Abstract
Oncolytic viruses are emerging as a potential new way of treating cancers. They are selectively replication-competent viruses that propagate only in actively dividing tumor cells but not in normal cells and, as a result, destroy the tumor cells by consequence of lytic infection. At least six different oncolytic herpes simplex viruses (oHSVs) have undergone clinical trials worldwide to date, and they have demonstrated an excellent safety profile and intimations of efficacy. The first pivotal Phase III trial with an oHSV, talimogene laherparepvec (T-Vec [OncoVexGM-CSF]), is almost complete, with extremely positive early results reported. Intuitively, therapeutically beneficial interactions between oHSV and chemotherapeutic and targeted therapeutic drugs would be limited as the virus requires actively dividing cells for maximum replication efficiency and most anticancer agents are cytotoxic or cytostatic. However, combinations of such agents display a range of responses, with antagonistic, additive, or, perhaps most surprisingly, synergistic enhancement of antitumor activity. When synergistic interactions in cancer cell killing are observed, chemotherapy dose reductions that achieve the same overall efficacy may be possible, resulting in a valuable reduction of adverse side effects. Therefore, the combination of an oHSV with “standard-of-care” drugs makes a logical and reasonable approach to improved therapy, and the addition of a targeted oncolytic therapy with “standard-of-care” drugs merits further investigation, both preclinically and in the clinic. Numerous publications report such studies of oncolytic HSV in combination with other drugs, and we review their findings here. Viral interactions with cellular hosts are complex and frequently involve intracellular signaling networks, thus creating diverse opportunities for synergistic or additive combinations with many anticancer drugs. We discuss potential mechanisms that may lead to synergistic interactions.
Introduction
Using viruses to treat cancer is not a new idea. For more than 100 years there have been clinical observations that cancer patients who contracted viral infections would enter periods of remission.Citation1 During the 1950s and 1960s, there was considerable activity using wild-type viruses as anticancer treatments, but many of these trials were limited by the toxicity of the wild-type virus (for a historical perspective see Kelly and RussellCitation1). Progress has only recently been possible as advances in virology and molecular biology have allowed either the identification of naturally occurring viruses with intrinsic tumor selectivity or by genetically engineering oncolytic viruses.
Oncolytic herpes simplex virus (oHSV)
Oncolytic herpes viruses are attenuated, replication competent, herpes simplex type 1 viruses that selectively infect, replicate within, and lyse cancer cells. One of the first reports of an oncolytic virus being used for cancer therapy was in the early 1990s when Martuza et alCitation2 showed that a replication competent thymidine kinase negative herpes simplex virus (HSV)-1 mutant effectively prolonged survival of nude mice bearing intracranial glioma. Since then, numerous oHSVs have been described, most of which have deletions in either RL1, UL39, or both.
ICP34.5, the protein product of the γ34.5 gene, is a specific determinant of neurovirulence. It plays a key role by facilitating escape from a major host defense mechanism involving the protein kinase R-mediated innate immune response pathway by directly interacting with protein phosphatase 1α to dephosphorylate eIF2α ().
Figure 1 HSV-1 can overcome normal cells protective block in protein synthesis: 1. HSV-1 enters the host cell and begins replication. 2. Complementary RNA anneal to produce dsRNA. 3. PKR binds dsRNA, dimerizes resulting in activation and autophosphorylation. 4. Phosphorylated PKR selectively phosphorylates elF2α. 5. Phosphorylated elF2α causes the host cell to shutdown translation thereby preventing viral replication. 6. HSV produced ICP34.5 which forms a protein complex with PP1α. 7. The ICP34.5 PP1α complex dephosphorylates elF2α so the viral replication (8) can continue unchecked.
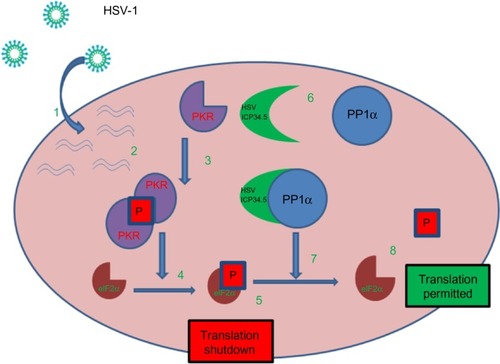
In contrast, oncolytic HSV, which lacks functional ICP34.5 protein, cannot dephosphorylate eIF2α. Thus, infection with an ICP34.5 null virus causes the host cell to shut down protein synthesis, hence, preventing the virus from replicating in normal cells. Cancer cells, however, in the course of transforming to malignant cells have impaired antiviral mechanisms that permit unimpeded viral replication.Citation3
UL39 is the HSV gene encoding for the large subunit of ribonucleotide reductase (RR), the main rate limiting enzyme for viral DNA synthesis and replication, controlling the nucleotide substrate pool by regulating the conversion of ribonucleotides to deoxyribonucleotides. HSV RR is required for growth in nondividing cells but not in rapidly dividing cells, in which there is ample cellular RR for the virus to utilize. Oncolytic HSV with a defective UL39 gene exclusively replicates in and lyses rapidly dividing cancer cells, as such cells provide sufficient levels of RR activityCitation4 (for comprehensive review of oHSV see Cassady and Parker,Citation5 Manservigi et al,Citation6 and Varghese and RabkinCitation7).
Modified (armed and targeted) oHSV
The concept of using viral vectors to deliver therapeutic genes to tumors is well established. Many studies have evaluated both the oncolytic and antitumor activity, and the antitumor immune response of oncolytic viruses engineered to express either immunostimulatory genes or therapeutic genes, including those that can activate prodrugs.
The therapeutic efficacy of oncolytic HSV vectors encompasses two modes of action: direct oncolysis by the virus itself and indirect induction of an antitumor response. By arming viruses with genes that encode for immunomodulatory proteins such as IL(interleukin)-12,Citation8–Citation10 IL-2,Citation11 soluble B7.1-Ig,Citation12 or granulocyte macrophage colony-stimulating factor (GM-CSF)Citation13–Citation16 to help promote the antitumor immune response, the modified viruses are more efficacious.
Virus-directed enzyme prodrug therapy systems have also been utilized with oncolytic HSV. There are numerous reports of viruses that have been modified to code for enzymes that catalyze prodrugs into active substrates, such as HSV1yCD, a modified HSV coding for the yeast cytosine deaminase (CD) enzyme. HSV1yCD converts the nontoxic 5-fluorocytosine into fluorouracil (5-FU), a highly toxic chemotherapeutic agent,Citation17 rRp450 carrying rat cytochrome P450 (CYP2B1) (which converts cyclophosphamide into the alkylating toxin phosphoramide mustard),Citation18 and nitroreductase, which converts the prodrug CB1954 to an active alkylating agent.Citation19 The extensive field of oncolytic HSV vectors modified for enhanced efficacy is beyond the scope of this review; the major approaches are detailed here but reviewed in greater detail by Varghese and Rabkin.Citation7
lists the principal oHSV in clinical development. At least six different oHSV have undergone clinical trials worldwide to date. oHSV have demonstrated excellent safety profiles and, in numerous studies, signals of efficacy. The first Phase III trial with an oHSV, talimogene laherparepvec (T-Vec [OncoVexGM-CSF]) has almost been completed. Initial extremely encouraging findings of the trial have been reported, with T-Vec demonstrating a statistically significant improvement in durable response rate.Citation20
Table 1 Oncolytic HSVs in clinical trials
Oncolytic viruses in combination with chemotherapy
The use of many chemotherapeutic agents is limited by severe dose limiting toxicities and the emergence of resistant disease.Citation21 In comparison, the mode of action of oncolytic viruses (lytic infection) means that cancer cells are unlikely to become resistant to them. Furthermore, oncolytic viruses have a high therapeutic index (ie, the comparison of the amount of a therapeutic agent that causes the therapeutic effect, to the amount that causes toxicity) with very limited toxicities. summarizes the potential advantages of oncolytic virotherapy.
Table 2 Advantages of oncolytic virotherapy
Viral infection initiates many complex host defense pathways;Citation22 however, viruses have coevolved equally complex countermeasures to circumvent these activities.Citation23,Citation24 Many of these countermeasures are retained by their oncolytic variants ( outlines the main cellular and viral pathways activated upon viral infection). As chemotherapeutic and targeted anticancer agents target key cellular processes that also involve complex intracellular signaling networks, there are extensive opportunities for antagonistic and synergistic interactions with oncolytic viruses, and these need to be explored and understood as the clinical acceptance of oncolytic HSV looks increasingly likely.Citation25
Table 3 Main cellular and viral pathways activated upon viral infection
Combining these two very different modalities in order to increase cancer cell killing is a rational approach. The clinical implications of this combination therapy are not limited to enhanced efficacy. The dose reduction index, the most relevant clinical parameter derived by Chou and Talalay analysis,Citation26 reveals the potential for significant dose reductions without compromising tumor cell kill. Reducing the dose of drugs such as chemotherapeutics would minimize the toxicity and may allow patients to remain on an otherwise intolerable regime, or increase their quality of life whilst still receiving treatment for their disease.
Since the initial groundbreaking studies by Toyoizumi et alCitation27 with HSV1716 and four standard chemotherapeutic drugs, methotrexate, cisplatin, mitomycin C, and doxorubicin, there have been many reports of the increased efficacy of oHSV in combination with a wide range of existing and potentially new anticancer drugs. – present the wide variety of different combinations that have been examined, and also summarize the results. The aim of this review is not to discuss the individual results presented in these tables, but to attempt a mechanistic overview that relates to their findings. Crucially, there are a number of reasons why oncolytic virus therapy in combination with chemotherapeutic agents, or other anticancer treatments, will be beneficial. Firstly, the mode of action of oncolytic viruses is completely different from chemotherapeutics and they are not, therefore, in direct competition. Secondly, oncolytic cell killing is independent of the many genomic alterations that lead to drug-resistant tumors and so may be effective even in drug-resistant cells.
Table 4 Oncolytic viruses and chemotherapeutic agent
Table 5 Oncolytic viruses and mTOR inhibitors
Table 6 Oncolytic viruses and PI3K inhibitors
Table 7 Oncolytic viruses and HDAC inhibitors
Table 8 Oncolytic viruses and others
The most widely used method of studying drug/drug (or virus/drug) interactions between two modalities in vitro is using the methods of Chou and Talalay.Citation26,Citation28 This type of analysis is one of the few available that identifies beneficial interactions based on an extrapolated equation. The possibility of predicting a false positive is minimized as the analysis takes into account both the potency (the IC50 [half the maximal inhibitory concentration] or the LD50 [median lethal dose]) and the slope of the dose effect curves (m-value) in the precise analysis of two therapeutic combinations. The method defines the expected additive effect of two (or more) agents and quantifies synergy or antagonism by way of how different the measured effect is from the expected additive effect. The equations are detailed elsewhere.Citation26,Citation28,Citation29 Interpretation of the combination index (CI) values are defined as: CI =1 indicates an additive effect; a CI of <1 indicates synergy; and a CI >1 indicates antagonism. Synergy is the working together of two agents to produce a result greater than the sum of their individual effects, while antagonism is less than that of an additive effect.
Chou and TalalayCitation26 analysis can also be used effectively in vivo, but it is more common practice, as reported in the literature, to look for differences in tumor growth between treatment groups and to use analysis of variance or t-tests to determine if the differences (often either tumor volume or length of survival) between groups are significant. Information on synergy and/or enhanced efficacy of combinations will also come from clinical studies. Most patients that take part in new cancer therapy trials have already had, or are currently being treated with, the standard treatment for their particular disease, and it will be interesting to see if any group treated with oHSV and another agent respond better or worse than predicted. There are a number of different ways in which an oHSV in combination with an anticancer drug can be synergistic and these are discussed below.
Compounds that increase the replicative capacity of the virus
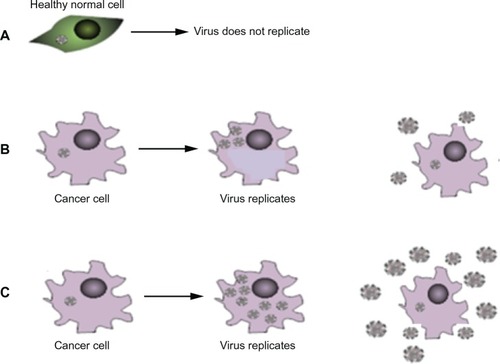
Oncolytic HSV have selective replication competence in cancer cells and, by increasing the replicative capacity of the virus within those cells, the number of progeny viruses produced during a cycle of infection could be increased ().
Figure 2 Increasing replicative capacity of the virus: (A) in normal cells the virus does not replicate. (B) In a cancer cell the virus replicates, lyses the cell and produces viral progeny that go on to infect further cancer cells. (C) In the presence of certain drugs the virus can produce more viral progeny. Upon lysis more progeny virus are released – potentially increasing the number of cells that can be infected.
Differentiating inducing agent hexamethylene bisacetamide (HMBA) has been shown to improve viral yield, with up to a 10,000-fold increase in vitro for an ICP34.5 null virus, R849, at low MOI (multiplicity of infection). HSV immediate early gene expression ( shows the basic HSV replication cycle) was also increased with HMBA.Citation30 Mice treated with both HMBA and R849 virus had significantly smaller tumor burden and survived longer than either virus or HMBA treatment alone, with increased levels of HSV transcripts of immediate early, early, and late genes in the combination treatment group. This suggests HMBA may increase and/or activate cellular proteins such as transcription factors, which act to improve viral yield. HMBA is a drug that was thought to have some potential as a stand-alone anticancer agent; however, the level of drug required for such anticancer activity could not be achieved in patients.Citation31 In the study with oHSV, a much lower dose of drug was able to be used; one which could easily be achieved in patients and potentially would act as a promoting agent for oncolytic therapy.
Figure 3 Anti-viral host response mediated by IFN (interferon) induces apoptosis of surrounding cells. By using drug to block innate antiviral defence mechanism the infected cell will not signal other nearby cells to ‘warn’ them about the virus, hence viral replication will occur.
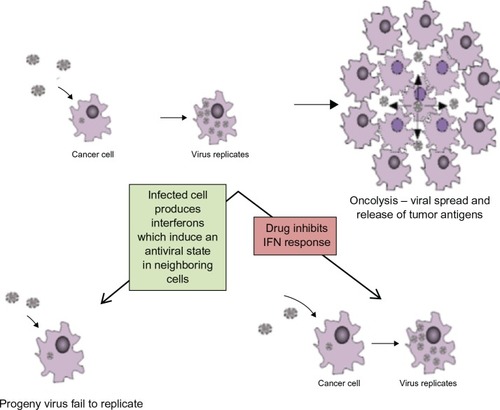
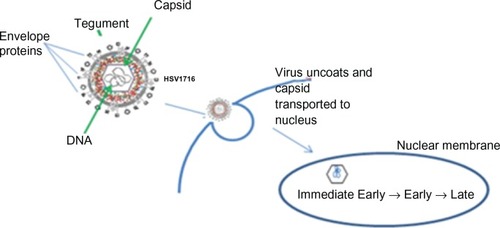
Figure 4 Herpes simplex virus (HSV) replication cycle HSV-1 is a double stranded DNA virus which encodes for around 100 transcripts and contains three main structural components. The central capsid (or nucleocapsid) contains the viral DNA. This is surrounded by an envelope. The tegument is located between the envelope and the capsid. HSV enters the host cell at either the cell surface or via pH dependent endocytosis through a process involving envelope glycoproteins. The tegument proteins are released into the cell and the capsid is transported to the nucleus where viral DNA is released into the nucleus. There are three classes of viral genes that are transcribed and translated in a specific order: Immediate Early (IE) genes, which encode for proteins that promote expression of viral genes and also have a role in innate immune invasion, Early (E) are responsible for the replication of viral DNA and lastly Late (L) genes which include capsid, tegument and envelope proteins.
Another mechanism for increasing viral yields may be to temporarily block apoptosis. Upon viral infection, one of the cellular host responses is to induce apoptosis in infected cells and in cells surrounding infected cells () in order to limit the ability of the virus to replicate and spread. Therefore, by blocking apoptosis temporarily, there is the potential for improving the propagation of viral progeny, maximizing the lateral spread of virus and increasing tumor destruction. Wood and ShillitoeCitation32 reported on increased viral replication in the presence of zVAD-fmk; a pan caspase inhibitor that has previously been shown to prevent HSV-1-induced apoptosis.Citation33 The authors showed that the inhibitor increased levels of replication in an ICP34.5 null mutant back to the levels of wild type HSV-1. Stanziale et alCitation34 also reported increased apoptosis in cells that neighbored NV1066-infected cells and could mitigate this effect with treatment with an inhibitor of apoptosis: N-acetylcysteine. This suggests that the increased viral yield seen with the caspase inhibitors is likely to be due to neighboring noninfected but alarmed cells being prevented from initiating apoptosis and, therefore, become lytically infected with virus.Citation32,Citation34 Eisenberg et alCitation35 reported that hyperthermia potentiates oncolytic viral killing. After hyperthermic insult, the heat shock protein Hsp72 is upregulated, which inhibits cellular apoptosis, thereby allowing increased viral replication and, in turn, enhanced tumor kill. This finding has great potential as, in a clinical setting, the application of heat is likely to be noninvasive and relatively toxicity free.
Compounds that increase cell permissiveness to oHSV
Many chemotherapeutic drugs are DNA damaging agents and, following exposure to such agents, cells upregulate their DNA damage repair pathways. Such upregulation appears to be beneficial for oncolytic viral replication; mitomycin C,Citation36 temozolomide,Citation37,Citation38 and 5FUCitation39 have all been shown to increase oncolytic HSV replication.
Growth arrest and DNA damage-inducible protein GADD34 is induced by stressful growth arrest conditions and treatment with DNA-damaging agents. The carboxyl terminal of GADD34 bears significant homology with the virulence factor ICP34.5, which is deleted in some oHSV, eg, HSV1716, NV1066, R3613, and T-Vec (). Previous studiesCitation40 have shown that the carboxyl terminus of GADD34 can substitute for ICP34.5 in preventing premature shutoff of protein synthesis, and ICP34.5 null mutants can use the host cell GADD34 protein for viral replication. Thus, the presence of GADD34 in tumor cells following treatment with a DNA damaging agent would increase the number of cells permissive to oHSV infection and increase the viral spread through the tumor. Indeed, when GADD34 small interfering RNAs (siRNAs) were added to block GADD34 expression after treatment with a DNA damaging agent (cisplatin), the previously observed synergy with the oHSV NV1066 and cisplatin was abolished.Citation41
Another potential mechanism for synergy with some oHSV is upregulation of cellular RR by DNA-damaging chemotherapeutic agents.Citation42 High throughput screening has been reported to identify small-molecule compounds that augment the replication of HSV G47Δ,Citation43 and, of the 2,460 compounds screened, six compounds were identified and subsequently validated for enhanced G47Δ replication. Two of these compounds, dipyridamole and dilazep, interfered with nucleotide metabolism by potently and directly inhibiting the equilibra-tive nucleoside transporter-1 and were dependent on HSV mutations in ICP6, the large subunit of RR. Equilibrative nucleoside transporter-1 antagonists are thought to augment oHSV replication in tumor cells by increasing cellular RR activity.Citation43 As oHSV with UL39 deletions can only replicate in cells with active cellular RR, increasing cellular RR will improve viral replication.
Nakano et alCitation44 reported an upregulation in RR in tumors mediated by 5FU that augmented the therapeutic effect of G207. 5FU was also found to be synergistic both in vitro and in vivo with oHSV NV1020 (an oHSV with intact ICP6),Citation45 suggesting the effects of 5FU are not limited to upregulation of RR. The authors speculated that the synergy was in part due to the cells being sensitized to 5FU as the virus caused the cells to arrest in S phase of the cell cycle. They further speculated that the reduction in viral progeny could be due to the immune IFN (interferon)-γ response as well as the 5FU-induced upregulation of cell death via molecules such as TRAIL (TNF [tumor necrosis factor] related apoptosis-inducing ligand) and Fas ligand.
Rapamycin markedly increased the yield and dissemination of oHSV in semipermissive tumor cells both in vitro and in vivo but had no additional effect in cell lines that are permissive to the ICP34.5 null mutant oHSV Baco1.Citation46 The reason behind the observation is still unclear; however, inhibitors of the mTOR (mammalian target of rapamycin) signaling pathway increase permissiveness of resistant tumor cells to oncolytic myxoma virus,Citation47 vesicular stomatitis virus,Citation48 adenovirus,Citation49 and cytomegalovirus,Citation50 suggesting that the mTOR signaling pathway has an important role to play in virotherapy.
Compounds that modulate the immune system
The immune response to oncolytic viral therapy is an essential factor determining the success of oHSV as an antitumor agent; it can be a hindrance if it causes premature viral clearance, or could be seen as a positive, with the virally infected tumor becoming a target for clearance by the immune system.
The immune response to viral infection is beyond the scope of this review, but for an excellent insight into this field see Paludan et al.Citation22 Briefly, the immune reaction to a viral infection (oncolytic or otherwise) is a multipronged response. Very quickly upon infection, the innate immune response recruits natural killer (NK) cells, macrophages, and neutrophils to the site of infection and mediates a nonspecific viral clearance. NK cells appear to be an important player in the response to viral infection; patients with naturally occurring NK cell deficiencies (despite there being numerous different mutations that cause such deficiencies) have severe and recurrent herpes virus infections.Citation51 NK cells, activated by macrophages secreting IL-12, mediate the lysis of virally infected cells by releasing cytotoxic granules containing lytic enzymes and by binding to apoptosis-inducing receptors on the infected cell. In addition, NK cells secrete IFN-γ, which activates further macrophages and, consequently, orchestrates the downstream adaptive immune response.
The oncolytic HSV rQNestin34.5 (ICP34.5 expression controlled by the nestin promoter) has been shown to induce a rapid recruitment of NK cells to orthotopic human glioblastoma xenografts with subsequent killing of the oHSV-infected xenograft cells by activated macrophages. Depletion of NK cells improved the oHSV efficacy in these glioblastoma models, further indicating the importance of the NK cells.Citation52 Previous studies have demonstrated that inhibition of the innate immune response using cyclophosphamideCitation53–Citation56 or macrophage depletionCitation57 enhances oHSV replication and efficacy. An oHSV variant, rRp450, with deleted ICP6 and incorporated cytochromeP450 transgene for direct cyclophosphamide activation has been described, and the virus enhances the antitumor effects of cyclophosphamide.Citation18,Citation54,Citation58,Citation59
Another key event in the immune response to viral infection is the secretion of IFN-γ (for an extensive review see RoizmanCitation40 and Bazan-Peregrino et alCitation60). The cytokine IFN-γ, or type II interferon, is critical for innate and adaptive immune response to viral infection, partly from its ability to inhibit viral replication directly, but, more importantly, also from its immunostimulatory and immunomodulatory effects. IFN-γ is produced predominantly by NK cells as part of the innate immune response, and by cluster of differentiation (CD)4+ T helper (Th)1 and CD8 cytotoxic T lymphocyte (CTL) effector cells once antigen-specific immunity develops.
Histone deacetylase inhibitors (HDIs) are a class of compounds that appear to benefit HSV oncolysis, possibly via suppression of innate immune responses. Histone deacetylases (HDACs) have pleiotropic effects on cells through deacetylation of proteins, including histones, which then alter the epigenome and transcription profiles. Numerous HDACs have been targeted for drug discovery for cancer therapies, either for use as a single agent or in combination with chemotherapeutic agents. Pretreatment with the HDI valproic acid was shown to enhance the oncolytic virus MGH2 and rQNestin34.5 replication and spread in tumors, and extended the survival of mice bearing intracerebral tumors.Citation52,Citation61 The authors attributed the synergy between HDIs and oHSV to inhibition of type I interferon responses that would usually restrict viral gene expression and replication.
Drugs that cause downregulation of the innate immune response can be synergistic with oncolytic viruses but there is also evidence of the immune response enhancing tumor clearance.Citation62 Benencia et alCitation63 reported that oHSV therapy was less effective in murine metastatic melanoma models lacking NK and T cell subsets. Similarly, HSV1716-induced expression of IFN-γ inducible chemokines was accompanied by a significant increase in the number of NK and CD8+ cells in the tumor microenvironment in a syngeneic ovarian carcinoma model.Citation59,Citation63
Synergy has also been reported with oHSV and compounds that increase IFN-γ production.Citation64 The authors found that pretreating tumor cells with gemcitabine before oHSV significantly reduced tumor growth in vivo. Pretreatment was necessary as the drug itself induces early termination of DNA synthesis, which prevents replication of oncolytic viruses.Citation39,Citation64–Citation66 Gemcitabine selectively kills myeloid-derived suppressor cells, which inhibit IFN-γ production by CD8+ cells. So, when myeloid-derived suppressor cells themselves are killed, CD8+ T cells will secrete higher levels of IFN-γ, thus directing more T cells to tumor sites, which results in an improved antitumor response. In addition, IFN-γ can change the tumor microenvironment in terms of macrophages phenotype. Macrophages are classified as m1 (classically activated) or m2 (alternatively activated). During tumor progression there is a switch from m1- to m2-like phenotype that is believed to allow the tumor cells to avoid the immune system. Higher levels of IFN-γ can change the macrophage phenotype back to m1, resulting in the cancer cells being more likely to be tagged for destruction by the immune system.Citation64
Recently, a number of immunotherapeutic agents have been approved as cancer treatments. Ipilimumab, a monoclonal antibody that blocks the CTL-associated antigen 4 receptor, which would normally inhibit cytotoxic T lymphocyte, for example, is approved for use in advance metastatic melanoma.Citation67,Citation68 It is by blocking the CTL-associated antigen 4 receptor that CTLs are activated and can recognize and destroy cancer cells. As the presence of an oncolytic virus within a tumor will make the tumor more antigenic, there is good reason to think that the combination of oncolytic virus and immunotherapy will be synergistic and, indeed, there are many reports of improved efficacy of oHSV engineered to express genes that make immunomodulatory proteins including IL-12, IL-24, IL-4, RANTES (Regulated on Activation, Normal T cell Expressed and Secreted), CD80, and IFNα.Citation68 Granulocyte-macrophage colony-stimulating factor, which generates an antitumor response by the recruitment and differentiation of activating dendritic cells in the tumor microenvironment, has been inserted successfully into T-Vec,Citation69,Citation70 and a clinical study investigating T-Vec in combination with ipilimumab is underway,Citation71 with primary results expected in summer 2016.
Immunomodulatory drugs highlight the complexities of potential interactions between oHSV and anticancer agents, with synergy reported with drugs that inhibit or upregulate the immune system. It is likely that drugs that inhibit the very early innate immune response will allow the virus longer to enter cells and undergo initial viral replication, increasing the spread of the virus. Drugs that act by boosting later immune responses, such as upregulating T cells, mean that the infected tumor cells and potentially uninfected neighboring tumor cells are more likely to be targeted for destruction by the immune system. It will be interesting to see if downregulating innate immunity by HDIs, for example, and upregulating T cells by gemcitabine, would result in further synergistic effects when combined with an oncolytic virus. To date, no triple combinations have been reported in the literature, probably due to the increasing complexity of such experiments.
Compounds that alter the tumor microenvironment
Angiogenesis is the formation of new blood vessels and, as tumors need blood vessels to grow and spread, inhibitors of angiogenesis, which prevent the formation of new blood vessels, could potentially prevent or slow the growth or spread of tumors. Unlike chemotherapeutic agents, angiogenesis inhibitors will not kill cancer cells directly but instead prevent tumors from growing, so potentially, in order to completely eradicate a tumor, an antiangiogenic drug would have to be given in combination with a modality that kills cancer cells, such as an oncolytic virus.
Vascular endothelial growth factor (VEGF) is a key component in tumor angiogenesis and is overexpressed in many human tumors. It has numerous effects on tumor vasculature such as increased vasodilation and permeabilization, and inhibitors of VEGF, such as Avastin®, sorafenib, and sunitinib, appear to “normalize” tumor vasculature, potentially enhancing localization of systemic oncolytic virus. ICP34.5 null oHSV infectivity and cytotoxicity were diminished under hypoxic conditions (when the cells are deprived of oxygen) in several glioblastoma xenolines, which are cell lines maintained by xenograft passage.Citation69 Normalization of the blood vessels by antiangiogenic agents may reduce hypoxia within the tumor microenvironment and potentially improve oHSV replication. However, other studies have shown improved oHSV replication in hypoxic conditions.Citation70–Citation73 Bevacizumab (Avastin®), a monoclonal antibody against VEGF A, had no effect on the spread or replication of oHSV in vitro. However, in vivo, in several studies using different xenograft models,Citation74,Citation75 groups of mice receiving the dual therapy of both oHSV and Avastin® had tumors that were significantly smaller than tumors from either treatment alone. Results from these studies indicated that Avastin® improved replication and spread of the oHSV within the xenograft microenvironment. Although cytotoxic in vitro, in some xenograft models rRp450 had only mild antitumor effects.Citation76 The host inflammatory response to rRp450 therapy was found to induce an acute neutrophil infiltrate, a relative decrease of intratumoral macrophages, and a myeloid cell-dependent upregulation of host-derived VEGF. Bevacizumab and r84 (which selectively inhibit binding to VEGF receptor 2 but not VEGF receptor 1) enhanced the antitumor effects of rRp450 therapy, in part due to decreased angiogenesis. However, although neither bevacizumab nor r84 increased virus production or affected neutrophil infiltration, both partially mitigated virus-induced depletion of macrophages. Therefore, the enhancement in efficacy with the combination of oHSV therapy and anti-VEGF antibodies appears to be in part due to modulation of host inflammatory reaction to virus.
Vinblastine, a microtubule disrupting agent that has been shown to inhibit angiogenesis in humansCitation77 and, in combination with the oHSV NV1042, showed increased anti-tumor and antiangiogenic effects in vivo in prostate cancer models,Citation78 provides further evidence that the combination of an antiangiogenic agent and an oncolytic virus may have clinical benefit. However, to the best of our knowledge, there are no preclinical published studies of oHSV in combination with small molecule VEGF receptor inhibitors such as sorafenib or sunitinib.
HSV DNA replication occurs in discrete compartments in the nucleus that assemble as prereplicative sites with viral DNA and the HSV DNA binding protein ICP8. HSV DNA polymerase and cellular factors are then recruited to these compartments for use in viral replication. The DNA damage and repair pathways repair the damage to the cancer cell DNA caused by treatment with DNA-damaging drugs such as temozolomide (TMZ). However, in the presence of oHSV infection, key components of these pathways are sequestered into discrete compartments for use in viral replication, hence are not available to repair the damage caused by drugs. Thus, the damage, in terms of number of cancer cells killed by a specific amount of drug, is greater in the presence of oHSV.Citation37
Cellular kinases play a key role in the regulation of signaling events that govern multiple pathways affecting growth, proliferation, migration, and angiogenesis. These include PI3K (phosphatidylinositide 3-kinases)-Akt-mTOR and mitogen-activated protein kinases pathways, which are often mutated in cancer cells to support unchecked cellular replication. Inhibition of these pathways could potentially reduce tumor growth, and this is reflected in the intensive drug development looking for PI3K-Akt-mTOR and mitogen-activated protein kinases inhibitors. For example, 80% of glioblastomas are having genetic alterations in the PI3K-Akt-mTOR pathways and there are at least 10 different inhibitors in development.Citation79 However, due to the high level of redundancy and cross regulatory feedback loops, monotherapy may be unlikely to have significant clinical efficacy;Citation80 for example, rapamycin only reduces mTOR activity for 12 hours before another kinase substitutes and reengages the mTOR network.Citation81 Furthermore, such inhibitors are likely to be cytostatic: they will stop the cancer cells from growing or dividing but will not eradicate them.
The PI3K-Akt-mTOR pathway is also important in viral replication (for a full review see Terada et alCitation61 and Buchkovich et alCitation82). Upon infection, viruses frequently activate this pathway to benefit from the survival signaling associated with Akt activation. One of the downstream effectors of activated Akt is the mTOR kinase, a component of the mTOR complexes (mTORC) 1 and 2. Activated mTORC1 is crucial for the maintenance of cap-dependent translation which is required by most mammalian DNA viruses and many RNA viruses. mTORC2 is less well understood, but is thought to have roles in Akt phosphorylation and the organization of the actin cytoskeleton. It would therefore seem reasonable to assume that inhibitors that block the function of mTOR or PI3K would not only block translation of cellular proteins but would drastically reduce the ability of viruses to replicate by virtue of stopping their cap-dependent translation. Theoretically, PI3K and mTOR inhibitors would be antagonistic if used in combination with oncolytic viruses. The literature, however, reveals diverse results that vary depending on the specific virus, the specific inhibitor, and the status of the cells used.
Breitbach et alCitation83 found that compounds such as rapamycin, which blocks the activation of mTOR, and PD098059, which blocks the activation of MAP (mitogen-activated protein) kinase, did not affect the ability of oHSV R3616 to replicate in pancreatic tumor cells. Treatment with the inhibitor LY294002, which inhibits the PI3K pathway, prevented the replication of R3616. Similarly, synergy was not observed between LY294002 and the ICP34.5 null oHSV, but was observed with oHSV mutants with a Us3 mutation.Citation84 The gene product of Us3 protects virus-infected cells from apoptosis; a cellular pathway that is often dysfunctional in tumors. Thus, Us3 mutants, whose replication would be inhibited by apoptosis in normal cells, would be selective for tumor cells, and the combination treatment of LY294002 and Us3-null oHSV is synergistic due to enhanced apoptosis in the combination treated cells.Citation85
Compounds that affect the cell cycle
Strong synergy between oHSV and trichostatin A (an HDAC inhibitor) was observed in a wide range of cancer and proliferating endothelial cell lines but not in normal prostate or quiescent epithelial cells.Citation86 Unlike other HDIs, the synergy was seen regardless of the dosing sequence of the oHSV (G47Δ) or trichostatin A. The synergy was attributed to reduced cyclin D1 expression in cells that normally have a high level of cyclin D (ie, cancer cells). The combination also inhibited secretion of the angiogenic factor VEGF, which correlated with the decreased vascularity within the tumor in vivo.
Another combination that appears to affect the cell cycle occurs between the oHSV G207 and paclitaxel. Paclitaxel is an approved cancer therapy that stabilizes microtubules and, as a result, interferes with the normal breakdown of microtubules during cell division. In the presence of paclitaxel, chromosomes are unable to achieve metaphase spindle configuration. This inability to form the correct formation blocks the progression of mitosis which in turn triggers apoptosis or the cell to revert to the G phase of the cell cycle without dividing. Despite the G207/paclitaxel combination being synergistic, oncolysis or viral replication was not increased.Citation87 The authors concluded that they differentially affected cell cycle progression, either by the cells arresting in G1 (virus-mediated) or mitosis (paclitaxel-mediated), a combination that served to increase apoptosis further. Paclitaxel also showed synergy with other oHSV, HF10, and G47Δ, both in vitro and in vivo.Citation88,Citation89 The oHSV HF10 has been studied alone and in combination with paclitaxel in colon cancer models.Citation88 In vivo, the combination of HF10 and paclitaxel prolonged survival of mice bearing carcinomatous dissemination of CT26 tumors compared with the control groups. G47Δ also synergized with paclitaxel and the closely related docetaxel to enhance the in vitro killing of LNCap and DU145 prostate cancer cells.Citation89 Docetaxel-induced accumulation of the phosphospecific mitotic markers op18/stathmin or histone H3 was significantly reduced by G47Δ, and this correlated with enhanced apoptosis and required active virus replication. Another microtubule inhibitor, vincristine, was also shown to be synergistic with oHSV in rhabdomyosarcoma xenografts.Citation90
Cheema et alCitation91 reported synergy with etoposide, an inhibitor of topoisomerase II, and oHSV G47Δ in glioma stem cell xenografts. Gutermann et alCitation45 found synergy with SN38 (the active metabolite of irinotecan, a topoisomerase I inhibitor) and NV1020 in a panel of human colon carcinoma cell lines in vitro. Synergy with irinotecan and MGH2 (an oHSV with UL39 and -γ34.5 deletions) was also reported in glioma, both in vitro and in vivo.Citation59
Other compounds where synergy and/or enhancement is seen but the mechanism is unclear
Although not using an oHSV, Heo et alCitation92 reported on the first clinical signs of positive interactions between oncolytic virotherapy and standard of care drugs with JX-594 (an oncolytic pox virus) and sorafenib, a small molecule inhibitor of the signaling oncoprotein B-raf and VEGF receptor, which is licensed as a treatment for hepatocellular carcinoma. The authors reported that a number of patients treated with JX-594 and then sorafenib up to 8 weeks later had objective tumor responses (ie, tumor shrinkage) compared to zero in 15 untreated patients matched for age, stage, and sex. Furthermore, they also reported a complete cure in one patient treated with sunitinib, another inhibitor similar to sorafenib, 8 weeks after JX-594 treatment. As the virus is likely to be cleared from the patient by 8 weeks, the mechanism by which the oncolytic virus can sensitize tumors to these inhibitors is unclear. Interestingly, the patients who have the best responses to sorafenib are those patients who have hepatitis C related hepatocellular carcinoma,Citation93 suggesting that there may be a therapeutic class effect where viruses sensitize tumors to VEGF receptor inhibitors.
Erlotinib, an epidermal growth factor receptor inhibitor, combined additively with two oHSV, G207, and hrR3 in order to enhance cytotoxicity in vitro in human malignant peripheral nerve sheath tumor cells often associated with Ras/epidermal growth factor receptor hyperactivation; however, this effect did not translate into an in vivo malignant peripheral nerve sheath tumor xenograft model.Citation94 Thalidomide, which is now approved for use in multiple myeloma patients, was found to have significant benefit in reducing tumor burden in combination with OncdSyn (an NV1020-like oHSV) than either OncdSyn or thalidomide alone in a murine breast cancer model,Citation95 though the mechanism is unclear.
Conclusion
Oncolytic viruses are a new and emerging treatment for cancer. As they become an established therapy, much attention will have to be paid to the interaction between current standard of care drugs and oncolytic viruses. So far, the signs are encouraging; not only can oHSV be given alongside other cancer treatments, but can actually result in an enhancement of efficacy in reducing tumor burden and improving survival. The majority of virus–drug combinations listed in – show synergistic, enhanced, or additive effects, but this may in part reflect the fact that antagonistic combinations might not be submitted for publication. Recently, Kulu et alCitation96 reported on the inhibition of HSV oncolysis in colon and pancreatic cancer cell lines in vitro when combined with 5-FU, irinotecan, or methotrexate. Their studies showed that replication of both ICP6 and/or ICP34.5 deleted oHSV was significantly reduced in HT29 and SW620 (colon) and Capan-2 (pancreatic) cell lines. Others have reported additive/synergistic interactions (with respect to cell killing) between 5-FU, irinotecan, and methotrexate () with oHSV in diverse cell lines, including both colon and pancreatic lines. It is conceivable that the drugs can inhibit virus replication but the combined effects of virus and drug act in concert to enhance cell death, and seemingly conflicting results serve to illustrate our poor understanding of such interactions.
Furthermore, the sequence in which the drug and oHSV are given may impact on cell killing. For example, gemcitabine and HDIs such as valproic acid are synergistic when given as a pretreatment to the virus, thus sensitizing the tumor to virus, whereas sorafenib appeared to work better given after oncolytic virus; thus the virus is acting as the sensitizer. Similarly, when oHSV rRp450 was given before Avastin® (bevacizumab) there was a significantly prolonged survival compared to the same combination in reverse order.Citation74
Many of the published combination studies examined the effects of combinations in vitro. These identify combinations that enhance cancer cell cytotoxicity. However, many of the interactions between oHSV and drugs either affect the tumor or host biology, and these interactions will only be seen in vivo. The immune system is a key player in the efficacy of any combination treatment; it appears that initial suppressing of the innate immune response in order to allow the virus to undergo replication, then an upregulation of the immune system to clear the virus and tumor, would be a rational strategy in terms of reducing tumor burdens.
The use of patient-derived tumor xenografts, where primary human tumors are transplanted into immune deficient mice within hours after the sample is collected, are increasingly being used to predict the effectiveness of chemotherapeutic drugs in patients. To our knowledge, such models have not been reported for testing combinations of oncolytic HSV together with chemotherapy or targeted drugs, but are likely to be valuable and should provide data that will improve decision making and accelerate development programs for virus/drug combinations.
As preclinical studies progress into the clinical setting, major progress in the understanding of oHSV in combination with other treatments is likely to occur. Early clinical trials usually involve patients who have already exhausted all the available standard treatment options, and even later Phase III trials will often compare standard of care versus standard of care plus oHSV. Such studies should help confirm preclinical findings on useful virus/drug combinations and hopefully bring benefit to cancer sufferers.
Disclosure
Lynne Braidwood, Joe Conner, and Alex Graham are employees of Virttu Biologics Ltd. The authors report no other conflicts of interest in this work.
References
- KellyERussellSJHistory of oncolytic viruses: genesis to genetic engineeringMol Ther200715465165917299401
- MartuzaRLMalickAMarkertJMRuffnerKLCoenDMExperimental therapy of human glioma by means of a genetically engineered virus mutantScience199125250078548561851332
- Campadelli-FiumeGDe GiovanniCGattaVNanniPLolliniPLMenottiLRethinking herpes simplex virus: the way to oncolytic agentsRev Med Virol201121421322621626603
- MinetaTRabkinSDMartuzaRLTreatment of malignant gliomas using ganciclovir-hypersensitive, ribonucleotide reductase-deficient herpes simplex viral mutantCancer Res19945415396339668033122
- CassadyKAParkerJNHerpesvirus vectors for therapy of brain tumorsOpen Virol J2010410310820811578
- ManservigiRArgnaniRMarconiPHSV Recombinant Vectors for Gene TherapyOpen Virol J2010412315620835362
- VargheseSRabkinSDOncolytic herpes simplex virus vectors for cancer virotherapyCancer Gene Ther200291296797812522436
- TodaMMartuzaRLKojimaHRabkinSDIn situ cancer vaccination: an IL-12 defective vector/replication-competent herpes simplex virus combination induces local and systemic antitumor activityJ Immunol19981609445744649574551
- VargheseSRabkinSDLiuRNielsenPGIpeTMartuzaRLEnhanced therapeutic efficacy of IL-12, but not GM-CSF, expressing oncolytic herpes simplex virus for transgenic mouse derived prostate cancersCancer Gene Ther200613325326516179929
- ParkerJNMelethSHughesKBGillespieGYWhitleyRJMarkertJMEnhanced inhibition of syngeneic murine tumors by combinatorial therapy with genetically engineered HSV-1 expressing CCL2 and IL-12Cancer Gene Ther200512435936815678154
- CarewJFKoobyDAHaltermanMWKimSHFederoffHJFongYA novel approach to cancer therapy using an oncolytic herpes virus to package amplicons containing cytokine genesMol Ther20014325025611545616
- TodoTMartuzaRLDallmanMJRabkinSDIn situ expression of soluble B7-1 in the context of oncolytic herpes simplex virus induces potent antitumor immunityCancer Res200161115316111196154
- HuJCCoffinRSDavisCJA phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factorClin Cancer Res200612226737674717121894
- MalhotraSKimTZagerJUse of an oncolytic virus secreting GM-CSF as combined oncolytic and immunotherapy for treatment of colorectal and hepatic adenocarcinomasSurgery2007141452052917383529
- KaufmanHLBinesSDOPTIM trial: a Phase III trial of an oncolytic herpes virus encoding GM-CSF for unresectable stage III or IV melanomaFuture Oncol20106694194920528232
- KaufmanHLKimDWDeRaffeleGMitchamJCoffinRSKim-SchulzeSLocal and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanomaAnn Surg Oncol201017371873019915919
- NakamuraHMullenJTChandrasekharSPawlikTMYoonSSTanabeKKMultimodality therapy with a replication-conditional herpes simplex virus 1 mutant that expresses yeast cytosine deaminase for intratumoral conversion of 5-fluorocytosine to 5-fluorouracilCancer Res200161145447545211454690
- ChaseMChungRYChioccaEAAn oncolytic viral mutant that delivers the CYP2B1 transgene and augments cyclophosphamide chemotherapyNat Biotechnol19981654444489592392
- BraidwoodLDunnPDHardySEvansTRBrownSMAntitumor activity of a selectively replication competent herpes simplex virus (HSV) with enzyme prodrug therapyAnticancer Res20092962159216619528476
- SheridanCAmgen announces oncolytic virus shrinks tumorsNat Biotechnol201331647147223752412
- FisherRPusztaiLSwantonCCancer heterogeneity: implications for targeted therapeuticsBr J Cancer2013108347948523299535
- PaludanSRBowieAGHoranKAFitzgeraldKARecognition of herpesviruses by the innate immune systemNat Rev Immunol201111214315421267015
- LilleyCEChaurushiyaMSBoutellCEverettRDWeitzmanMDThe intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0PLoS Pathog201176e100208421698222
- ZachosGKoffaMPrestonCMClementsJBConnerJHerpes simplex virus type 1 blocks the apoptotic host cell defense mechanisms that target Bcl-2 and manipulates activation of p38 mitogen-activated protein kinase to improve viral replicationJ Virol20017562710272811222695
- HeoJReidTRuoLRandomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancerNat Med201319332933623396206
- ChouTCTalalayPQuantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitorsAdv Enzyme Regul19842227556382953
- ToyoizumiTMickRAbbasAEKangEHKaiserLRMolnar-KimberKLCombined therapy with chemotherapeutic agents and herpes simplex virus type 1 ICP34.5 mutant (HSV-1716) in human non-small cell lung cancerHum Gene Ther199910183013302910609661
- ChouTCTalalayPGeneralized equations for the analysis of inhibitions of Michaelis-Menten and higher-order kinetic systems with two or more mutually exclusive and nonexclusive inhibitorsEur J Biochem198111512072167227366
- ChouTCTheoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studiesPharmacol Rev200658362168116968952
- NaitoSObayashiSSumiTEnhancement of antitumor activity of herpes simplex virus gamma(1)34.5-deficient mutant for oral squamous cell carcinoma cells by hexamethylene bisacetamideCancer Gene Ther200613878079116645620
- EgorinMJSigmanLMVan EchoDAForrestAWhitacreMYAisnerJPhase I clinical and pharmacokinetic study of hexamethylene bisacetamide (NSC 95580) administered as a five-day continuous infusionCancer Res19874726176233791246
- WoodLWShillitoeEJEffect of a caspase inhibitor, zVADfmk, on the inhibition of breast cancer cells by herpes simplex virus type 1Cancer Gene Ther2011181068569421701533
- AubertMPomeranzLEBlahoJAHerpes simplex virus blocks apoptosis by precluding mitochondrial cytochrome c release independent of caspase activation in infected human epithelial cellsApoptosis2007121193517080326
- StanzialeSFPetrowskyHAdusumilliPSBen-PoratLGonenMFongYInfection with oncolytic herpes simplex virus-1 induces apoptosis in neighboring human cancer cells: a potential target to increase anticancer activityClin Cancer Res20041093225323215131064
- EisenbergDPCarpenterSGAdusumilliPSHyperthermia potentiates oncolytic herpes viral killing of pancreatic cancer through a heat shock protein pathwaySurgery2010148232533420633729
- BennettJJAdusumilliPPetrowskyHUp-regulation of GADD34 mediates the synergistic anticancer activity of mitomycin C and a gamma134.5 deleted oncolytic herpes virus (G207)FASEB J20041891001100315059970
- KanaiRRabkinSDYipSOncolytic virus-mediated manipulation of DNA damage responses: synergy with chemotherapy in killing glioblastoma stem cellsJ Natl Cancer Inst20121041425522173583
- HadjipanayisCGFellows-MayleWDelucaNATherapeutic efficacy of a herpes simplex virus with radiation or temozolomide for intracranial glioblastoma after convection-enhanced deliveryMol Ther200816111783178818728637
- EisenbergDPAdusumilliPSHendershottKJ5-fluorouracil and gemcitabine potentiate the efficacy of oncolytic herpes viral gene therapy in the treatment of pancreatic cancerJ Gastrointest Surg20059810681077 discussion 1077–107916269377
- RoizmanBThe function of herpes simplex virus genes: a primer for genetic engineering of novel vectorsProc Natl Acad Sci U S A1996932111307113128876131
- AdusumilliPSChanMKChunYSCisplatin-induced GADD34 upregulation potentiates oncolytic viral therapy in the treatment of malignant pleural mesotheliomaCancer Biol Ther200651485316294031
- PetrowskyHRobertsGDKoobyDAFunctional interaction between fluorodeoxyuridine-induced cellular alterations and replication of a ribonucleotide reductase-negative herpes simplex virusJ Virol200175157050705811435585
- PasserBJCheemaTZhouBIdentification of the ENT1 antagonists dipyridamole and dilazep as amplifiers of oncolytic herpes simplex virus-1 replicationCancer Res201070103890389520424118
- NakanoKTodoTZhaoGEnhanced efficacy of conditionally replicating herpes simplex virus (G207) combined with 5-fluorouracil and surgical resection in peritoneal cancer dissemination modelsJ Gene Med20057563864815754306
- GutermannAMayerEvon Dehn-RothfelserKEfficacy of oncolytic herpesvirus NV1020 can be enhanced by combination with chemotherapeutics in colon carcinoma cellsHum Gene Ther200617121241125317117895
- FuXTaoLRiveraAZhangXRapamycin enhances the activity of oncolytic herpes simplex virus against tumor cells that are resistant to virus replicationInt J Cancer201112961503151021128236
- StanfordMMBarrettJWNazarianSHWerdenSMcFaddenGOncolytic virotherapy synergism with signaling inhibitors: Rapamycin increases myxoma virus tropism for human tumor cellsJ Virol20078131251126017108021
- AlainTLunXMartineauYVesicular stomatitis virus oncolysis is potentiated by impairing mTORC1-dependent type I IFN productionProc Natl Acad Sci U S A201010741576158120080710
- MishraRMiyamotoMYoshiokaTAdenovirus-mediated eukaryotic initiation factor 4E binding protein-1 in combination with rapamycin inhibits tumor growth of pancreatic ductal adenocarcinoma in vivoInt J Oncol20093451231124019360336
- MoormanNJShenkTRapamycin-resistant mTORC1 kinase activity is required for herpesvirus replicationJ Virol201084105260526920181700
- HalfordWPMaenderJLGebhardtBMRe-evaluating the role of natural killer cells in innate resistance to herpes simplex virus type 1Virol J200525616022737
- Alvarez-BreckenridgeCAYuJKaurBCaligiuriMAChioccaEADeciphering the Multifaceted Relationship between Oncolytic Viruses and Natural Killer CellsAdv Virol2012201270283922312364
- FulciGBreymannLGianniDCyclophosphamide enhances glioma virotherapy by inhibiting innate immune responsesProc Natl Acad Sci U S A200610334128731287816908838
- CurrierMAGillespieRASawtellNMEfficacy and safety of the oncolytic herpes simplex virus rRp450 alone and combined with cyclophosphamideMol Ther200816587988518388918
- WakimotoHFulciGTyminskiEChioccaEAAltered expression of antiviral cytokine mRNAs associated with cyclophosphamide’s enhancement of viral oncolysisGene Ther200411221422314712306
- FriedmanATianJPFulciGChioccaEAWangJGlioma virotherapy: effects of innate immune suppression and increased viral replication capacityCancer Res20066642314231916489036
- FulciGDmitrievaNGianniDDepletion of peripheral macrophages and brain microglia increases brain tumor titers of oncolytic virusesCancer Res200767199398940617909049
- PawlikTMNakamuraHMullenJTProdrug bioactivation and oncolysis of diffuse liver metastases by a herpes simplex virus 1 mutant that expresses the CYP2B1 transgeneCancer20029551171118112209705
- TyminskiELeroySTeradaKBrain tumor oncolysis with replication-conditional herpes simplex virus type 1 expressing the prodrug-activating genes, CYP2B1 and secreted human intestinal carboxylesterase, in combination with cyclophosphamide and irinotecanCancer Res200565156850685716061668
- Bazan-PeregrinoMArvanitisCDRifaiBSeymourLWCoussiosCCUltrasound-induced cavitation enhances the delivery and therapeutic efficacy of an oncolytic virus in an in vitro modelJ Control Release2012157223524221982902
- TeradaKWakimotoHTyminskiEChioccaEASaekiYDevelopment of a rapid method to generate multiple oncolytic HSV vectors and their in vivo evaluation using syngeneic mouse tumor modelsGene Ther200613870571416421599
- MillerCGFraserNWRequirement of an integrated immune response for successful neuroattenuated HSV-1 therapy in an intracranial metastatic melanoma modelMol Ther20037674174712788647
- BenenciaFCourregesMCConejo-GarciaJROncolytic HSV exerts direct antiangiogenic activity in ovarian carcinomaHum Gene Ther200516676577815960607
- EsakiSGoshimaFKimuraHMurakamiSNishiyamaYEnhanced antitumoral activity of oncolytic herpes simplex virus with gemcitabine using colorectal tumor modelsInt J Cancer201313271592160122949155
- SimpsonGRHorvathAAnnelsNECombination of a fusogenic glycoprotein, pro-drug activation and oncolytic HSV as an intravesical therapy for superficial bladder cancerBr J Cancer2012106349650722240799
- WatanabeDGoshimaFMoriITamadaYMatsumotoYNishiyamaYOncolytic virotherapy for malignant melanoma with herpes simplex virus type 1 mutant HF10J Dermatol Sci200850318519618226503
- HodiFSO’DaySJMcDermottDFImproved survival with ipilimumab in patients with metastatic melanomaN Engl J Med2010363871172320525992
- HodiFSObleDADrappatzJCTLA-4 blockade with ipilimumab induces significant clinical benefit in a female with melanoma metastases to the CNSNat Clin Pract Oncol20085955756118665147
- FriedmanGKHaasMCKellyVMMarkertJMGillespieGYCassadyKAHypoxia Moderates gamma(1)34.5-Deleted Herpes Simplex Virus Oncolytic Activity in Human Glioma Xenoline Primary CulturesTransl Oncol20125320020722741039
- AghiMKLiuTCRabkinSMartuzaRLHypoxia enhances the replication of oncolytic herpes simplex virusMol Ther2009171515618957963
- AmgenIpilimumab With or Without Talimogene Laherparepvec in Unresected Melanoma Available from: http://clinicaltrials.gov/show/NCT01740297. NLM identifier: NCT01740297Accessed September 26, 2013
- FasulloMBurchADBrittonAHypoxia enhances the replication of oncolytic herpes simplex virus in p53-breast cancer cellsCell Cycle20098142194219719502787
- SgubinDWakimotoHKanaiRRabkinSDMartuzaRLOncolytic herpes simplex virus counteracts the hypoxia-induced modulation of glioblastoma stem-like cellsStem Cells Transl Med20121432233223197811
- EshunFKCurrierMAGillespieRAFitzpatrickJLBairdWHCripeTPVEGF blockade decreases the tumor uptake of systemic oncolytic herpes virus but enhances therapeutic efficacy when given after virotherapyGene Ther201017792292920508601
- DeguchiTShikanoTKasuyaHCombination of the tumor angiogenesis inhibitor bevacizumab and intratumoral oncolytic herpes virus injections as a treatment strategy for human gastric cancersHepatogastroenterology2012591181844185022172413
- CurrierMAEshunFKShollAVEGF Blockade Enables Oncolytic Cancer Virotherapy in Part by Modulating Intratumoral Myeloid CellsMol Ther20132151014102323481323
- AlbertssonPLennernasBNorrbyKDose effects of continuous vinblastine chemotherapy on mammalian angiogenesis mediated by VEGF-AActa Oncol200847229330018210302
- PasserBJCheemaTWuSWuCLRabkinSDMartuzaRLCombination of vinblastine and oncolytic herpes simplex virus vector expressing IL-12 therapy increases antitumor and antiangiogenic effects in prostate cancer modelsCancer Gene Ther2013201172423138870
- Cancer Genome Atlas Research NComprehensive genomic characterization defines human glioblastoma genes and core pathwaysNature200845572161061106818772890
- EngelmanJATargeting PI3K signalling in cancer: opportunities, challenges and limitationsNat Rev Cancer20099855056219629070
- KudchodkarSBYuYMaguireTGAlwineJCHuman cytomegalovirus infection induces rapamycin-insensitive phosphorylation of downstream effectors of mTOR kinaseJ Virol20047820110301103915452223
- BuchkovichNJYuYZampieriCAAlwineJCThe TORrid affairs of viruses: effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathwayNat Rev Microbiol20086426627518311165
- BreitbachCJReidTBurkeJBellJCKirnDHNavigating the clinical development landscape for oncolytic viruses and other cancer therapeutics: no shortcuts on the road to approvalCytokine Growth Factor Rev2010212–3858920472490
- LiuTCWakimotoHMartuzaRLRabkinSDHerpes simplex virus Us3(-) mutant as oncolytic strategy and synergizes with phosphatidylinositol 3-kinase-Akt targeting molecular therapeuticsClin Cancer Res200713195897590217908985
- KanaiRWakimotoHMartuzaRLRabkinSDA novel oncolytic herpes simplex virus that synergizes with phosphoinositide 3-kinase/Akt pathway inhibitors to target glioblastoma stem cellsClin Cancer Res201117113686369621505062
- LiuTCCastelo-BrancoPRabkinSDMartuzaRLTrichostatin A and oncolytic HSV combination therapy shows enhanced antitumoral and antiangiogenic effectsMol Ther20081661041104718388912
- LinSFGaoSPPriceDLSynergy of a herpes oncolytic virus and paclitaxel for anaplastic thyroid cancerClin Cancer Res20081451519152818316577
- ShimoyamaSGoshimaFTeshigaharaOEnhanced efficacy of herpes simplex virus mutant HF10 combined with paclitaxel in peritoneal cancer dissemination modelsHepatogastroenterology200754761038104217629034
- PasserBJCastelo-BrancoPBuhrmanJSVargheseSRabkinSDMartuzaRLOncolytic herpes simplex virus vectors and taxanes synergize to promote killing of prostate cancer cellsCancer Gene Ther200916755156019197321
- CinatlJJrCinatlJMichaelisMPotent oncolytic activity of multi-mutated herpes simplex virus G207 in combination with vincristine against human rhabdomyosarcomaCancer Res20036371508151412670897
- CheemaTAKanaiRKimGWEnhanced antitumor efficacy of low-dose Etoposide with oncolytic herpes simplex virus in human glioblastoma stem cell xenograftsClin Cancer Res201117237383739321976549
- HeoJBreitbachCJMoonASequential therapy with JX-594, a targeted oncolytic poxvirus, followed by sorafenib in hepatocellular carcinoma: preclinical and clinical demonstration of combination efficacyMol Ther20111961170117921427706
- CabreraRLimayeARHornePThe anti-viral effect of sorafenib in hepatitis C-related hepatocellular carcinomaAliment Pharmacol Ther2013371919723094860
- MahllerYYVaikunthSSCurrierMAOncolytic HSV and erlotinib inhibit tumor growth and angiogenesis in a novel malignant peripheral nerve sheath tumor xenograft modelMol Ther200715227928617235305
- DiasJDHemminkiODiaconuITargeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4Gene Ther2012191098899822071969
- KuluYKawasakiHDonahueJMConcurrent chemotherapy inhibits herpes simplex virus-1 replication and oncolysisCancer Gene Ther201320213314023348635
- LiuBLRobinsonMHanZQICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour propertiesGene Ther200310429230312595888
- HarringtonKJHingoraniMTanayMAPhase I/II study of oncolytic HSV GM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neckClin Cancer Res201016154005401520670951
- KemenyNBrownKCoveyAPhase I, open-label, dose-escalating study of a genetically engineered herpes simplex virus, NV1020, in subjects with metastatic colorectal carcinoma to the liverHum Gene Ther200617121214122417107303
- KellyKJWongJFongYHerpes simplex virus NV1020 as a novel and promising therapy for hepatic malignancyExpert Opin Investig Drugs200817711051113
- GeevargheseSKGellerDAde HaanHAPhase I/II study of oncolytic herpes simplex virus NV1020 in patients with extensively pretreated refractory colorectal cancer metastatic to the liverHum Gene Ther20102191119112820486770
- SzeDYIagaruAHGambhirSSDe HaanHAReidTRResponse to intra-arterial oncolytic virotherapy with the herpes virus NV1020 evaluated by [18F]fluorodeoxyglucose positron emission tomography and computed tomographyHum Gene Ther2012231919721895536
- YazakiTManzHJRabkinSDMartuzaRLTreatment of human malignant meningiomas by G207, a replication-competent multimutated herpes simplex virus 1Cancer Res19955521475247567585498
- MinetaTRabkinSDYazakiTHunterWDMartuzaRLAttenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomasNat Med1995199389437585221
- HunterWDMartuzaRLFeigenbaumFAttenuated, replication-competent herpes simplex virus type 1 mutant G207: safety evaluation of intracerebral injection in nonhuman primatesJ Virol19997386319632610400723
- TodoTRabkinSDMartuzaRLEvaluation of ganciclovir-mediated enhancement of the antitumoral effect in oncolytic, multimutated herpes simplex virus type 1 (G207) therapy of brain tumorsCancer Gene Ther20007693994610880026
- MarkertJMMedlockMDRabkinSDConditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trialGene Ther200071086787410845725
- MarkertJMLiechtyPGWangWPhase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBMMol Ther200917119920718957964
- AghiMKChioccaEAPhase ib trial of oncolytic herpes virus G207 shows safety of multiple injections and documents viral replicationMol Ther20091718919116635
- HarrowSPapanastassiouVHarlandJHSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survivalGene Ther200411221648165815334111
- PapanastassiouVRamplingRFraserMThe potential for efficacy of the modified (ICP 34.5(−)) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: a proof of principle studyGene Ther20029639840611960316
- RamplingRCruickshankGPapanastassiouVToxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant gliomaGene Ther200071085986610845724
- McKieEAMacLeanARLewisADSelective in vitro replication of herpes simplex virus type 1 (HSV-1) ICP34.5 null mutants in primary human CNS tumours – evaluation of a potentially effective clinical therapyBr J Cancer19967457457528795577
- MaceATHarrowSJGanlyIBrownSMCytotoxic effects of the oncolytic herpes simplex virus HSV1716 alone and in combination with cisplatin in head and neck squamous cell carcinomaActa Otolaryngol2007127888088717763002
- NakaoAKasuyaHSahinTTA phase I dose-escalation clinical trial of intraoperative direct intratumoral injection of HF10 oncolytic virus in non-resectable patients with advanced pancreatic cancerCancer Gene Ther201118316717521102422
- NakaoATakedaSShimoyamaSClinical experiment of mutant herpes simplex virus HF10 therapy for cancerCurr Cancer Drug Targets20077216917417346108
- KimataHImaiTKikumoriTPilot study of oncolytic viral therapy using mutant herpes simplex virus (HF10) against recurrent metastatic breast cancerAnn Surg Oncol20061381078108416865590
- NakaoAKimataHImaiTIntratumoral injection of herpes simplex virus HF10 in recurrent breast cancerAnn Oncol200415698898915151960
- KohnoSILuoCNawaAOncolytic virotherapy with an HSV amplicon vector expressing granulocyte-macrophage colony-stimulating factor using the replication-competent HSV type 1 mutant HF10 as a helper virusCancer Gene Ther2007141191892617693992
- KohnoSLuoCGoshimaFNishiyamaYSataTOnoYHerpes simplex virus type 1 mutant HF10 oncolytic viral therapy for bladder cancerUrology20056651116112116286150
- FujimotoYMizunoTSugiuraSIntratumoral injection of herpes simplex virus HF10 in recurrent head and neck squamous cell carcinomaActa Otolaryngol2006126101115111716923721
- SahinTTKasuyaHNomuraNImpact of novel oncolytic virus HF10 on cellular components of the tumor microenviroment in patients with recurrent breast cancerCancer Gene Ther201219422923722193629
- ChahlaviATodoTMartuzaRLRabkinSDReplication-competent herpes simplex virus vector G207 and cisplatin combination therapy for head and neck squamous cell carcinomaNeoplasia19991216216910933051
- SimpsonGRHanZLiuBWangYCampbellGCoffinRSCombination of a fusogenic glycoprotein, prodrug activation, and oncolytic herpes simplex virus for enhanced local tumor controlCancer Res20066694835484216651439
- WatanabeIKasuyaHNomuraNEffects of tumor selective replication-competent herpes viruses in combination with gemcitabine on pancreatic cancerCancer Chemother Pharmacol200861587588217726607
- MulleradMBochnerBHAdusumilliPSHerpes simplex virus based gene therapy enhances the efficacy of mitomycin C for the treatment of human bladder transitional cell carcinomaJ Urol2005174274174616006968
- NaganoSPerentesJYJainRKBoucherYCancer cell death enhances the penetration and efficacy of oncolytic herpes simplex virus in tumorsCancer Res200868103795380218483263
- AghiMRabkinSMartuzaRLEffect of chemotherapy-induced DNA repair on oncolytic herpes simplex viral replicationJ Natl Cancer Inst2006981385016391370
- Children’s Hospital Medical Center, CincinnatiHSV1716 in Patients With Non-Central Nervous System (Non-CNS) Solid Tumors Available from: http://clinicaltrials.gov/show/NCT00931931. NLM identifier: NCT00931931Accessed September 27, 2013
- Virttu Biologics LimitedIntrapleural Administration of HSV1716 to Treat Patients With Malignant Pleural Mesothelioma. (1716–1712) Available from: http://clinicaltrials.gov/show/NCT01721018. NLM identifier: NCT01721018Accessed September 27, 2013
- KennethKTanabeMDrRp450-Phase I Trial in Liver Metastases and Primary Liver Tumors Available from: http://clinicaltrials.gov/show/NCT01071941. NLM identifier: NCT01071941Accessed September 27, 2013
- KatsuraTIwaiSOtaYShimizuHIkutaKYuraYThe effects of trichostatin A on the oncolytic ability of herpes simplex virus for oral squamous cell carcinoma cellsCancer Gene Ther20091623724518949013
- OtsukiAPatelAKasaiKHistone deacetylase inhibitors augment antitumor efficacy of herpes-based oncolytic virusesMol Ther2008161546155518648350
- IsrayelyanAShannonEJBaghianAKearneyMTKousoulasKGThalidomide suppressed the growth of 4T1 cells into solid tumors in Balb/c mice in a combination therapy with the oncolytic fusogenic HSV-1 OncdSynCancer Chemother Pharmacol2009641201121019308409