycythemia vera myelofibrosis (PPV-MF), or post-essential thrombocythemia-myelofibrosis (PET-MF) [abstract]J Clin Oncol201533 abstract LBA7006, ASCO
- Cell TherapeuticsOral pacritinib versus best available therapy to treat myelofibrosis with thrombocytopenia (PAC326) Available from: http://www.clinicaltrials.gov/ct2/show/NCT02055781. NLM identifier: NCT02055781Accessed June 5, 2015
- VerstovsekSTamCSWadleighMPhase I evaluation of XL019, an oral, potent, and selective JAK2 inhibitorLeuk Res201438331632224374145
- SanofiSanofi discontinues clinical development of investigational JAK2 agent fedratinib (SAR302503)Paris, France2013 Available from: http://en.sanofi.com/Images/34935_20131118_JAK-2-FEDRATINIB_en.pdfAccessed June 6, 2015
- SantosFPKantarjianHMJainNPhase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post-polycythemia myelofibrosisBlood201011561131113620008298
- PardananiALabordeRRLashoTLSafety and efficacy of CYT387, a JAK1 and JAK2 inhibitor, in myelofibrosisLeukemia20132761322132723459451
- GileadMomelotinib versus ruxolitinib in subjects with myelofibrosis (simplify 1) Available from: http://www.clinicaltrials.gov/ct2/show/NCT01969838. NLM Identifier: NCT01969838Accessed June 5, 2015
- BeerPADelhommeauFLeCouédicJPTwo routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasmBlood2010115142891290020008300
- PardananiADLevineRLLashoTMPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patientsBlood2006108103472347616868251