
Abstract
The thiopurine S-methyltransferase (TPMT) gene encodes for the TPMT enzyme that plays a crucial role in the metabolism of thiopurine drugs. Genetic polymorphisms in this gene can affect the activity of the TPMT enzyme and have been correlated with variability in response to treatment with thiopurines. Advances in the pharmacogenetics of TPMT allowed the development of dosing recommendations and treatment strategies to optimize and individualize prescribing thiopurine in an attempt to enhance treatment efficacy while minimizing toxicity. The influence of genetic polymorphisms in the TPMT gene on clinical outcome has been well-documented and replicated in many studies. In this review, we provide an overview of the evolution, results, conclusions and recommendations of selected studies that investigated the influence of TPMT pharmacogenetics on thiopurine treatment in acute lymphoblastic leukemia, inflammatory bowel disease and autoimmune disorders. We focus mainly on prospective studies that explored the impact of individualized TPMT-based dosing of thiopurines on clinical response. Together, these studies demonstrate the importance of preemptive TPMT genetic screening and subsequent dose adjustment in mitigating the toxicity associated with thiopurine treatment while maintaining treatment efficacy and favorable long-term outcomes. In addition, we briefly address the cost-effectiveness of this pharmacogenetics approach and its impact on clinical practice as well as the importance of recent breakthrough advances in sequencing and genotyping techniques in refining the TPMT genetic screening process.
Introduction
Thiopurine S-methyltransferase (TPMT) is an important cytoplasmic enzyme that catalyses the rate-limiting step in the metabolism of thiopurine drugs. It is coded by the TPMT gene and exerts its effect via S-adenosyl-L-methionine as the S-methyl donor and S-adenosyl-L-homocysteine as a by-product.Citation1–Citation3 Thiopurine drugs, mainly 6-mercaptopurine (6-MP), and its prodrug azathioprine (AZA), are implicated as anti-metabolite cytotoxic and immunosuppressive agents in the treatment of malignancies such as acute lymphoblastic leukemia (ALL), inflammatory disorders like inflammatory bowel disease (IBD) and many autoimmune disorders, including rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), autoimmune hepatitis (AIH) and generalized eczematous disorders.Citation3–Citation5 However, gastrointestinal disturbances (like nausea and vomiting), rashes, as well as more serious adverse drug reactions (ADRs) like bone marrow toxicity, hepatotoxicity and pancreatitis can lead to discontinuation of therapy in up to one-third of patients;Citation6 these factors limit the use of these drugs.Citation2
AZA is an inactive compound that must be converted into 6-MP via a glutathione-dependent process and both drugs eventually produce 6-thioguanine nucleotides (6-TGNs), a mechanism through which thiopurines exert both their cytotoxic and therapeutic effects.Citation7,Citation8 Numerous studies have demonstrated that the efficacy and toxicity of thiopurine drugs are correlated to the activity of the TPMT enzyme as this enzyme competes with xanthine-oxidase and hypoxanthine-guanine-phosphoribosyltransferase to determine the amount of 6-MP metabolized to 6-TGNs.Citation1,Citation7–Citation10 6-TGNs then either incorporate directly into DNA, which triggers delayed cytotoxicity, or they inhibit intracellular signaling pathways that ultimately promote cell death via apoptosis.Citation11 Furthermore, 6-MP is also metabolized to methyl-thioinosine-monophosphate that provokes an additional cytotoxic effect by inhibiting de novo purine synthesis.Citation12
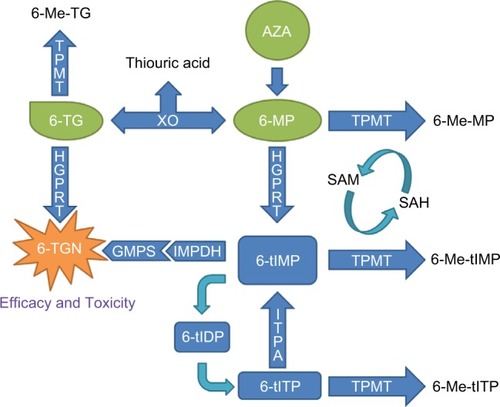
Thioguanine (TG) is also a prodrug that belongs to the thiopurines family (2-amino-6-mercaptopurine) and is also partly metabolized by TPMT. Like AZA and 6-MP, it exerts its effect through mechanisms that involve the production of 6-TGNs, but they differ in the pathways implicated as depicted in . However, due to its more pronounced toxicity profile and lack of additional benefit, its use became somewhat limited to the intensification phase of some anti-leukemia protocols.Citation13
Figure 1 Metabolic pathways involved in the mechanism of action of thiopurines.
Abbreviations: 6-MP, 6-mercaptopurine; 6-Me-MP, 6-methyl-mercaptopurine; 6-Me-TG, 6-methyl-thioguanine; 6-Me-tIMP, 6-methyl-thioinosine-monophosphate; 6-Me-tITP, 6-methyl-thioinosine-triphosphate; 6-TG, thioguanine; 6-TGN, 6-thioguanine nucleotides; 6-tIDP, 6-thio-inosine diphosphate; 6-tIMP, 6-thio-inosine monophosphate; 6-tITP, 6-thio-inosine triphosphate; AZA, azathioprine; GMPS, guanosine monophosphatase synthetase; HGPRT, hypoxanthine guanine phosphoribosyl transferase; IMPDH, inosine monophosphate dehydrogenase; ITPA, inosine triphosphate pyrophosphatase; SAH, S-adenosyl-L-homocysteine; SAM, S-adenosyl-L-methionine; TPMT, thiopurine S-methyltransferase; XO, xanthine oxidase.
TPMT deficiency was first described around 3 decades ago and it is currently well established that homozygous or compound heterozygous carriers of TPMT-deficient alleles have a significantly higher risk of early severe myelosuppression than patients homozygous for the wild-type.Citation14,Citation15 Patients with absent or reduced TPMT activity accumulate high doses of 6-TGNs, resulting in thiopurine-induced myelotoxicity that is characterized by early onset of severe neutropenia when such patients are treated with standard doses of thiopurine drugs. This toxicity is particularly evident in patients carrying two nonfunctional alleles and requires treatment cessation or dose adjustment.Citation1,Citation5,Citation11,Citation16–Citation21 Bone marrow suppression has been linked to higher cumulative incidence of infections, mortality and death.Citation5,Citation22,Citation23 Conversely, myelosuppression can be induced by a number of factors independent of TPMT in individuals taking thiopurines, that is co-medications, viral infections, underlying disease and idiosyncratic reactions,Citation24 as well as genetic polymorphisms in genes other than TPMT encoding enzymes involved in thiopurines metabolism like inosine triphosphate pyrophosphatase (ITPA)Citation25 and Nudix hydrolase 15 (NUD15)Citation26 genes. On the other end of the spectrum, some studies indicated that high TPMT activity has been linked to poor treatment response and that an elevated dose is needed in order to achieve therapeutic effect.Citation27,Citation28
TPMT pharmacogenetics
Enzymatic activity of TPMT can be indirectly assessed through red blood cell enzyme activity assay (phenotype) or can be inferred from the genetic profile of the white blood cells.Citation1,Citation11,Citation29 Genetic polymorphisms in the TPMT gene can affect the enzymatic activity of TPMT and have been studied extensively. To date, over 38 variant alleles have been identified.Citation2,Citation15,Citation30–Citation32 They have been correlated with variability in response to thiopurine drugs, which provides an important example of the clinical importance of pharmacogenetics. Nonetheless, only a few of these polymorphisms are considered in clinical settings that can identify the most frequently reduced-activity TPMT alleles and account for ≥95% of variant TPMT alleles.Citation12,Citation18,Citation31–Citation33 The wild-type allele is defined as TPMT*1. The mutant TPMT*2 allele is defined by the G238C transversion whereas the TPMT*3 family alleles are defined by the G460A and A719G transitions (i.e., TPMT*3A[G460A and A719G], TPMT*3B[G460A] and TPMT*3C[A719G]).Citation12,Citation18,Citation31–Citation33 The prevalence of TPMT variants is much higher among Caucasians (8.1%–10.1%) than Asian populations (2.3%–4.2%)Citation15 and it is well established that TPMT*3A is the most prevalent mutant allele in Caucasians, making up to (85%) of all observed mutant alleles,Citation14,Citation18 while TPMT*3C is the most frequently found allele in African and Southeast Asian populations.Citation14,Citation34
Other than variants in the coding region of TPMT, it is being increasingly acknowledged that variants in the non-coding regions such as the TPMT-promoter and introns can also affect the activity of the TPMT enzyme, possibly by influencing the transcription of its gene.Citation3 One well-studied example of such polymorphisms is the variable number of tandem repeats (VNTR) region, which is a rare microsatellite region in the TPMT gene promoter. Interestingly, studies have shown that the architecture of this region can modulate TPMT transcription and possibly enzyme activity. For example, higher TPMT promoter activity was shown to be associated with a region that contains five or seven GCC repeats rather than six. Thus, studies suggest the use of VNTR architecture as a pharmacogenomic biomarker to refine the TPMT genetic screening process currently used prior to the introduction of thiopurine therapy to enhance the treatment outcome in ALL.Citation3 However, contrary to the results of ALL studies, the expression of the TPMT gene seems to decrease in IBD patients treated with thiopurine drugs and thus VNTR genotype cannot predict the TPMT activity, which seems to be influenced by the treated condition, the protocol used and the concomitant administration of other drugs.Citation3
Across all ethnic groups, ~1 in 300 individuals are homozygous (or compound heterozygous) for a mutant TPMT allele and have very low or absent TPMT activity while around 4%–11% of individuals are heterozygous and are generally considered to have intermediate enzymatic activity.Citation1,Citation18,Citation31 Nonetheless, such genotype-based classification is not always representative of the actual state of enzymatic activity. In literature, conflicting data were obtained by studies that addressed the concordance between the genetic and phenotypic tests, as results ranged from 100% or almost perfect match in the majority of studies to as low as 77% concordance in a few of them.Citation1,Citation3,Citation9–Citation11,Citation19,Citation33,Citation35–Citation38 This discordance was particularly observed in patients with intermediate activity in whom the estimated probability of obtaining matching results varied from 70% to 86%.Citation11,Citation33,Citation36,Citation39
Many factors influence TPMT enzyme activity and eventually affect this genotype–phenotype concordance such as the age and gender of the patient, coadministration of drugs that could potentially interfere with the disease condition or TPMT activity (e.g., methotrexate),Citation40 levels of TPMT cofactor S-adenosyl-methionine,Citation41 recent blood transfusion,Citation42 life span of red blood cells,Citation43 as well as untested rare or novel variants in the coding and regulatory regions of the TPMT gene (e.g., TPMT*38 and the VNTR architecture).Citation3,Citation37,Citation44 Furthermore, interethnic variability in the TPMT enzymatic activity levels have been observed in people of Afro-Caribbean descent having lower activity than Caucasians and South Asians.Citation1,Citation45,Citation46 Taken together, there is always a risk of misclassifying patients if the decision was based on only one of the two abovementioned methods, but it is also unreasonable to perform both tests for all patients. A recent randomized clinical trial (RCT) concluded that there was no advantage or disadvantage of TPMT genotyping compared with phenotyping,Citation16 whereas a more recent study concluded that genotyping was superior to phenotyping and should be considered as the primary choice for pretreatment evaluation of TPMT function.Citation35 Nonetheless, phenotype testing supplemented by genotyping can be a useful strategy in specific circumstances (e.g., after recent blood transfusion and for confirmation of intermediate activity in known high-risk patients).Citation16
Recent advances in the pharmacogenetics of TPMT have allowed for the development of dosing recommendations and treatment strategies to optimize and individualize therapy with thiopurines to obtain maximum treatment benefit with minimal toxicity.Citation47 However, the implementation of pharmacogenetic tests in clinical practice is still somewhat limited due to the lack of robust evidence stemming directly from large-scale RCTs and proving the clinical utility of such strategy.Citation1,Citation16,Citation22 Nonetheless, given the indisputable influence of pharmacogenetics on TPMT activity and the seriousness of thiopurines-induced toxicities, particularly myelosuppression, several regulatory agencies and clinical guidelines such as the US Food and Drug Administration, British National Formulary and Clinical Pharmacogenetics Implementation Consortium (CPIC) recommend pretreatment TPMT activity testing either by genotyping or phenotyping.Citation11,Citation47 In general, most guidelines suggest that the initial dose of thiopurines should be reduced to 10% of the standard dose when administered to homozygous carriers of TPMT-deficient alleles, as well as a reduction in administration frequency. For heterozygous patients, the recommendation differs slightly depending on the type of thiopurine used, as CPIC guidelines suggest an initial dose of AZA and 6-MP that is 30%–70% of the standard protocol dose while the recommendation for TG is 30%–50% of that dose.Citation47
As new data is being continuously generated by RCTs and studies about the long-term outcome of previous treatment protocols, the strength of the clinical evidence should be constantly revised and the recommendations of the guidelines should be reevaluated and modified when deemed necessary. In this review, we provide an overview of the evolution, results, conclusions and recommendations of studies that investigated the influence of TPMT pharmacogenetics on clinical response to thiopurines in ALL, IBD and autoimmune disorders.
Acute lymphoblastic leukemia
Childhood ALL is the most frequent pediatric cancer. The survival rate currently exceeds 85% in favorable settings. 6-MP is coadministered with methotrexate as key components in the maintenance therapy for pediatric ALL and their use is associated with significant reduction in disease relapse.Citation33
An early study reported that TPMT activity was significantly higher in blood samples of ALL patients on long-term 6-MP treatment compared with controls.Citation48 They also noted a relationship between low TPMT activity and the risk of developing severe myelosuppression in patients treated with thiopurine drugs, plausibly due to elevated 6-TGN concentrations.Citation48,Citation49 Others reported that higher TPMT activity was linked to an elevated risk of relapse.Citation50 These findings led to the suggestion that genetic screening of TPMT activity could play a role in influencing treatment response to childhood ALL.Citation48,Citation50 Indeed, one study of childhood ALL suggested that prospectively screening for major TPMT coding region polymorphisms followed by selective administration of an initially reduced dose of 6-MP to heterozygous patients and a subsequent gradual increase to a target range of blood cell count allowed these patients to eventually achieve the full drug dose without experiencing any toxicity.Citation51 Many clinical trials have investigated the impact of TPMT gene polymorphisms on treatment outcome, with most of them demonstrating the benefit of preemptive TPMT screening, but results were somewhat inconsistent ().
Table 1 Summary of selected studies that investigated the influence of TPMT pharmacogenetics on thiopurine treatment response in childhood acute lymphoblastic leukemia
The Total Therapy Study XII explored the impact of 6-MP dose reduction from the standard protocol dose to a maximum tolerable dose subsequently to the development of myelosuppression and investigated the association between the maximum tolerable doses and TPMT genotypes in a total of 188 patients. The results showed that TPMT genotype was an important predictor of 6-MP toxicity in ALL patients, as the cumulative incidence of dose reduction or treatment interruption was significantly different across the 3 groups (P<0.001); the results also showed that wild-type patients had the lowest incidence (7%) followed by heterozygous carriers (35%) and homozygous carriers of TPMT-deficient alleles (100%).Citation10,Citation23 They also concluded that administering lower doses of 6-MP in these patients was successful in maintaining adequately high levels of 6-TGN while allowing the administration of other agents at full protocol doses.Citation10 Furthermore, the investigators pointed out that the reduced activity patients tended to improve event-free survival (EFS) compared with wild-type patients (P=0.096) and that higher dose intensity of 6-MP was the most significant predictor of that outcome (P=0.020).Citation23 However, the authors also observed a nonsignificant trend for patients with low TPMT activity to have higher incidence and shorter onset of secondary acute myeloid leukemia as well as higher cumulative incidence of brain tumors when compared with patients with wild-type.Citation52,Citation53 In their later trial, Total Therapy Study XIIIB, which included 247 patients and pioneered the implementation of pharmacogenetics in leukemia therapy, they continued to administer a standard initial dose of 6-MP at the start of the continuation therapy but then selectively decreased the dose when deemed necessary based on a strategy that involved up-front knowledge of TPMT status combined with clinical tolerance and measurement of thiopurine metabolite levels.Citation54,Citation55 They eventually reported that TPMT genotype was not associated with the risk of hematologic relapse and that the long-term outcome showed no association with TPMT status (5-year cumulative incidences of 13.2%±2.3% and 6.7%±6.7% for wild-type and low-activity genotypes, respectively; P=0.46), further confirming that considering pharmacogenetics of TPMT for dose adjustment of 6-MP dosage in ALL can help to reduce treatment-associated toxicity while not compromising its efficacy.Citation54–Citation57
In the NOPHO-ALL-92 study of The Nordic Society of Paediatric Haematology and Oncology, a higher risk of relapse was observed in patients homozygous for wild-type (P=0.02) and/or high TPMT activity (P=0.002).Citation36,Citation58,Citation59 However, the authors also observed that patients with low TPMT activity, although at lower risk of relapse, had a higher risk of developing second myeloid neoplasms (SMN) associated with high levels of 6-TGN and methylated metabolites, probably leading to DNA damage and subsequent malignancies. The authors believe that this theory explains why low TPMT activity patients did not have a superior overall survival (OS) compared with those with wild-type activity (P=0.82) despite their lower risk of relapse.Citation38,Citation60 These observations, together with the ones from the Total Therapy Study XII, led the NOPHO to adopt TPMT genotype-dependent initial dosing of 6-MP in their later protocols, ALL-2000 and ALL-2008.Citation36,Citation58 Indeed, the long-term survival results from the ALL-2000 trial indicate that selecting the initial 6-MP dose based on TPMT genotype did reduce the risk of SMN in heterozygous patients, but at the expense of an increased risk of relapse. This explains why although a slight nonsignificant improvement in EFS was achieved by the new protocol, it had no difference in overall EFS or OS from its predecessor (5-year results: NOPHO-ALL-92: EFS=77.4%±1.0%, OS=87.6%±0.8%, n=1654; and NOPHO-ALL-2000: EFS=79.4%±1.5% and OS=89.1%±1.1%, n=1023).Citation58,Citation61
In the UK, ALL97and ALL97/99 trials, wild-type and heterozygous patients on the 6-MP arm initially received full dose of the drug, which was later adjusted to clinical hematologic toxicity whereas TPMT-deficient patients received 10% of the dose also adjusted for toxicity. While investigators observed a finding similar to the aforementioned studies in that patients with the TPMT*1/*3A genotype (n=99, EFS=88%) had better outcome at 5 years compared with TPMT wild-type patients (n=1206, EFS=80%; P=0.05), paradoxically, patients with the TPMT*1/*3C genotype also had lower EFS than those with *1/*3A genotype (n=17, EFS=53%; P=0.002). Furthermore, patients with heterozygous genotypes were found to experience more myelosuppression, accumulated higher 6-TGN concentrations and required dose reduction more frequently. However, no association between the risk of secondary malignancy and TPMT genotype was found.Citation13,Citation33,Citation62 In their subsequent trial, ALL-2003, which used minimal residual disease (MRD) to guide risk stratification and treatment intensity, the protocol prospectively observed the influence of TPMT genotype on treatment outcome by applying pretreatment genetic screening of the most common TPMT polymorphisms to 2387 of the study patients. The dosing regimen for 6-MP was similar to that of ALL97 in the sense that TPMT-deficient patients received 10% of the dose while the others received a standard dose subsequently adjusted according to a target cell count. The results showed that overall EFS – all TPMT genotypes confounded – was significantly higher than that of the previous protocol, which was attributed to the improved survival in the TPMT wild-type and TPMT*1/*3C genotype groups (EFS at 5 years=88%, 88% and 94% for TPMT wild-type, *1/*3A and *1/*3C, respectively). However, within this protocol, no significant differences in OS, EFS or relapse-free survival were observed with respect to TPMT genotypes. Thus, it was concluded that the improved risk-adapted protocol had reduced the influence of TPMT genotypes on treatment outcome and that the only factor that affected the outcome was MRD. Furthermore, there was no difference in survival within each MRD risk group with respect to TPMT genotypes.Citation33 Overall, the cumulative experience of the many UK ALL trials led to mandating preemptive TPMT screening for all children and young adults who start the ALL-2011 trial protocol.Citation11
In the Berlin-Frankfurt-Münster-2000 (BFM-2000) trial, the 6-MP dose was reduced by 10-fold from the standard starting dose for TPMT-deficient patients, but no dose adjustment was carried for heterozygous carriers who were given doses equal to the homozygous carriers of the wild-type. The investigators assessed the genotypes of 814 patients and used MRD for risk-stratification. The results showed no difference in the rate of hematopoietic toxicity between TPMT heterozygous variant carriers and homozygous wild-type carriers or between TPMT status and the risk of developing secondary cancers. Interestingly, it was observed that TPMT genotype had a significant impact on MRD during induction consolidation treatment as heterozygous patients had better MRD response (2.9-fold reduction), indicating an increased clearance of disease likely due to higher intensity of 6-MP effect (Relative risk=0.34; 95% confidence interval [CI], 0.13–0.86; P=0.02).Citation63,Citation64
In summary, all these trials demonstrate the importance of preemptive TPMT genetic screening and subsequent dose adjustment in mitigating the toxicity associated with thiopurine treatment while maintaining, if not enhancing, treatment efficacy and favorable long-term outcome.
Inflammatory bowel disease
IBD is a polygenic chronic, relapsing and remitting disease of the gastrointestinal tract that can be divided into two major clinical subtypes, Crohn’s disease and ulcerative colitis.Citation2,Citation65 Thiopurines, particularly AZA, are proven effective in inducing and maintaining long-term remission in IBD patients.Citation5 More than 20% of patients experience severe ADRs that lead to dose modification, treatment interruption or cessation.Citation66,Citation67 Bone marrow toxicity represented by leukopenia is one of the most serious thiopurine-related ADRs. Many studies investigated the influence of TPMT genotype on the efficacy and toxicity of thiopurines and most suggested a significant impact on clinical response ().
Table 2 Summary of selected studies which investigated the influence of TPMT pharmacogenetics on thiopurine treatment response in inflammatory bowel disease
A systematic review followed by a meta-analysis that eventually combined the results of 47 studies that investigated the risk of myelosuppression with respect to intermediate TPMT activity demonstrated a 4.19-fold increase in odd-ratio of leukopenia (95% CI: 3.20–5.48) in IBD patients with reduced TPMT activity compared with wild-type. One critic of this meta-analysis is that it combined rather smaller studies with sample sizes of <100 patients in most cases and the majority having retrospective cohort designs. However, in a sub-analysis of this study that combined 834 patients coming only from the 11 studies that had a prospective cohort design, the significant association of reduced TPMT activity with the risk of myelosuppression had an odd-ratio of 4.3 (95% CI: 2.53–7.29).Citation1 Among these prospective studies, an observational study with preemptive TPMT genetic testing for all patients and a relatively large sample size of 207 participants found that heterozygous TPMT genotype strongly predicted treatment withdrawal due to early-onset of ADRs following a conventional fixed-dosing regimen (79% vs 35% in heterozygous and wild-type, respectively; P<0.001). They highlighted that gastric intolerance (GI) was the most frequent reason for withdrawal among this group of patients and that myelotoxicity and GI occurred significantly more frequently among heterozygous (26% and 37%, respectively) than wild-type patients (0.5% and 7%, respectively). Interestingly, they had a 100% concordance of genotype to phenotype activity and found that TPMT activity was strongly predictive of clinical response as it was significantly higher in nonresponders.Citation68 Other prospective studies with preemptive TPMT genetic screening component reported that overall thiopurine-related ADRs were significantly more common among patients with low-to-intermediate TPMT activity when doses were not adjusted;Citation67 particularly myelotoxicity, which was more profound in TPMT-deficient genotype.Citation67,Citation69
Two independent meta-analysis further investigated the impact of pharmacogenetics on treatment response by exclusively combining studies (14 and 9 studies, respectively) that investigated the association between TPMT polymorphisms and ADRs in IBD patients, regardless of the study design (i.e., cross-sectional cohort, prospective cohort and case–control studies).Citation2,Citation65 They involved 2206 and 1309 patients respectively, and both concluded that TPMT polymorphisms were significantly associated with thiopurine-induced overall ADRs and bone marrow toxicity (around 3- and 6-fold increase in the odd-ratios, respectively) but not with hepatotoxicity, pancreatitis, flu-like symptoms, GI or skin reactions.Citation2,Citation65
The TPMT: Azathioprine Response to Genotyping and Enzyme Testing (TARGET) trial is a pragmatic RCT that prospectively investigated the impact of genotype-guided initial dosing of AZA followed by upward-titration to the maximum tolerable dose of the full protocol dose compared to no genotyping and full standard dose administration to all participants. It included 333 patients with inflammatory diseases and the primary aim was to see if this strategy would result in a significant reduction in the rate of ADRs-induced treatment cessation. No differences were found between the conventional and pharmacogenetics arms with respect to the frequency of treatment interruption due to ADRs (frequency: 27.7% vs 28.8%; odds ratio [OR]: 1.1; 95% CI: 0.66–1.8; P=0.74). On the other hand, the study did not find any difference in the rate of remission between the intervention and control groups indicating that the adjustment did not affect treatment efficacy. However, the investigators did not provide a stratified analysis addressing the differences in outcomes according to genotype groups within each study arm or between the two arms, probably due to small sample size. Moreover, it is worth mentioning that a single patient with homozygous variant genotype in the study who was on the nongenotyping arm and subsequently received the full dose of AZA developed severe neutropenia, which underlines the importance of genetic testing to identify this group of patients.Citation16
A larger and more recent prospective RCT, which involved 783 IBD patients, the Thiopurine response Optimization by Pharmacogenetic testing in Inflammatory bowel disease Clinics (TOPIC) trial, similarly showed no significant overall impact of TPMT genotype-guided dosing of thiopurines on treatment efficacy or on the risk of hematologic ADRs (i.e., leukopenia and thrombocytopenia) between the genotyped and nongenotyped arms (frequency: 7.4% vs 7.9%; relative risk: 0.93; 95% CI: 0.57–1.52). The efficacy results of this study further advocate that a reduced thiopurine dose does not result in under-treatment. Moreover, a subgroup analysis of this study, which compared only carriers of TPMT variants between the two arms, revealed that the pharmacogenetic approach was able to significantly decrease the risk of hematologic ADRs by 10-fold in carriers of at least one genetic variant (frequency: 2.6% vs 22.9%; relative risk: 0.11; 95% CI: 0.01–0.85).Citation22 The results of the secondary aim of this study excluded any significant association between TPMT genotypes and anemia, hepatotoxicity, pancreatitis, skin rash, GI and general malaise, which is consistent with the results of the aforementioned meta-analysis and other results in the literature.Citation2,Citation22,Citation65 It also suggested that factors other than TPMT genotype play an important role in the development of thiopurine-induced ADRs.Citation24
Autoimmune disorders
Autoimmune diseases are a group of heterogeneous conditions that involve a destructive attack against the host’s tissues launched by a deregulated immune system as in SLE, RA, AIH and generalized-eczematous disorders. Thus, treatment strategies are usually based on the use of immunosuppressants that act by modifying the activity of the immune system. AZA is widely used as an immunosuppressive agent in autoimmune diseases but its use is limited by its ADRs.Citation14 Similar to the meta-analysis that focused on IBD, another meta-analysis, which included 651 patients with autoimmune diseases coming from 11 studies demonstrated that overall ADRs and AZA-induced bone marrow toxicity are significantly associated with TPMT polymorphisms with OR of 3.12 (95% CI: 1.48–6.56) and 3.76 (95% CI: 1.97–7.17), respectively. The results remained significant in two analysis, one that grouped the homozygous and heterozygous carriers into one reduced-activity group and the other that focused on heterozygous carriers only. The study also showed a significant association with GI with OR of 6.43 (95% CI: 2.04–20.25), but the authors suspect that the observed association might have been driven by a single study, since after excluding this study, the association was no longer significant with OR of 2.1 (95% CI: 0.36–12.42).Citation14 The study also excludes the association of TPMT polymorphisms with hepatotoxicity. The sub-analysis that examined the association with myelosuppression according to the type of disease found significant results in SLE, RA and AIH subgroups. They also concluded that the risk prediction of bone marrow toxicity and overall ADRs based on TPMT variant-positive genotypes has high specificity (94.10% and 92.93%, respectively) but at the expense of sensitivity (16.30% and 14.85%, respectively).Citation14 Furthermore, in a prospective study that investigated the impact of TPMT-activity guided AZA dosing on the treatment response in patients with atopic eczema, the investigators concluded that TPMT-based dosing was able to maintain the drug efficacy while reducing the predicted toxicity.Citation70
Cost-effectiveness
Most of the above studies concluded that TPMT testing could lead to improved prescribing of thiopurines, which would ultimately result in an increased treatment efficacy and a reduction in the rate and intensity of ADRs. Nonetheless, the cost-effectiveness of such an intervention is still open to debate. Only a few studies have addressed the cost-effectiveness of TPMT pharmacogenetics interventions. In an effort toward this evaluation, a case study examined the cost-effectiveness of prospective TPMT genotyping in children with ALL treated with thiopurines and suggested positive results manifested in financial savings and a gain in life-years in the most favorable settings of the sensitivity analysis.Citation71 Similarly, another study established a model based on a theoretical IBD population treated with AZA and found that pretreatment screening for TPMT genotype would be cost-effective in avoiding patient mortality due to myelosuppression.Citation29 However, data coming from RCTs do not necessarily support this conclusion as one perspective study found that such a technique incurred excessive cost associated with genotyping but did not predict AZA-induced toxicity in IBD patients.Citation72 Nonetheless, these studies were too small and not adequately powered to answer this question.Citation16 A systematic review came to the conclusion that screening for TPMT activity either by genotyping or phenotyping was a cost-effective strategy that can be used to reduce health-care costs while improving clinical effectiveness.Citation73 Another study aimed at the evaluation of the added-value of genetic screening of TPMT followed by dose adjustment of AZA prior to the initiation of treatment found that genetic-based dosing dominated the standard dosing strategy in patients with rheumatological disease by reducing the treatment cost and the frequency of AZA-induced side effects.Citation74 In a more recent prospective economic evaluation that was conducted alongside the TARGET study, in which the study aim was to test the cost-effectiveness of theTPMT genotyping approach in autoimmune diseases, the researchers concluded that the genetic approach had up to 71% probability of being cost-effective depending on the cost of the genetic test. The results, however, were not conclusive as the observed economic advantage in the intervention group owing to lower use of resources was accompanied by a slight (almost negligible) reduction in the quality of life.Citation75
Impact on clinical practice
Over the past decade, TPMT enzyme testing has gained a lot of acceptance, as reflected by the rapid increase in the number of tests performed in clinical practice.Citation16,Citation76 This sudden increase was the inevitable result of multiple factors supporting this approach, which include the increase in the available knowledge about the role of TPMT in treatment outcome, the stronger recommendations coming from clinical guidelines like the CPIC and the wider accessibility to genetic testing (i.e., larger availability, reduced cost, faster turnaround of results and shorter interpretation time).Citation1,Citation16 This shift in clinical practice was evaluated in the TARGET study, which observed that the physicians did follow the recommendations coming from British clinical guidelines (e.g., British Association of Dermatologists Therapy and British Society for Rheumatology) for TPMT heterozygous patients and chose a lower initial dose of AZA for those patients but the investigators also noted that the physicians used overall lower starting doses for wild-type patients as well.Citation16 This “safe” practice reflects the physicians’ reservation regarding the sensitivity and specificity of this test, which stems from the fact that being homozygous carrier of TPMT wild-type, although predictive of a reduced risk of AZA-induced myelosuppression, does not completely eliminate the possibility. Indeed, it was mentioned earlier that a fraction of TPMT wild-type patients can still have intermediate TPMT-activity and that other factors play a role in the development of this ADR.Citation16,Citation33,Citation36 Moreover, other side effects such as hepatotoxicity, pancreatitis, nausea and vomiting cannot be predicted by TPMT testing.Citation1,Citation14,Citation65 The adoption of pretreatment TPMT screening seems to vary according to discipline, as reported by one survey, with 94% of dermatologists, 60% of gastroenterologists and only 47% rheumatologists requesting it.Citation76 This could be related to the level of evidence available in the domain of practice and the strength of the recommendations of the respective guidelines and protocols used by each specialist (e.g., UK guidelines in dermatology and gastroenterology recommend genetic screening while ALL-2011 protocol mandates it).Citation1,Citation11,Citation76 However, from an evidence-based perspective, and besides the universally accepted association with hematotoxicity, the recommendations for preemptive genetic testing still have some margin to evolve. Plus, even in well-established scenarios, like in the case of myelosuppression in TPMT-deficient patients, strong evidence is still lacking to support that the pharmacogenetic approach would result in a significantly better outcome.Citation14,Citation65
TPMT in the new era of sequencing
The influence of genetic polymorphisms in the TPMT gene on treatment outcome has been well-documented and replicated in many studies. However, studies have also concluded that the genetic-based screening for TPMT activity should be interpreted with caution, as the activity of the TPMT enzyme can be co-influenced by other factors, and the development of thiopurine-induced ADRs is a multifactorial event.Citation14 For instance, most of the presented studies inferred TPMT activity by genotyping the most common nonfunctional TPMT alleles while results of a recent study that explored the sequencing data suggest that in certain populations, the inferred activity can be refined by incorporating the genotypes of other alleles. The study also identified a new variant in the TPMT gene, TPMT*38 (T514C), which had an allelic frequency of 0.11% and was predicted to be a damaging mutation.Citation15 Moreover, as increasingly reported by different studies, genetic variants in other genes involved in thiopurines metabolism like ITPA, HGPRT and MTHFR as well as variants in genes independent of TPMT can influence thiopurines treatment outcome.Citation14,Citation20,Citation25,Citation41,Citation70,Citation77 For example, genome-wide association studies have identified variants in the PACSIN2 gene that influence TPMT activity and were linked to 6-MP-related gastrointestinal toxicity in children with ALL, whereas variants in the NUDT15 gene were associated with thiopurine-induced leukopenia.Citation26,Citation41 However, since a lot of genes have significant differences in the frequencies of polymorphisms across major ethnic groups, it is important to evaluate the genetic profiles of patients in a global frame that considers all of the genes involved in a specific pathway to better understand the impact of ethnic diversity on drug response. One particularly interesting example of the role of pharmacoethnicity is the case of NUDT15 in Japanese population, in whom polymorphisms of this gene were associated with higher risk of toxicity and were more frequent than TPMT-deficient variants.Citation26 Studies also suggest that combining the effects of such polymorphisms with variants in TPMT gene could strengthen the predictive power of the risk of developing thiopurines-related toxicity.Citation14,Citation22 This should soon become feasible with the breakthrough advances in sequencing and genotyping techniques. Indeed, in a recent study that tested the sensitivity, specificity and predictive values of the imputation of TPMT-alleles, most values were over 90%, indicating that imputation of TPMT alleles can be used as a screening method for individuals with high-risk of developing serious thiopurine-induced ADRs.Citation46 Furthermore, nongenetic factors should be taken into consideration before thiopurine initiation as they can have a big influence on the outcome and might interfere with the genotype-guided dosing.Citation14
Conclusion
In conclusion, although it is currently well established that TPMT polymorphisms can explain a certain portion of thiopurine-induced ADRs, particularly hematotoxicity, it is surely not capable of predicting all of them. Indeed, many studiesCitation2,Citation14,Citation22,Citation65 have found that certain ADRs were not associated with a reduced TPMT activity such as pancreatitis and hepatotoxicity. This holds true in the context of ALL, IBD and the different types of autoimmune disorders. What is clear so far is that TPMT-deficient genotypes (homozygous variant carriers and compound heterozygous), and to lesser extent, heterozygous patients are predisposed to thiopurine-induced severe hematotoxicity.Citation1,Citation47 However, other factors such as disease progression and co-medications can also modulate the risk of myelosuppression, regardless of the genotype. While TPMT-deficient patients will definitely benefit from dose reduction of thiopurines, the validity of this approach for heterozygous carriers is still arguable since studies have shown that not all of these patients are intolerant to thiopurine; in fact, 30%–60% of heterozygous patients do tolerate it.Citation33,Citation47 Moreover, depending on the treated condition and treatment protocol used, TPMT wild-type patients also exhibit higher risks of worse outcome such as hematologic relapse in ALL and treatment failure in IBD, which adds an extra layer of complexity to the already troublesome process of finding the best therapeutic regimen that would ensure maximum efficacy and minimum toxicity.Citation47 Consequently, regular clinical testing and hematologic assessment remain the mainstay in the monitoring of thiopurine treatment while genetic testing adds the advantage of refining the initial dosing and patient-stratification processes, as well as suggesting customized monitoring for certain patient groups. One nice example backed with strong clinical evidence is the abovementioned scenario of myelosuppression. Preemptive TPMT genetic screening and tailored thiopurine initial dosing followed by upward/downward titration and hematological monitoring to a target level of myelosuppression can be considered a cost-effective approach which would allow the prevention and early detection of myelosuppression in this vulnerable population without compromising the efficacy of the treatment.Citation47
Perspective
While the goal of personalized medicine in general, and pharmacogenetics in particular, is to deliver patient-tailored treatments that would ensure maximum efficacy with minimum toxicity, the studies presented in this review make the argument that this is not an easy task. There is a balance to consider between treatment benefits and ADRs, which is controlled by multiple factors. This being said, what we can be sure of, for now, is that the more we get to know about the impact of pharmacogenetics on the variability of treatment response, the better we will be able to control the outcome to the advantage of the patient. Moreover, most pharmacoeconomic analyses have indicated that screening for TPMT pharmacogenetics promises to be cost-effective. With the advent of next-generation sequencing and the many breakthroughs in bioinformatics, the cost of analyzing the entire human genome is bound to drop, which would allow for greater accessibility to genetic data and a larger understanding of how their interactions with each other and with other factors influence the treatment. In the meantime, it is very promising to see that most major institutions have already incorporated preemptive TPMT screening in their treatment protocols to enhance treatment outcome and the continuously emerging long-term data proving the utility of doing so, which should encourage other institutions to follow.
Acknowledgments
Rachid Abaji is a scholar of the Cole Foundation and the Network of Applied Medical Genetics and acknowledges the support of both organizations.
Disclosure
The authors report no conflicts of interest in this work.
References
- HiggsJEPayneKRobertsCNewmanWGAre patients with intermediate TPMT activity at increased risk of myelosuppression when taking thiopurine medications?Pharmacogenomics201011217718820136357
- DongXWZhengQZhuMMTongJLRanZHThiopurine S-methyltransferase polymorphisms and thiopurine toxicity in treatment of inflammatory bowel diseaseWorld J Gastroenterol201016253187319520593505
- KoturNDokmanovicLJanicDTPMT gene expression is increased during maintenance therapy in childhood acute lymphoblastic leukemia patients in a TPMT gene promoter variable number of tandem repeat-dependent mannerPharmacogenomics201516151701171226411491
- ChouchanaLNarjozCBeaunePLoriotMARoblinXReview article: the benefits of pharmacogenetics for improving thiopurine therapy in inflammatory bowel diseaseAliment Pharmacol Ther2012351153622050052
- GisbertJPGomollonFThiopurine-induced myelotoxicity in patients with inflammatory bowel disease: a reviewAm J Gastroenterol200810371783180018557712
- FraserAGOrchardTRJewellDPThe efficacy of azathioprine for the treatment of inflammatory bowel disease: a 30 year reviewGut200250448548911889067
- LennardLThe clinical pharmacology of 6-mercaptopurineEur J Clin Pharmacol19924343293391451710
- LennardLTPMT in the treatment of Crohn’s disease with azathioprineGut200251214314612117866
- ChouchanaLNarjozCRocheDInterindividual variability in TPMT enzyme activity: 10 years of experience with thiopurinepharmacogenetics and therapeutic drug monitoringPharmacogenomics201415674575724897283
- RellingMVHancockMLRiveraGKMercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locusJ Natl Cancer Inst199991232001200810580024
- LennardLImplementation of TPMT testingBr J Clin Pharmacol201477470471423962279
- WangLPelleymounterLWeinshilboumRVery important pharmacogene summary: thiopurine S-methyltransferasePharmacogenet Genomics201020640140520154640
- VoraAMitchellCDLennardLToxicity and efficacy of 6-thioguanine vs. 6-mercaptopurine in childhood lymphoblastic leukaemia: a randomised trialLancet200636895441339134817046466
- LiuYPXuHQLiMAssociation between thiopurine S-methyl-transferase polymorphisms and azathioprine-induced adverse drug reactions in patients with autoimmune diseases: a meta-analysisPLoS One20151012e014423426633017
- KimHYLeeSHLeeMNComplete sequence-based screening of TPMT variants in the Korean populationPharmacogenet Genomics201525314314625564374
- NewmanWGPayneKTrickerKA pragmatic randomized controlled trial of thiopurinemethyltransferase genotyping prior to azathioprine treatment: the TARGET studyPharmacogenomics201112681582621692613
- EvansWEHornerMChuYQKalwinskyDRobertsWMAltered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurinemethyltransferase-deficient child with acute lymphocytic leukemiaJ Pediatr199111969859891960624
- YatesCRKrynetskiEYLoennechenTMolecular diagnosis of thiopurine S-methyltransferase deficiency: genetic basis for azathioprine and mercaptopurine intoleranceAnn Intern Med199712686086149103127
- EvansWEHonYYBomgaarsLPreponderance of thiopurine S-methyltransferase deficiency and heterozygosity among patients intolerant to mercaptopurine or azathioprineJ Clin Oncol20011982293230111304783
- Karas-KuzelickiNJazbecJMilekMMlinaric-RascanIHeterozy-gosity at the TPMT gene locus, augmented by mutated MTHFR gene, predisposes to 6-MP related toxicities in childhood ALL patientsLeukemia200923597197418987660
- Peregud-PogorzelskiJTetera-RudnickaEKurzawskiMThiopurine S-methyltransferase (TPMT) polymorphisms in children with acute lymphoblastic leukemia, and the need for reduction or cessation of 6-mercaptopurine doses during maintenance therapy: the Polish multicenter analysisPediatr Blood Cancer201157457858221319286
- CoenenMJde JongDJvan MarrewijkCJIdentification of patients with variants in TPMT and dose reduction reduces hematologic events during thiopurinetreatment of inflammatory bowel diseaseGastroenterology20151494907917.e726072396
- RellingMVHancockMLBoyettJMPuiCHEvansWEPrognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemiaBlood19999392817282310216075
- ColombelJFFerrariNDebuysereHGenotypic analysis of thiopurine S-methyltransferase in patients with Crohn’s disease and severe myelosuppression during azathioprine therapyGastroenterology200011861025103010833476
- StoccoGCheokMHCrewsKRGenetic polymorphism of inosine triphosphate pyrophosphatase is a determinant of mercaptopurine metabolism and toxicity during treatment for acute lymphoblastic leukemiaClin Pharmacol Ther200985216417218685564
- MoriyamaTNishiiRPerez-AndreuVNUDT15 polymorphisms alterthiopurine metabolism and hematopoietic toxicityNat Genet201648436737326878724
- DubinskyMCYangHHassardPV6-MP metabolite profiles provide a biochemical explanation for 6-MP resistance in patients with inflammatory bowel diseaseGastroenterology2002122490491511910342
- AnsariAHassanCDuleyJThiopurinemethyltransferase activity and the use of azathioprine in inflammatory bowel diseaseAliment Pharmacol Ther200216101743175012269967
- WinterJWalkerAShapiroDGaffneyDSpoonerRJMillsPRCost-effectiveness of thiopurinemethyltransferase genotype screening in patients about to commence azathioprine therapy for treatment of inflammatory bowel diseaseAliment Pharmacol Ther200420659359915352906
- AppellMLBergJDuleyJNomenclature for alleles of the thiopurinemethyltransferase genePharmacogenet Genomics201323424224823407052
- Collie-DuguidESPritchardSCPowrieRHThe frequency and distribution of thiopurinemethyltransferase alleles in Caucasian and Asian populationsPharmacogenetics199991374210208641
- KataraPKuntalHTPMT polymorphism: when shield becomes weaknessInterdiscip Sci20168215015526297310
- LennardLCartwrightCSWadeRVoraAThiopurinemethyltransferase and treatment outcome in the UK acute lymphoblastic leukaemia trial ALL2003Br J Haematol2015170455055825940902
- WinterJWGaffneyDShapiroDAssessment of thiopurinemethyltransferase enzyme activity is superior to genotype in predicting myelosuppression following azathioprine therapy in patients with inflammatory bowel diseaseAliment Pharmacol Ther20072591069107717439508
- HindorfUAppellMLGenotyping should be considered the primary choice for pre-treatment evaluation of thiopurinemethyltransferase functionJ Crohns Colitis20126665565922398041
- SchmiegelowKForestierEKristinssonJThiopurinemethyltransferase activity is related to the risk of relapse of childhood acute lymphoblastic leukemia: results from the NOPHO ALL-92 studyLeukemia200923355756418987654
- SchaeffelerEFischerCBrockmeierDComprehensive analysis of thiopurine S-methyltransferase phenotype-genotype correlation in a large population of German-Caucasians and identification of novel TPMT variantsPharmacogenetics200414740741715226673
- SchmiegelowKAl-ModhwahiIAndersenMKMethotrexate/6-mercaptopurine maintenance therapy influences the risk of a second malignant neoplasm after childhood acute lymphoblastic leukemia: results from the NOPHO ALL-92 studyBlood2009113246077608419224761
- BoothRAAnsariMTLoitEAssessment of thiopurine S-methyltransferase activity in patients prescribed thiopurines: a systematic reviewAnn Intern Med201115412814823W-295W-29821690596
- LevinsenMRosthojSNygaardUMyelotoxicity after high-dose methotrexate in childhood acute leukemia is influenced by 6-mercaptopurine dosing but not by intermediate thiopurinemethyltransferase activityCancer Chemother Pharmacol2015751596625347948
- TammRMagiRTremmelRPolymorphic variation in TPMT is the principal determinant of TPMT phenotype: a meta-analysis of three genome-wide association studiesClin Pharmacol Ther Epub20161022
- FordLProutCGaffneyDBergJWhose TPMT activity is it anyway?Ann Clin Biochem200441Pt 649850015588444
- LennardLChewTSLilleymanJSHuman thiopurinemethyltransferase activity varies with red blood cell ageBr J Clin Pharmacol200152553954611736862
- YanLZhangSEiffBThiopurinemethyltransferase polymorphic tandem repeat: genotype-phenotype correlation analysisClin Pharmacol Ther200068221021910976552
- LauJIoannidisJPTerrinNSchmidCHOlkinIThe case of the misleading funnel plotBMJ2006333756859760016974018
- AlmogueraBVazquezLConnollyJJImputation of TPMT defective alleles for the identification of patients with high-risk phenotypesFront Genet201459624860591
- RellingMVGardnerEESandbornWJClinical pharmacogenetics implementation consortium guidelines for thiopurinemethyltransferase genotype and thiopurine dosing: 2013 updateClin Pharmacol Ther201393432432523422873
- LennardLVan LoonJALilleymanJSWeinshilboumRMThiopurinepharmacogenetics in leukemia: correlation of erythrocyte thiopurinemethyltransferase activity and 6-thioguanine nucleotide concentrationsClin Pharmacol Ther198741118253467886
- LennardLGibsonBENicoleTLilleymanJSCongenital thiopurinemethyltransferase deficiency and 6-mercaptopurine toxicity during treatment for acute lymphoblastic leukaemiaArch Dis Child19936955775798257179
- LennardLLilleymanJSVan LoonJWeinshilboumRMGenetic variation in response to 6-mercaptopurine for childhood acute lymphoblastic leukaemiaLancet199033687092252291973780
- DokmanovicLUrosevicJJanicDAnalysis of thiopurine S-methyltransferase polymorphism in the population of Serbia and Montenegro and mercaptopurine therapy tolerance in childhood acute lymphoblastic leukemiaTher Drug Monit200628680080617164697
- RellingMVRubnitzJERiveraGKHigh incidence of secondary brain tumours after radiotherapy and antimetabolitesLancet19993549172343910406363
- RellingMVYanishevskiYNemecJEtoposide and antimetabolite pharmacology in patients who develop secondary acute myeloid leukemiaLeukemia19981233463529529129
- PuiCHPeiDSandlundJTLong-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemiaLeukemia201024237138220010620
- RellingMVPuiCHChengCEvansWEThiopurinemethyltransferase in acute lymphoblastic leukemiaBlood2006107284384416401827
- RochaJCChengCLiuWPharmacogenetics of outcome in children with acute lymphoblastic leukemiaBlood2005105124752475815713801
- MoriyamaTRellingMVYangJJInherited genetic variation in childhood acute lymphoblastic leukemiaBlood20151253988399525999454
- SchmiegelowKForestierEHellebostadMLong-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemiaLeukemia201024234535420010622
- SchmiegelowKBjorkOGlomsteinAIntensification of mercaptopurine/methotrexate maintenance chemotherapy may increase the risk of relapse for some children with acute lymphoblastic leukemiaJ Clin Oncol20032171332133912663723
- ThomsenJBSchrøderHKristinssonJPossible carcinogenic effect of 6-mercaptopurine on bone marrow stem cellsCancer19998661080108610491537
- LevinsenMRotevatnEØRosthøjSPharmacogenetically based dosing of thiopurines in childhood acute lymphoblastic leukemia: influence on cure rates and risk of second cancerPediatric Blood Cancer201461579780224395436
- LennardLCartwrightCSWadeRVoraAThiopurine dose intensity and treatment outcome in childhood lymphoblastic leukaemia: the influence of thiopurine methyltransferase pharmacogeneticsBr J Haematol2015169222824025441457
- StanullaMSchaeffelerEFlohrTThiopurine methyltransferase (TPMT) genotype and early treatment response to mercaptopurine in childhood acute lymphoblastic leukemiaJAMA2005293121485148915784872
- StanullaMSchaeffelerEMorickeAThiopurine methyltransferase genetics is not a major risk factor for secondary malignant neoplasms after treatment of childhood acute lymphoblastic leukemia on Berlin-Frankfurt-Munster protocolsBlood200911471314131819535798
- LiuYPWuHYYangXAssociation between thiopurine S-methyltransferase polymorphisms and thiopurine-induced adverse drug reactions in patients with inflammatory bowel disease: a meta-analysisPLoS One2015103e012174525799415
- de JongDJDerijksLJNaberAHHooymansPMMulderCJSafety of thiopurines in the treatment of inflammatory bowel diseaseScand J Gastroenterol Suppl2003239697214743886
- HindorfULindqvistMPetersonCPharmacogenetics during standardised initiation of thiopurine treatment in inflammatory bowel diseaseGut200655101423143116543290
- AnsariAArenasMGreenfieldSMProspective evaluation of the pharmacogenetics of azathioprine in the treatment of inflammatory bowel diseaseAliment Pharmacol Ther200828897398318616518
- DerijksLJGilissenLPEngelsLGPharmacokinetics of 6-mercaptopurine in patients with inflammatory bowel disease: implications for therapyTher Drug Monit200426331131815167634
- MeggittSJGrayJCReynoldsNJAzathioprine dosed by thiopurinemethyltransferase activity for moderate-to-severe atopic eczema: a double-blind, randomised controlled trialLancet2006367951383984616530578
- van den Akker-van MarleMEGurwitzDDetmarSBCost-effectiveness of pharmacogenomics in clinical practice: a case study of thiopurinemethyltransferase genotyping in acute lymphoblastic leukemia in EuropePharmacogenomics20067578379216886902
- SayaniFAProsserCBaileyRJJacobsPFedorakRNThiopurine methyltransferase enzyme activity determination before treatment of inflammatory bowel disease with azathioprine: effect on cost and adverse eventsCan J Gastroenterol200519314715115776134
- PayneKNewmanWGGurwitzDIbarretaDPhillipsKATPMT testing in azathioprine: a ‘cost-effective use of healthcare resources’?Personal Med200861103113
- MarraCAEsdaileJMAnisAHPractical pharmacogenetics: the cost effectiveness of screening for thiopurine S-methyltransferase polymorphisms in patients with rheumatological conditions treated with azathioprineJ Rheumatol200229122507251212465143
- ThompsonAJNewmanWGElliottRARobertsSATrickerKPayneKThe cost-effectiveness of a pharmacogenetic test: a trial-based evaluation of TPMT genotyping for azathioprineValue Health2014171223324438714
- FargherEATrickerKNewmanWCurrent use of pharmacogenetic testing: a national survey of thiopurine methyltransferase testing prior to azathioprine prescriptionJ Clin Pharm Ther200732218719517381669
- ZelinkovaZDerijksLJStokkersPCInosine triphosphate pyro-phosphatase and thiopurine s-methyltransferase genotypes relationship to azathioprine-induced myelosuppressionClin Gastroenterol Hepatol200641444916431304