Abstract
The new era of personalized medicine, which integrates the uniqueness of an individual with respect to the pharmacokinetics and pharmacodynamics of a drug, holds promise as a means to provide greater safety and efficacy in drug design and development. Personalized medicine is particularly important in oncology, whereby most clinically used anticancer drugs have a narrow therapeutic window and exhibit a large interindividual pharmacokinetic and pharmacodynamic variability. This variability can be explained, at least in part, by genetic variations in the genes encoding drug metabolizing enzymes, transporters, or drug targets. Understanding of how genetic variations influence drug disposition and action could help in tailoring cancer therapy based on individual’s genetic makeup. This review focuses on the pharmacogenomics of drug metabolizing enzymes and drug transporters, with a particular highlight of examples whereby genetic variations in the metabolizing enzymes and transporters influence the pharmacokinetics and/or response of chemotherapeutic agents.
Introduction
The new era of personalized medicine, which integrates the uniqueness of an individual with respect to the pharmacokinetics and pharmacodynamics of a drug, holds promise as a means to provide greater safety and efficacy in drug design and development. Personalized medicine is particularly important in oncology whereby most clinically used anticancer drugs have a narrow therapeutic window and exhibit a large interindividual pharmacokinetic and pharmacodynamic variability. This variability can lead to therapeutic failure or severe toxicity. Understanding of how genetic variations influence drug disposition and action could help in tailoring cancer therapy based on individual’s genetic makeup. Pharmacogenomics is the study of how variations in the human genome affect the response to medications. Each drug, after it enters the body, interacts with numerous proteins, such as carrier proteins, transporters, metabolizing enzymes, and multiple types of receptors. These protein interactions determine drug pharmacokinetics (ie, drug absorption, distribution, metabolism, and excretion) and pharmacodynamics (ie, target site of action, pharmacological and toxicological effects). Moreover, drugs trigger downstream secondary events which may impact additional gene or protein expression responses and can also vary among patients. As a result, the overall response to a drug is determined by the interplay of multiple genes that are involved in the pharmacokinetic and pharmacodynamic pathways of a drug. In general, important genetic variation in drug effect can be envisioned at the level of drug metabolizing enzymes, drug transporters, and drug targets. This review provides an overview on the commonly occurring, functionally and/or clinically relevant genetic polymorphisms within the genes encoding important drug metabolizing enzymes and drug transporters, with a particular highlight of examples whereby genetic variations in these genes influence the pharmacokinetics and/or response of chemotherapeutic agents.
Drug metabolizing-enzyme pharmacogenomics
Drug metabolizing enzymes are proteins that catalyze the biochemical modifications of xenobiotics (eg, drugs) and endogenous chemicals (eg, hormones, neurotransmitters). Broadly, drug metabolizing enzymes are divided into two categories: Phase I (functionalizing) enzymes that introduce or remove functional groups in a substrate through oxidation, reduction, or hydrolysis; and Phase II (conjugating) enzymes that transfer moieties from a cofactor to a substrate. Essentially all of the major human metabolizing enzymes exhibit genetic polymorphisms at the genomic level, and many of these enzymes have clinically relevant genetic polymorphisms.Citation1 A gene is considered to be polymorphic when the frequency of a variant allele in a specific population is at least 1%.
Phase I enzymes
Phase I metabolizing enzymes include those involved in:
Oxidation – cytochrome P450, alcohol dehydrogenase, aldehyde dehydrogenase, dihydropyrimidine dehydrogenase, monoamine oxidase, and flavin-containing monooxygenase;
Reduction – nicotinamide adenine dinucleotide phosphate (NADPH)-cytochrome P450 reductase and reduced cytochrome P450;
Hydrolysis – epoxide hydrolase, esterases, and amidases.
The most important Phase I enzymes that exhibit clinical relevant genetic polymorphisms are the cytochrome P450 (CYP) superfamily. The human CYP superfamily represents the most important system responsible for catalyzing the oxidation of a large number of endogenous and exogenous compounds including drugs, toxins, and carcinogens. In this superfamily, 57 genes and 58 pseudogenes have been identified, which are divided into 18 families and 43 subfamilies (http://drnelson.utmem.edu/cytochromeP450.html). Among them, three subfamilies of CYPs, including CYP1, CYP2, and CYP3, contribute to the oxidative metabolism of more than 90% of clinically used drugs.
The human CYP genes are highly polymorphic. The polymorphisms within the CYP genes, which include gene deletions, missense mutations, deleterious mutations creating splicing defects or premature stop codon, and gene duplications, could produce abolished, reduced, normal, or enhanced enzyme activity. As a result, patients can be classified into four phenotypes based on the level of a CYP enzyme activity: poor metabolizer (abolished activity), intermediate metabolizer (reduced activity), extensive metabolizer (normal activity), and ultrarapid metabolizer (enhanced activity). It is expected that poor metabolizers would have higher concentrations of a drug that is inactivated by that enzyme pathway and therefore require a lower dose to avoid adverse reactions, whereas ultrarapid metabolizers would require a higher dose to achieve therapeutic effective drug concentrations. The opposite pattern of reactions is expected for a prodrug that undergoes metabolic activation. A prodrug may have little therapeutic effect in poor metabolizers, while producing a toxic level of active form in ultrarapid metabolizers. Substantial evidence suggests that genetic polymorphisms within the CYP genes have significant impact on drug disposition and/or response. The common functional polymorphisms in the major human CYP genes and their clinical relevance are summarized in . Notably, the most pharmacologically and clinically relevant CYP polymorphisms are found in CYP2D6, CYP2C9, and CYP2C19. Of the Food and Drug Administration (FDA)-approved drug labels referring human genomic biomarkers, 62% are pertained to polymorphisms in the CYP enzymes, with CYP2D6 (35%), CYP2C19 (17%), and CYP2C9 (7%) being the most common.Citation2
Table 1 Most common naturally occurring functional polymorphisms in the major human CYP genes: allele frequency, functional effect, and highlights of clinical relevance
CYP2D6 is not inducible and therefore, the variations in the enzyme expression and activity are largely attributable to genetic polymorphisms. The CYP2D6 gene is highly polymorphic with more than 63 functional variants identified to date (http://www.cypalleles.ki.se). These alleles result in abolished, decreased, normal, or ultrarapid CYP2D6 enzyme activity. The most important null alleles are CYP2D6*4 (splicing defect) and CYP2D6*5 (gene deletion); the common alleles with severely reduced enzyme activity are represented by CYP2D6*10, *17, and *41; duplication or multiduplications of active CYP2D6 genes (eg, CYP2D6*1 × N (N ≥ 2)) result in ultrarapid enzyme activity (). The distributions of CYP2D6 alleles exhibit notable interethnic differences. The nonfunctional allele CYP2D6*4 is prevalent in Caucasians (allelic frequency, ∼25%), while the reduced function allele CYP2D6*10 and CYP2D6*17 is common in Asians (allelic frequency, ∼40%) and Africans (allelic frequency, ∼34%).Citation3 As a result, poor metabolizers of CYP2D6, mainly resulted from null allele CYP2D6*4, has a higher frequency in Caucasians (5%–14%) compared with Africans (0%–5%) and Asians (0%–1%). Ultrarapid metabolizers of CYP2D6, resulted from gene duplication or multiduplications, have a higher frequency in Saudi Arabians (20%) and black Ethiopians (29%) compared with Caucasians (1%–10%).Citation3 The inter-ethnic difference in the CYP2D6 genotypes may contribute to the inter-ethnic variations in the disposition and response of substrate drugs. CYP2D6 is involved in the metabolism of ∼25% of all drugs in clinical use, although it accounts for ∼2% of total hepatic CYP content. CYP2D6 genotype is of great importance for the pharmacokinetics and response of many drugs, including tricyclic antidepressants, antiarrhytmics, neuroleptics, analgesics, antiemetics, and anticancer drugs.Citation4
The human CYP2C9 and CYP2C19 genes are highly homologous at the nucleotide level. The most common nonsynonymous CYP2C9 polymorphisms, CYP2C9*2 and CYP2C9*3, produce enzyme with differing affinity or intrinsic clearance for different substrates. While CYP2C9*2 effects appear to be more substrate specific, CYP2C9*3 variant exhibits reduced catalytic activity towards the majority of CYP2C9 substrates. The clinical importance of CYP2C9 polymorphisms is exemplified by the dose adjustment of an oral anticoagulant warfarin based on CYP2C9 genotype. The patients carrying either CYP2C9*2 or CYP2C9*3 require a significantly smaller daily dose of warfarin to maintain desired therapeutic effects while avoiding severe toxicity, compared with patients carrying the wild-type CYP2C9.Citation5 With respect to CYP2C19, a splice site mutation in exon 4 (CYP2C19*2) and a premature stop codon in exon 4 (CYP2C19*3) represent the two most predominant null alleles. By genotyping for CYP2C19*2 and *3, one could detect ∼84%, ∼100%, and >90% of poor CYP2C19 metabolizers in Caucasians, Asians, and Africans, respectively. By also including CYP2C19*4 and *6 alleles, ∼92% of poor metabolizers in Caucasians can be detected. Generally, the poor metabolizer phenotype of CYP2C19 occurs in 12%–23% of the Asian population, in 1%–6% of Caucasians, and in 1%–7.5% of black Africans. Polymorphisms in CYP2C19 are known to affect the pharmacokinetics and/or response of several classes of drugs, including proton pump inhibitors (eg, omeprazole), barbiturates, and anticancer drugs.Citation4
Phase II enzymes
The most important Phase II enzymes that exhibit functional and clinical relevant genetic polymorphisms are uridine diphosphate glucuronosyltransferase (UGT), sulfotransferase (SULT), glutathione S-transferases (GST), N-acetyltransferase (NAT), and thiopurine methyltransferase (TPMT).Citation1 summarizes the most common functional polymorphisms in these Phase II enzymes and highlights their clinical significance.
Table 2 Most common naturally occurring functional polymorphisms in major human Phase II drug-metabolizing enzymes: allele frequency, functional effect, and highlights of clinical relevance
Uridine diphosphate glucuronosyltransferases (UGTs)
The human UGT superfamily is a group of conjugating enzymes that catalyze the transfer of the glucuronic acid group of uridine diphosphoglucuronic acid to the functional group (eg, hydroxyl, carboxyl, amino, sulfur) of a specific substrate.Citation6 Glucuronidation increases the polarity of the substrates and facilitates their excretion in bile or urine. UGTs are membrane-bound enzymes localized in the endoplasmic reticulum of liver and many other extrahepatic tissues. Seventeen human UGT genes have been identified thus far, and classified into two subfamilies (ie, UGT1 and UGT2). Genetic polymorphisms have been identified for almost all the UGT family members. Genetic variations in the UGT genes could alter the function or expression of the protein, and potentially modify the glucuronidation capacity of the enzyme towards a given drug, carcinogen or endogenous compounds. It is evident that genetic variations in the UGT genes contribute to differential susceptibility to diseases (eg, cancer) as well as influence the pharmacokinetics and clinical outcome of substrate drugs.Citation6,Citation7 The most common functional polymorphisms within the major UGT enzymes and their clinical relevance are summarized in . A representative example is that the UGT1A1 low promoter activity alleles (ie, UGT1A1*28) is associated with decreased glucuronidation of SN-38 (an active metabolite of irinotecan), thereby resulting in increased risk for irinotecan-induced toxicity.
Sulfotransferases (SULTs)
Cytosolic SULTs are Phase II enzymes that catalyze the transfer of the sulfonyl group from the cofactor 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to the nucleophilic sites of a variety of substrates including hormones and xenobiotics. Sulfo conjugation of xenobiotics can lead to the formation of polar, excretable products as well as reactive, potentially mutagenic and carcinogenic metabolites.Citation8 A total of 11 SULT proteins encoded by 10 genes have been identified in humans. They differ in substrate specificity and tissue distribution. Single nucleotide polymorphisms (SNPs) have been identified in most of the human SULT genes. Functional SNPs in SULTs that are associated with altered enzymatic activity have potential to influence therapeutic response and to modify cancer susceptibility.Citation8,Citation9 One widely studied functional SNP is SULT1A1*2 (Arg213His) that exhibits reduced enzymatic activity and thermal stability ().
Glutathione S-transferases (GST)
The super family of human GST catalyzes the conjugation of glutathione (GSH) to a wide range of endogenous metabolites and xenobiotics including alkylating and free radical generating anticancer drugs.Citation10 Human GSTs are categorized into three main families: cytosolic/nuclear, mitochondrial, and microsomal. The cytosolic GSTs are further divided into seven classes: alpha, mu, omega, pi, sigma, theta, and zeta. Besides their enzymatic function, GSTs also possess nonenzymatic functions, in which they act as regulators of cell signaling and post-translational modification pathway in response to stress, growth factors, and DNA damage, and in cell proliferation, cell death, and other processes that ultimately lead to tumor growth and drug resistance. These multiple functionalities establish the importance of GSTs as determinants of cancer susceptibility, therapeutic response, and prognosis.Citation10,Citation11 Most human GSTs have SNPs and, less frequently, deletions. The association of GST polymorphisms with cancer incidence, cancer treatment, and prognosis is highlighted in .
N-acetyltransferase (NAT)
The human NATs catalyze the transfer of an acetyl group from acetyl coenzyme A to arylamines, arylhydroxylamines, and arylhydrazines.Citation12 Two human NAT genes, NAT1 and NAT2, carry functional polymorphisms that influence the enzyme activity. Based on the level of NAT activity, patients can be classified into two phenotypes: fast acetylator (wild-type NAT acetylation activity) and slow acetylator (reduced NAT enzyme activity). For example, polymorphisms or haplotype in the NAT1 (ie, NAT1*14, *15, *17, *19, and *22) and NAT2 (eg, NAT2*5, *6, *7, *10, *14, and *17) lead to slow acetylation phenotype.Citation13 A comprehensive list of the NAT1/2 alleles is presented on the website http://louisville.edu/medschool/pharmacology/NAT.htlm. NAT2 plays an important role in the activation and/or deactivation of a large and diverse number of aromatic amine and hydrazine drugs used in clinic, and therefore the NAT2 genotype is particular relevant to the response to these drugs. One representative example is the association of the slow-acetylator NAT2 phenotype with increased risk for an antituberculosis drug (isoniazid)-induced hepatitis.Citation14 In addition, because NAT1 and NAT2 catalyze the bioactivation (via O-acetylation) of aromatic and hetercyclic amine carcinogens, genetic variations in the NAT1/2 genes may modify the cancer risk related to exposure to these carcinogens.Citation15 For instance, the slow-acetylator NAT2 phenotype is known to relate to a higher risk for bladder cancer.Citation16,Citation17
Thiopurine S-methyltransferase (TPMT)
TPMT is best known for its key role in the metabolism of the thiopurine drugs (eg, 6-mercaptopurine, azathiopurine, and 6-thioguanine) by catalyzing the S-methylation of thiopurine drugs via S-adenosyl-L-methionine as the S-methyl donor. These drugs are clinically used to treat cancers or as immunosuppressants. The TPMT gene exhibits significant genetic polymorphisms across all ethnic groups studied, with 18 TMPT alleles identified to date. Three main TPMT alleles, namely TPMT*2 (reduced activity), *3A (abolished activity), and *3C (reduced activity), account for 80%–95% of the intermediate and poor metabolizers.Citation18 Patients who inherit defect TPMT alleles or TPMT deficiency (ie, two nonfunctional alleles) are at significantly increased risk for thiopurine-induced toxicity (eg, myelosuppression). Indeed, patients with absent TMPT activity (∼0.3% prevalence) or low activity (∼10% prevalence) may tolerate only 5%–50% of the average mercaptopurine dose. Clinical diagnostic tests are now available for the detection of the SNPs in the human TPMT gene that lead to decreased or abolished enzyme activity. On the FDA-approved drug labels, TPMT variant pharmacogenetic test is recommended before treating patients with azathiopurine, mercaptopurine, and thioguanine.Citation19
Drug-transporter pharmacogenomics
In addition to drug metabolizing enzymes, uptake and efflux transporters that facilitate the movement of drugs in or out of the cell are important determinants of drug disposition and response. Broadly, drug transporters are classified into two families, namely efflux transporters of the adenosine triphosphate (ATP)-binding cassette (ABC) family and uptake transporters of the solute carrier (SLC) family. In the ABC transporter family, 49 genes have been identified and classified into seven subfamilies from ABCA through ABCG based on the sequence homology (http://nutrigene.4t.com/humanabc.htm). The ABC transporters are responsible for transport of diverse substrates out of the cell using ATP as an energy source. Among these, ABCB1, ABCC1/2, and ABCG2 have been well characterized for their roles in drug disposition and response. In the SLC family, 360 genes have been identified and classified into 46 subfamilies (http://www.bioparadigms.org/slc/menu.asp). Of particular relevance to drug disposition are members of the organic anion transporting polypeptides (OATP), organic cation transporter (OCT), and organic anion transporter (OAT) subfamilies.
summarizes the pharmacologically most important efflux ABC transporters (including ABCB1, ABCC1/2, and ABCG2) and uptake SLC transporters (including OATP, OCT, and OAT families), their tissue distributions, and representative drug substrates. These transporters play crucial roles in the intestinal absorption, biliary excretion, renal excretion, and tissue/cellular penetration of a wide variety of therapeutic drugs, and therefore they are important determinants of drug exposure in the system and at the site of action.Citation20 Genetic polymorphisms may influence the expression, subcellular localization, substrate specificity, and/or intrinsic transport activity of the transporter proteins and therefore, influence the disposition and response of drug substrates. The following sections serve to highlight the functional and clinical significance of the most commonly naturally occurring genetic polymorphisms within the pharmacologically most important ABC and SLC transporters with respect to drug disposition and response. A comprehensive list of genetic variants in the ABC and SLC transporters and related information are available in Pharmacogenetics Research Network databases at http://www.pharmGKB.org.
Table 3 The pharmacologically most important efflux and uptake drug transporters, tissue distribution, and representative substrate drugsTable Footnotea
ABCB1, ABCC1/2, and ABCG2 efflux transporters
ABCB1 gene
The ABCB1 gene, also named as the multidrug resistance 1 (MDR1) gene, encodes a polypeptide (P-glycoprotein) that has two halves, each containing six hydrophobic transmembrane domains and an ATP-binding domain. ABCB1, located on the apical or luminal surface of the epithelial cells, functions as an efflux transporter in restricting intestinal absorption, facilitating hepatobiliary excretion and renal excretion, and protecting the brain and fetus from xenobiotics. In addition, ABCB1 overexpression in cancer cells is implicated in multidrug resistance to chemotherapeutic agents.Citation21 ABCB1 transports a broad spectrum of structurally and functionally diverse drugs, including anticancer agents, antibiotics, immunosuppresants, cardiac drugs, calcium channel antagonists, and HIV protease inhibitors (). Of note, there is a strong overlap in substrate specificity and tissue distribution for ABCB1 and CYP3A4/5.Citation22
More than 50 SNPs have been identified in the human ABCB1 coding region. The most common SNPs are the synonymous 1236C>T and 3435C>T and the nonsynonymous 2677G>T (Ala899Ser). The allele frequencies of these three SNPs vary in different ethnic populations (). The 3435C>T SNP has strong linkage disequilibrium with other SNPs in the ABCB1 gene, creating common haplotypes consisting of 3435C>T combined with 2677G>T and/or 1236C>T.
Table 4 Most common functional polymorphisms in human ABCB1, ABCC1/2, and ABCG2: allele frequency and functional effects
Given the important role of ABCB1 in drug absorption and disposition, genetic polymorphisms in the ABCB1 gene may influence the outcome of pharmacotherapy. The first investigation of the functional and clinical effect of ABCB1 polymorphism was reported for a silent SNP 3435C>T, which was found to be associated with decreased duodenal expression of ABCB1 and thereby increased plasma concentration of digoxin after oral administration in humans.Citation23 A recent study demonstrates that the 3435C>T SNP affects the timing of cotranslational folding and insertion of ABCB1 into the membrane, thereby altering substrate specificity.Citation24 In the past decade, a number of preclinical and clinical studies have been conducted investigating the association of ABCB1 genotype with its tissue expression and function, and with pharmacokinetics and pharmacodynamics of substrates drugs ().Citation25 However, data reported on the functional and clinical impacts of ABCB1 polymorphisms are often inconsistent.Citation20,Citation25–Citation28 The desrepancies may be partly explained by lack of standardized methodology and assays among different studies. In addition, SNPs of ABCB1 may often result in very subtle functional outcomes. For example, a recent study demostrates that the haplotype 1236C>T/2677G>T/3435C>T does not change the substrate transport per se but instead affects the inhibition of transport by a small subset of modulators.Citation24 Conflicting results on the clinical impact of ABCB1 polymorphisms may reflect the complex disposition pathway of the substrate drugs. For example, the commonly used in-vivo ABCB1 probe drugs such as digoxin, fexofenadine, and talinolol were found to be the dual substrates for both ABCB1 and OATP transporters; cyclosporine is not only transported by ABCB1 but also metabolized by CYP3A4. This means that potential ABCB1 effect may be marked by the activity of OATP transporters or CYP3A4. Hence, a systemic analysis of polymorphisms in multiple genes known or suspected to contribute to drug disposition and response would provide better insights on the genetic impact on pharmacotherapy. In addition, the ABCB1 has multiple polymorphisms, some of which are in linkage disequilibrium, and therefore a haplotype approach would allow a more accurate prediction of clinical phenotypes.
ABCC1 and ABCC2
ABCC1/2, also called multidrug resistance-related proteins (MRP1/2), plays an essential role in transport and excretion of organic anions including physiological metabolites, carcinogens, and drugs. They are also believed to confer multidrug resistance to chemotherapeutic agents.Citation29 ABCC1 and ABCC2 have overlapping substrate specificities, typically glutathione, glucuronate, or sulfate conjugated and unconjugated drugs, including many anticancer agents (eg, vincristine and doxorubicin), HIV protease inhibitors (eg, ritonavir and saquinavir), and antibiotics (eg, difloxacin and grepafloxacin) (). Both ABCC1 and ABCC2 require co-transport of reduced glutathione (GSH) to transport some of their substrates.Citation30 ABCC1 is located in basolateral membranes of polarized cells, whereas ABCC2 is located to the apical domain. While ABCC1 is ubiquitously expressed, ABCC2 is mainly expressed in hepatocytes, renal proximal tubule cells, intestine, and brain ().
The human ABCC1 appears to be a conserved gene because many of the naturally occurring genetic variants in ABCC1 are relatively rare. Of the identified SNPs in the non-oding and coding region of ABCC1, 16 are known to result in amino acid changes, and some of them exhibit functional effects on either expression or function of the protein ().Citation31 Data on the role of ABCC1 polymorphisms in terms of in-vivo physiology and clinical drug resistance or toxicity are rather limited. Interestingly, one study has identified significant associations of ABCC1 2012G>T (Gly671Val) and a haplotype of ABCC2 with anthracycline-induced cardiotoxicity among non-Hodgkin lymphoma patients treated with doxorubicin.Citation32
Mutations in the ABCC2 gene have been initially identified in Dubin–Johnson Syndrome (DJS), a relatively rare recessive disorder characterized by conjugated hyperbilirubinemia resulting from loss of expression and function of ABCC2 in the liver. However, the impact of this loss of hepatic ABCC2-medicated transport on the pharmacokinetics of drug substrates in humans is unknown. Of the more commonly occurring ABCC2 SNPs, 1249G>A (Val417Ile) has been extensively studied. The effect of this SNP on ABCC2 expression varies depending on the tissue examined. For example, 1249G>A SNP was associated with lower ABCC2 mRNA and protein levels in preterm placenta, but not in duodenum and liver.Citation33,Citation34 One study demonstrated a possible association of 1249G>A variant with tenofovir-induced renal proximal tubulopathy, suggesting this SNP may influence renal excretion of some ABCC2 substrates.Citation35 In addition, 1249G>A SNP has been associated with changes in the ABCC2 localization in neuroepithelial tumors.Citation36 A number of other nonsynonymous and synonymous SNPs have been studied for their potential functional influence on the ABCC2 expression and transport activity (). It appears that ABCC2 SNPs have varied functional influence on different organs, or different substrates, or between in vitro and in vivo studies.Citation31
ABCG2
The ABCG2 (also known as BCRP, ABCP, or MXR) protein is an ABC half-transporter that bears six transmembrane domains and one ATP-binding domain. The protein actively extrudes a wide variety of chemically unrelated hydrophobic or partially hydrophobic compounds from the cells, including cytotoxic compounds (eg, mitoxantrone, topotecan, SN-38, flavopiridol, and methotrexate), fluorescent dyes (eg, Hoechst 33342), and toxic compounds found in normal food (eg, pheophorbide A) (). ABCG2 is expressed in the canalicular membrane of hepatocytes, in the epithelia of small intestine, colon, placenta, lung, kidney, adrenal and sweat glands, as well as in the endothelia of the central nerve system vasculature. It is responsible for host detoxification and protection against potentially toxic xenobiotics.Citation37–Citation39 ABCG2 transporter-mediated efflux has been increasingly recognized to not only confer drug resistance but also significantly modulate drug absorption, distribution, metabolism, and excretion.Citation40–Citation44
More than 80 polymorphisms in the ABCG2 gene have been identified in different ethnic populations.Citation45–Citation48 Several naturally occurring SNPs in ABCG2 have been found to affect the function and/or expression of its encoded protein.Citation46,Citation49–Citation51 In particular, a functional SNP in exon 5 of the ABCG2 gene, in which a C→A nucleotide transition at position 421 (ABCG2 421C>A), results in a nonsynonymous variant protein with a glutamine to lysine amino acid substitution in codon 141 (Q141K).Citation46 The ABCG2 421C>A variant has been associated with low ABCG2 expression levels and altered substrate specificity,Citation46 and has been found to alter the pharmacokinetics of diflomotecan and topotecan.Citation40,Citation52 In addition, recent studies have demonstrated that the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors such as gefitinib and erlotinib are ABCG2 substrates, and the ABCG2 421C>A variant is associated with greater gefitinib accumulation at steady-state and related to higher incidence of gefitinib-induced grade 1 or 2 diarrhea in cancer patients compared with the wild-type ABCG2.Citation53,Citation54
OATP, OCT, and OAT uptake transporters
Organic anion transporting polypeptides (OATPs)
OATPs are membrane influx transporters that facilitate cellular uptake of a wide range of endogenous compounds (eg, bile salts, hormones, and steroid conjugates) and clinically important drugs (eg, HMG-CoA-reductase inhibitors, cardiac glycosides, anticancer agents, and antibiotics) (). Of the 11 human OATP transporters, OATP1A2, OATP1B1, OATP1B3, and OATP2B1 are best characterized for their roles in drug pharmacokinetics. OATP1A2 is expressed on the luminal membrane of small intestinal enterocytes and at the blood–brain barrier and may facilitate the intestinal absorption and brain penetration of its substrates. OATP1B1, OATP1B3, and OATP2B1 are mainly expressed on the sinusoidal membrane of hepatocytes and can facilitate the hepatic uptake of their substrate drugs for further metabolism or biliary excretion.Citation55
A number of SNPs and other genetic variations have been identified in the SLCO1B1 gene (encoding OATP1B1), and their allele frequencies vary markedly across different populations ().Citation56 Some of SLCO1B1 SNPs and haplotypes have been associated with impaired transport activity in vitro towards different substrates.Citation57–Citation59 These functional impaired OATP1B1 variants may limit the uptake of the substrate drugs into the hepatocytes, thereby resulting in decreased biliary excretion or hepatic metabolism and thus increased systemic exposure. For example, a common variant allele, 521T>C, is associated with increased systemic exposure (eg, AUC) of several OATP1B1 drug substrates, including repaglinide and statins such as pravastatin.Citation60,Citation61 OATP1B1*15 (a haplotype of 388A>G and 512T>C) is associated with increased plasma concentrations of pravastatinCitation62 and increased concentrations of SN-38.Citation63,Citation64 OATP1B1*17 (a haplotype of −11187G>A, 388A>G and 512T>C) is associated with increased effect of pravastatin on rate of cholesterol synthesis. A recent genome-wide association study has demonstrated that a noncoding rs4363657 SNP, which is in nearly complete linkage disequilibrium with the SLCO1B1 521T>C SNP, is the only strong marker associated with simvastatin-induced myopathy.Citation65
Table 5 Most commonly naturally occurring nonsynonymous SNPs in genes encoding human OATP, OCT, and OAT transporters: allele frequency and functional effects
With respect to the SLCO1A2 gene (encoding OATP1A2), several nonsynonymous polymorphisms have been identified, some of which demonstrate decreased in-vitro transport activity towards OATP1A2 substrates ().Citation66 The impacts of these functional SNPs on the pharmacokinetics and clinical outcome of clinical used drugs need further studies. With respect to OATP1B1 and OATP2B1, there are few data on the clinical relevance of SLCO1B3 and SLCO2B1 polymorphisms, although some genetic variations within these two genes have been associated with altered in-vitro transport activity of the protein ().Citation67,Citation68
Organic cation transporter (OCT)
The OCTs belong to the solute carrier SLC22A family that mediate intracellular uptake of a broad range of structurally diverse small organic cations (molecular weight < 400). Three isoforms, OCT1, OCT2, and OCT3, with partially overlapping substrate spectrum, are identified in humans (). OCT1 is primarily expressed in the sinusoidal membrane of hepatocytes, whereas OCT2 is predominantly expressed in the basolateral membrane of the kidney proximal tubules; OCT3 is expressed in many tissues including placenta, heart, liver, and skeletal muscle (). The expression of OCTs was also detected in several cancer cell lines and tumor tissue samples.Citation69,Citation70
A number of nonsynonymous SNPs have been identified in the SLC22A1 (encoding OCT1) and SLC22A2 (encoding OCT2) from different ethnic groups; some of them have demonstrated altered (mostly impaired) transport function in vitro ().Citation71 With respect to the SLC22A3 (encoding OCT3), several synonymous SNPs have been identified, but their functional consequence is unknown. The functional polymorphisms in the OCT genes may influence the clinical pharmacokinetics and response of drug substrates. For example, functional polymorphisms of OCT1 and OCT2 impact the clinical effects and pharmacokinetics of metaformin, a drug used as a primary therapy for type 2 diabetes mellitus. Metaformin is eliminated predominantly by renal excretion in humans.Citation72 Because of the high expression of OCT2 in the kidney and the active renal secretion of metaformin via OCT2, defect function in OCT2 transport would result in decreased renal clearance of this drug. There is evidence that carriers of homozygous for low activity OCT2 variant 270S have a significant lower renal clearance and higher plasma concentration of metformin than those homozygous for the active variant (270A).Citation73,Citation74 On the other hand, low-function OCT1 variants including R61C, G401S, M420del, and G465R have been associated with significantly higher renal clearance of metaformin.Citation75 In addition to the effect on metformin pharmacokinetics, low-function OCT1 variants (R61C, G401S, M420del, and G465R) have been also associated with significantly decreased glucose-lowering response of metaformin in healthy volunteers probably by reducing the metaformin uptake in hepatocytes, which is the major target site of metformin’s action.Citation76
Organic anion transporter (OAT)
The OATs belong to the SLC22 family of solute carriers that mediate cellular uptake of a broad range of structurally diverse small hydrophilic organic anions. OAT substrates include many clinically important anionic drugs, such as β-lactam antibiotics, diuretics, nonsterioidal anti-inflammatory drugs, nucleoside/nucleotide antiviral drugs, and anticancer agents (). There are at least six OAT members (OAT1–6). OAT1–3 localize to the basolateral membrane of the renal proximal tubule mediating the uptake of drug substrates from blood into the proximal tubule cells, whereas OAT4 localizes to the apical side of the renal proximal tubule functioning in the secretion of drug substrates into urine. Collectively, these transporters are responsible for the movement of drug substrates from the blood to the urine. Thus, genetic variations in the genes encoding OATs may contribute to interindividual variability in the renal clearance of drug substrates. To date, a number of polymorphisms have been reported in the coding region and 5′ regulatory region of human SLC22A6 (encoding OAT1), SLC22A7 (encoding OAT2), SLC22A8 (encoding OAT3), and SLC22A11 gene (encoding OAT4) (); some of them resulted in altered in vitro transport activity of the protein.Citation77,Citation78 Nevertheless, the coding region polymorphisms in these genes are infrequent (∼1%). The regulatory region polymorphisms of these genes, particularly SLC22A8 (encoding OAT3), may be especially important in accounting for variation in the renal clearance of drug substrates.Citation78 The functional and clinical relevance of these coding and regulatory region polymorphisms need further study.
Pharmacogenomics in cancer therapy
5-fluorouracil (5-FU)
Since its introduction more than 50 years ago,Citation79 5-FU has remained the most frequently prescribed anticancer drug for the treatment of malignancies of the gastrointestinal tract including colorectal and gastric cancer. 5-FU, a fluoropyrimidine analog, is a prodrug that is converted to the active metabolite, 5-fluoro-2-deoxyuridine monophosphate (FdUMP) that leads to inhibition of thymidylate synthase (TS) and subsequently inhibition of DNA synthesis. 5-FU is converted to FdUMP through three pathways: oratate phosphoribosyltransferase (OPRT) (pathway 1), uridine phosphorylase (UP) (pathway 2), and thymidine phosphorylase (TP) (pathway 3) ().Citation80 The vast majority (∼80%–85%) of administered 5-FU is metabolized by the enzyme dihydropyrimidine dehydrogenase (DPD) in the liver into the inactive form, dihydrofluorouracil (FDHU), and excreted as a fluoro-β-alanine.
Figure 1 Pathways that affect 5-FU efficacy. Genetic polymorphisms within the genes that are involved in 5-FU metabolic activation (eg, OPRT), detoxification (eg, DPD), and target interaction (eg, TS) are important determinants of the efficacy and safety of 5-FU treatment.
Copyright © 2009. Nature Publishing Group. Adapted and reprinted with permission: Walther A, Johnstone E, Swanton C, Midgley R, Tomlinson I, Kerr D. Genetic prognostic and predictive markers in colorectal cancer. Nat Rev Cancer. 2009;9:489–499.Citation152
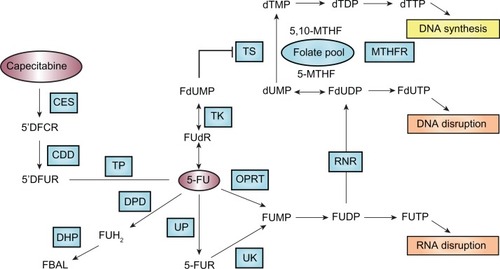
The Phase I metabolizing enzyme DPD plays the most important role in detoxification of 5-FU. Expression of DPD has been related to tolerance and response to 5-FU-based chemotherapy. Specifically, low expression or absence of DPD has been associated with accumulation of 5-FU, thereby exposing patients to increased risk of severe toxicities, while high expression of DPD has been associated with poor response to 5-FU.Citation81,Citation82 Patients lacking the DPD enzyme may experience severe to lethal toxicities when receiving 5-FU-based chemotherapy. Genetic aberration in the dihydropyrimidine dehydrogenase (DPYD) gene, such as exon skipping, deletion, and missense mutation, contributes to a DPD-deficiency phenotype. Approximately 3%–5% of the population is partially or completely deficient in DPD enzyme activity.Citation83 The most known DPYD SNPs associated with grade 3 and 4 toxicities are IVS14 + 1G>A, 2846A>T, 1679T>G, and 85T>C.Citation84 In particular, the exon 14-skipping mutation IVS14 + 1G>A, a G-to-A point mutation within the 5′-splicing site of intron 14, leads to a mutant DPD that lacks amino acids 581–635 and consequently lacks catalytic activity. The allele frequency of this mutation was 0.91% in a Dutch Caucasian population.Citation85 In patients heterozygous for the IVS14 + 1G>A allele, half of the mean normal activity of DPD is found, which is sufficient to lead to severe 5-FU toxicities. In patients homozygous for the IVS14 + 1G>A allele, DPD activity is completely lacking, and 5-FU toxicities become life-threatening and sometimes fatal.Citation86,Citation87
Orotate phosphoribosyltransferase (OPRT), an enzyme contributing to phosphorylation and activation of 5-FU (), may serve as a predictor of response to 5-FU-based chemotherapy. Overexpression and high levels of OPRT mRNA, as well as a high OPRT/DPD ratio have been associated with improved response to 5-FU-based chemotherapy in patients with metastatic colorectal cancer.Citation88 In a retrospective study examining the association of TS and OPRT genotypes with 5-FU related toxicity, co-presence of the OPRT Gly213Ala variant allele and TS 2R/2R genotype was related to grade 3 and 4 neutropenia and diarrhea.Citation89
In addition to pharmacogenetic influence of genes involved in 5-FU pharmacokinetics, genetic polymorphisms in the drug target also impact the clinical outcome of patients receiving 5-FU. 5-FU acts by inhibition of TS. TS expression varies considerably among tumors. The mechanism of the variability in TS expression is not fully understood; however, there is evidence that TS expression is modulated by three functional significant gemeline polymorphisms in the 5′ and 3′ untranslated regions (5′UTR and 3′UTR) of the TS gene. These include a polymorphic tandem repeat of a 28-base pair (bp) sequence that is present in either duplicate (2R) or in triplicate (3R) in the TS promoter enhancer region (TS 2R>3R polymorphism), a SNP (G>C) in the second repeat when three repeats are present (TS 3R G>C SNP), and a 6-bp deletion in the 3′UTR of the TS gene (TS 1494del6bp). The TS 2R>3R polymorphism is of clinical significance as patients with metastatic colorectal cancer homozygous for the triple repeat (TS 3R/3R) had significantly higher intratumoral TS gene expression.Citation90,Citation91 The 28-bp TS tandem repeats contain elements that bind upstream stimulating factor (USF) and thus act to enhance transcriptional activity of the TS gene. The presence of a G>C SNP within the second repeat of the 3R allele results in decreased transcriptional activity by abolishing the binding of USF within the repeat.Citation92 The 6-bp deletion in the 3′UTR of the TS gene could decrease TS mRNA stability, and has been associated with decreased intratumoral TS mRNA level in patients.Citation93 TS polymorphisms are not only prognostic factors of disease-free survival and overall survival but also predictors of chemotherapeutic benefit from 5-FU-based chemotherapy. In general, most studies support that patients with colorectal cancer who have a high-expression genotype (ie, TS-2R/3G, −3C/3G, −3G/3G, 3′-UTR +6bp/+6bp) showed a trend toward poor prognosis and worse response to 5-FU-based chemotherapy, but possibly less severe toxicities, compared with the low-expression group (ie, TS-2R/2R, −2R/3C, −3C/3C, 3′-UTR + 6bp/−6bp, −6bp/−bp).Citation94
In summary, genetic polymorphisms within the genes that are involved in 5-FU metabolic activation (eg, OPRT), detoxification (eg, DPD), and target interaction (eg, TS) are important determinants of the efficacy and safety of 5-FU treatment. Systemic assessment of the genotypes of these genes would allow tailoring 5-FU-based chemotherapy for individual patients.
Irinotecan
The topoisomerase I inhibitor irinotecan (CPT-11) is a water-soluble, semisynthetic derivative of camptothecin, a plant alkaloid isolated from Camptotheca acuminata (family Nyssaceae). It is widely used in the treatment of metastatic colorectal cancer, either in combination with 5-fluorouracil in the first-line treatment setting or as monotherapy in the second-line setting.Citation95 Irinotecan acts as a prodrug and undergoes complex disposition pathways in vivo (). Specifically, irinotecan is activated to 7-ethyl-10-hydroxycamptothecin (SN-38) by human carboxylesterase 1 and 2 (hCE1 and hCE2). SN-38 is subsequently detoxified by UGT1A1 to a β-glucuronide derivative, SN-38G.Citation96 In addition, irinotecan undergoes CYP3A4-mediated oxidation to form two inactive metabolites, 7-ethyl-10-(4-N-(5-aminopentanoic acid)-1-piperidino) carbonyloxycamptothecin (APC) and 7-ethyl-10-(4-amino-1-piperidino) carbonyloxycamptothecin (NPC), the latter of which also undergo a subsequent conversion by CES2 to SN-38.Citation97,Citation98 The pharmacological behavior of irinotecan can be additionally complicated by the substrate affinity of irinotecan and its metabolites (ie, SN-38 and SN-38G) for the ABC transporters including ABCB1, ABCC1/2, and ABCG2Citation99,Citation100 ().
Figure 2 Schematic illustration of irinotecan disposition pathway. Irinotecan is activated to 7-ethyl-10-hydroxycamptothecin (SN-38) by human carboxylesterase 1 and 2 (hCE1 and hCE2), and SN-38 is subsequently detoxified by UGT1A1 to a β-glucuronide derivative, SN-38G. In addition, irinotecan undergoes CYP3A4-mediated oxidation to form the inactive metabolites 7-ethyl-10-(4-N-(5-aminopentanoic acid)-1-peperidino) carbonyloxycamptothecin (APC) and 7-ethyl-10-(4-amino-1-peperidino) carbonyloxycamptothecin (NPC), and NPC also undergo a subsequent conversion by hCE2 to SN-38. Irinotecan and its metabolites (ie, SN-38 and SN-38G) are also transported by the ABC transporters including ABCB1, ABCC1/2, or ABCG2 or the organic anion transporting polypeptide 1B1 (OATP1B1).
Adapted and reprinted by permission from the American Association for Cancer Research: van Erp NP, Baker SD, Zhao M, et al. Effect of milk thistle (Silybum marianum) on the pharmacokinetics of irinotecan. Clin Cancer Res. 2005;11:7800–7806.
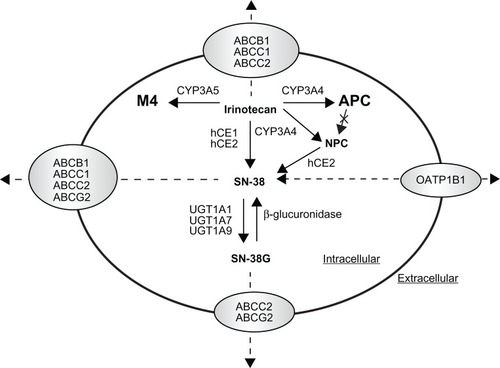
In clinical use, irinotecan exhibits substantial interindividual variability in its pharmacokinetics, efficacy, and toxicity profiles,Citation101 which is, in part, related to genetic polymorphisms in the metabolic enzymes and transporters involved in irinotecan disposition. It is evident that the genetic variant UGT1A1*28, characterized by the presence of an additional TA repeat in the TATA sequence of the UGT1A1 promoter ((TA)7TAA instead of (TA)6TAA), is associated with reduced SN-38 glucuronidation and greater susceptibility to irinotecan induced gastrointestinal and hematological toxicity.Citation102–Citation104 In addition, other polymorphisms in the UGT1A1 gene have also been associated with irinotecan-related toxicity. Of particular importance to East Asian population is UGT1A1*6 (Gly71Arg) with an allele frequency of ∼12%, which reduces UGT1A1 catalytic activity by 60% in homozygotes.Citation105 This variant allele has been related to a higher incidence of toxicity in a population of Korean patients treated with irinotecan and cisplatin for advanced nonsmall cell lung cancer.Citation106 Additionally, two promoter variants (ie, UGT1A1 −3263T>G and −3156G>A), which are in strong linkage disequilibrium with UGT1A1*28 in Caucasians while less apparent in African-Americans and Asians, have been associated with higher incidence of irinotecan-induced grade 4 neutropenia or diarrhea.Citation104,Citation107
Besides UGT1A1, associations between genetic polymorphisms within the genes encoding the drug transporters such as ABCB1, ABCC1, ABCG2, and OATP and irinotecan pharmacokinetics and/or toxicity have been reported, though the data is limited.Citation108 For ABCB1, ABCB1 3435C>T variant allele was related to higher irinotecan plasma concentration in Chinese,Citation109 and ABCB1 1236C>T variant allele resulted in a higher area under the plasma concentration-time curve (AUC) of irinotecan and SN-38 but lower AUC of SN-38G in a Caucasian population.Citation110 However, the true clinical relevance of a single SNP on ABCB1 to irinotecan treatment remains to be clarified. A haplotype approach may be more useful to estimate true genetic effects. A haplotype analysis in 49 Japanese patients indicated that the patients carrying the ABCB1*2 haplotype (containing 1236C>T, 2677G>T, and 3435C>T) exhibit significantly lower renal clearance of irinotecan, SN-38, and APC.Citation111 With respect to ABCC2, which is involved in bile excretion of irinotecan, a functional SNP ABCC2 3972C>T was found to have significant effect on the AUC of irinotecan, APC, and SN-38G, all being higher in patients carrying homozygous 3972T.Citation112 With respect to ABCG2, though irinotecan and SN-38 are good substrates of this transporter, no significant associations between ABCG2 polymorphisms and irinotecan pharmacokinetics or toxicity have been demonstrated up to now. With respect to OATP1B1 that is involved in the transport of SN-38 but not SN-38G, variants of this transporter including 521T>C, −11187G>A, 388A>G, and OATP1B1*15 haplotype have been associated with a lower clearance and higher systemic exposure of SN-38 and irinotecan.Citation63,Citation64
In addition to pharmacogenetic influence of genes involved in irinotecan pharmacokinetics, polymorphisms in the drug target of SN-38, topoisomerase I (TOP1), and cellular downstream effectors leading to DNA repair or cell death may influence patient outcomes to irinotecan treatment. A recent study in 107 advanced colorectal cancer patients showed that TOP1 and TDP1 haplotype tagging SNPs (htSNPs) were related to grade 3/4 neutropenia and response, respectively; and a DNA repair gene XRCC1 haplotype was associated with response.Citation113
Collectively, it is clear that UGT1A1*28 is associated with greater susceptibility to irinotecan induced gastrointestinal and hematological toxicity. This risk was emphasized by a warning added to the package insert of irinotecan, where a reduced initial dose is recommended for patients homozygous for the UGT1A1*28 allele. The true clinical significance of polymorphisms within other genes involved in the pharmacokinetics and pharmacodynamics of irinotecan with respect to patient outcome treated with this drug remains to be validated.
Tamoxifen
Tamoxifen, a selective estrogen receptor (ER) modulator, is a standard endocrine therapy for the treatment and prevention of ER-positive breast cancer. ER-positive breast cancers are often dependent on estrogen for growth. Selective ER modulators bind to the ligand-binding domain of an ER and block the binding of estrogen. This prevents conformational changes of the ER that it requires for its association with co-activators, thus blocking transcriptional activation functions of the ER and subsequently reducing or eliminating estrogen-driven proliferation of ER-postitive tumors.
Tamoxifen can be considered as a prodrug, which requires metabolic activation to exert its pharmacological activity. The metabolism of tamoxifen is complex and involves hepatic Phase I enzymes (including CYP3A4, CYP3A5, CYP2C9, CYP2C19, CYP1A2, CYP2B6, and CYP2D6, as well as flavin-containing monooxygenase 1 and 3) and Phase II enzymes (including SULT1A1 and UGTs) ().Citation114–Citation116 Specifically, tamoxifen is metabolized by hepatic CYP enzymes (mainly by CYP2D6 and CYP3A4/5) to form two main primary metabolites, 4-hydroxytamoxifen and N-desmethyltamoxifen. The formation of these metabolites accounts for ∼92% and ∼7% of primary tamoxifen oxidation, respectively.Citation115 Both of these metabolites are further converted to abundant and pharmacologically active 4-hydroxy-N-desmethyltamoxifen (endoxifen). Endoxifen formation from N-desmethyltamoxifen is almost exclusively catalysed by CYP2D6, and formation from 4-hydroxytamoxifen by CYP3A4/5.Citation115 In addition, tamoxifen and its metabolites undergo Phase II metabolism including sulphation and glucuronidation. Endoxifen and 4-hydroxytamoxifen show much greater affinity for the estrogen receptor than tamoxifen.Citation115–Citation117 While 4-hydroxytamoxifen and endoxifen have a similar anti-estrogen activity, endoxifen plasma concentrations are 6- to 12-fold higher than those of 4-hydroxytamoxifen,Citation118 suggesting endoxifen is the predominant and crucial active metabolite responsible for the in vivo pharmacological activity of tamoxifen.
Figure 3 Metabolism pathway of tamoxifen and its interaction with estrogen receptors.
Copyright © 2009. Nature Publishing Group. Adapted and reprinted with permission; Hoskins JM, Carey LA, McLeod HL. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat Rev Cancer 2009;9:576–586.Citation131
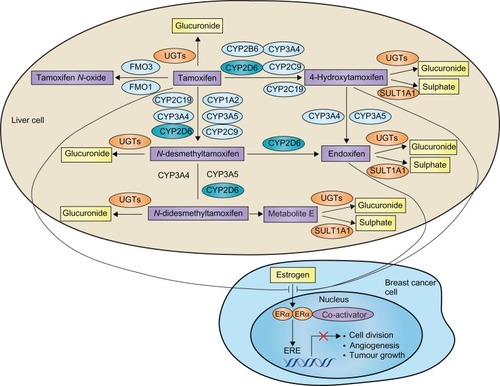
Given the key role of CYP2D6 in catalyzing the conversion of tamoxifen to its abundant active metabolite endoxifen, altered CYP2D6 activity due to either genetic or environmental (drug-induced or drug-inhibited) factor could directly affect endoxifen concentrations and likely the clinical outcome of patients treated with tamoxifen. As mentioned earlier, the functional alleles of CYP2D6 result in abolished, decreased, normal, or ultrarapid CYP2D6 enzyme activity (). As a result, patients can be classified as four phenotypes: poor metabolizer (PM) (abolished activity), intermediate metabolizer (IM) (reduced activity), extensive metabolizer (EM) (normal activity), and ultrarapid metabolizer (UM) (enhanced activity).
There is evidence that women with nonfunctional and reduced-function CYP2D6 alleles appear to have significantly lower circulating endoxifen concentrations than those with wild-type CYP2D6.Citation119,Citation120 Similarly, concomitant CYP2D6 inhibitors, such as certain selective serotonin reuptake inhibitors (SSRIs) (fluoretine and paroxetine) or selective noradrenaline reuptake inhibitors (SNRIs), have been shown to reduce the plasma concentrations of endoxifen.Citation118,Citation119 It has also been shown that use of CYP2D6 inhibitors such as SSRIs and SNRIs has a negative impact on the efficacy of tamoxifen.Citation119 Collectively, these data support the notion that low CYP2D6 activity, caused by genetic polymorphisms or drug interactions, leads to low levels of the active tamoxifen metabolite.
The effect of CYP2D6 activity on tamoxifen pharmacokinetics also translates into an effect on clinical outcome. Despite conflicting data in some instance, the majority of retrospective studies suggest that the presence of nonfunctional or reduced-function alleles of CYP2D6 is associated with worse outcome of patients receiving tamoxifen.Citation121,Citation122 Notably, a recent large retrospective analysis of 1325 patients with early-stage breast cancer treated with adjuvant tamoxifen suggests that compared with extensive metabolizer, those with decreased CYP2D6 activity (heterozygous extensive/intermedidate and poor metabolizers) have significantly increased risk of recurrence as well as worse event-free survival and disease-free survival.Citation123 In addition, CYP2D6 genotype has also been shown to influence the efficacy of tamoxifen as a chemopreventive agent, whereby tamoxifen-treated women with poor metabolizer phenotype was associated with a significantly higher incidence of breast cancer compared with controls.Citation124 Findings from these studies support a role for the CYP2D6 genotype in the activation of tamoxifen and likelihood of therapeutic benefit from testing for CYP2D6 genoptype.
Besides CYP2D6, genetic variations in genes encoding for other enzymes involved in tamoxifen metabolism as well as genes encoding for the drug target (ie, estrogen receptor) may influence the efficacy and toxicicity of tamoxifen. This may explain, at least in part, the descrepancies of results from different studies with respect to the role of CYP2D6 genotype in the clinical outcome of tamoxifen. Tamoxifen and its metabolites undergo Phase II metabolism by SULT1A1 and UGTs. Interindividual variation in the activity of Phase II enzymes may contribute to variability in circulating endoxifen levels and patient response to tamoxifen. It has been reported that women with high activity UGT2B15 genotype (UGT2B15*2/*2) had a worse recurrence-free survival than those with wild-type alleles.Citation125 Interestingly, a retrospective analysis of 337 tamoxifen-treated women with breast cancer found that those with low-activity SULT1A1 genotype (SULT1A1*2/*2) had approximately three times the risk of death as controls.Citation126 One possible explanation for this observation is that sulfation of tamoxifen metabolites (4-hydroxy-tamoxifen and endoxifen) by SULT1A1 forms highly reactive products leading to DNA adducts,Citation127,Citation128 and therefore, individuals with low-activity SULT1A1 genotype have poor clinical outcomes. In addition, genetic polymorphisms within the target genes (ESR1 and ESR2) have been associated with altered susceptibility to tamoxifen-induced hot flashes and hormone-resistance.Citation129,Citation130
In conclusion, tamoxifen can be considered as a prodrug. Its metabolism is complex and involves multiple Phase I and II enzymes. Genetic variations in these metabolizing enzymes likely contribute to the variability in tamoxifen active metabolite concentrations and patient outcome. It is generally agreed that women with reduced CYP2D6 activity genotype appear to have lower circulating endoxifen concentration and are less likely to derive therapeutic benefit from tamoxifen compared with those with normal CYP2D6 activity. However, because of the lack of concordant data, mandatory CYP2D6 genotyping test to guide the selection and dose of tamoxifen is premature. Idealy, large, prospective clinical studies should be conducted to systematically assess the impact of multiple genetic polymorphisms within multiple genes involved in the disposition and action of tamoxifen on the clinical outcome of patients receiving tamoxifen.
Conclusion
Pharmacogenomics provides a unique approach toward investigating, appreciating, and therapeutically serving the individual cancer patient. Continued investigation and adaptation of pharmacogenomics with respect to malignancy should likely provide improved risk versus benefit ratios with respect to therapeutic efficacy versus side-effect profiles. Further, effective re-evaluation of drug design toward the generation of novel and specific therapies focused on enzyme and transporter biology pertaining to malignancy may eventually be personalized and individualized to the patient for maximum efficacy.
Disclosure
The authors report no conflicts of interest in this work.
References
- EvansWERellingMVPharmacogenomics: translating functional genomics into rational therapeuticsScience199928648749110521338
- FruehFWAmurSMummaneniPPharmacogenomic biomarker information in drug labels approved by the United States food and drug administration: prevalence of related drug usePharmacotherapy20082899299818657016
- BradfordLDCYP2D6 allele frequency in European Caucasians, Asians, Africans and their descendantsPharmacogenomics2002322924311972444
- ZhouSFLiuJPChowbayBPolymorphism of human cytochrome P450 enzymes and its clinical impactDrug Metab Rev2009418929519514967
- HigashiMKVeenstraDLKondoLMAssociation between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapyJAMA20022871690169811926893
- GuillemetteCPharmacogenomics of human UDP-glucuronosyltransferase enzymesPharmacogenomics J20033313615812815363
- NagarSRemmelRPUridine diphosphoglucuronosyltransferase pharmacogenetics and cancerOncogene2006251659167216550166
- GlattHMeinlWPharmacogenetics of soluble sulfotransferases (SULTs)Naunyn Schmiedebergs Arch Pharmacol2004369556814600802
- NowellSFalanyCNPharmacogenetics of human cytosolic sulfotransferasesOncogene2006251673167816550167
- LoHWAli-OsmanFGenetic polymorphism and function of glutathione S-transferases in tumor drug resistanceCurr Opin Pharmacol2007736737417681492
- McIlwainCCTownsendDMTewKDGlutathione S-transferase polymorphisms: cancer incidence and therapyOncogene2006251639164816550164
- BlumMGrantDMMcBrideWHuman arylamine N-acetyltransferase genes: isolation, chromosomal localization, and functional expressionDNA Cell Biol199091932032340091
- SimELackNWangCJArylamine N-acetyltransferases: structural and functional implications of polymorphismsToxicology200825417018318852012
- HuangYSChernHDSuWJPolymorphism of the N-acetyltransferase 2 gene as a susceptibility risk factor for antituberculosis drug-induced hepatitisHepatology20023588388911915035
- HeinDWMolecular genetics and function of NAT1 and NAT2: role in aromatic amine metabolism and carcinogenesisMutat Res2002506–5076577
- CartwrightRAGlashanRWRogersHJRole of N-acetyltransferase phenotypes in bladder carcinogenesis: a pharmacogenetic epidemiological approach to bladder cancerLancet198228428456126711
- Garcia-ClosasMMalatsNSilvermanDNAT2 slow acetylation, GSTM1 null genotype, and risk of bladder cancer: results from the Spanish Bladder Cancer Study and meta-analysesLancet200536664965916112301
- ZhouSClinical pharmacogenomics of thiopurine S-methyltransferaseCurr Clin Pharmacol2006111912818666383
- RellingMVGardnerEESandbornWJClinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosingClin Pharmacol Ther20118938739121270794
- KerbRImplications of genetic polymorphisms in drug transporters for pharmacotherapyCancer Lett200623443316504381
- GottesmanMMFojoTBatesSEMultidrug resistance in cancer: role of ATP-dependent transportersNat Rev Cancer20022485811902585
- WacherVJWuCYBenetLZOverlapping substrate specificities and tissue distribution of cytochrome P450 3A and P-glycoprotein: implications for drug delivery and activity in cancer chemotherapyMol Carcinog1995131291347619215
- HoffmeyerSBurkOvon RichterOFunctional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivoProc Natl Acad Sci U S A2000973473347810716719
- Kimchi-SarfatyCOhJMKimIWA “silent” polymorphism in the MDR1 gene changes substrate specificityScience200731552552817185560
- IeiriITakaneHOtsuboKThe MDR1 (ABCB1) gene polymorphism and its clinical implicationsClin Pharmacokinet20044355357615217301
- Pauli-MagnusCKroetzDLFunctional implications of genetic polymorphisms in the multidrug resistance gene MDR1 (ABCB1)Pharm Res20042190491315212152
- MarzoliniCPausEBuclinTPolymorphisms in human MDR1 (P-glycoprotein): recent advances and clinical relevanceClin Pharmacol Ther200475133314749689
- SakaedaTMDR1 genotype-related pharmacokinetics: fact or fiction?Drug Metab Pharmacokinet20052039141416415525
- HinoshitaEUchiumiTTaguchiKIncreased expression of an ATP-binding cassette superfamily transporter, multidrug resistance protein 2, in human colorectal carcinomasClin Cancer Res200062401240710873092
- RothnieACallaghanRDeeleyRGRole of GSH in estrone sulfate binding and translocation by the multidrug resistance protein 1 (MRP1/ABCC1)J Biol Chem2006281139061391416565074
- GradhandUKimRBPharmacogenomics of MRP transporters (ABCC1-5) and BCRP (ABCG2)Drug Metab Rev20084031735418464048
- WojnowskiLKulleBSchirmerMNAD(P)H oxidase and multidrug resistance protein genetic polymorphisms are associated with doxorubicin-induced cardiotoxicityCirculation20051123754376216330681
- Meyer zu SchwabedissenHEJedlitschkyGGratzMVariable expression of MRP2 (ABCC2) in human placenta: influence of gestational age and cellular differentiationDrug Metab Dispos20053389690415821043
- MeierYPauli-MagnusCZangerUMInterindividual variability of canalicular ATP-binding-cassette (ABC)-transporter expression in human liverHepatology200644627416799996
- IzzedineHHulotJSVillardEAssociation between ABCC2 gene haplotypes and tenofovir-induced proximal tubulopathyJ Infect Dis20061941481149117083032
- VogelgesangSKunert-KeilCCascorbiIExpression of multi-drug transporters in dysembryoplastic neuroepithelial tumors causing intractable epilepsyClin Neuropathol20042322323115581025
- KrishnamurthyPSchuetzJDRole of ABCG2/BCRP in biology and medicineAnnu Rev Pharmacol Toxicol20064638141016402910
- JonkerJWBuitelaarMWagenaarEThe breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyriaProc Natl Acad Sci U S A200299156491565412429862
- JonkerJWSmitJWBrinkhuisRFRole of breast cancer resistance protein in the bioavailability and fetal penetration of topotecanJ Natl Cancer Inst2000921651165611036110
- SparreboomALoosWJBurgerHEffect of ABCG2 genotype on the oral bioavailability of topotecanCancer Biol Ther2005465065815908806
- BurgerHNooterKPharmacokinetic resistance to imatinib mesylate: role of the ABC drug pumps ABCG2 (BCRP) and ABCB1 (MDR1) in the oral bioavailability of imatinibCell Cycle200431502150515611623
- KondoCOnukiRKusuharaHLack of improvement of oral absorption of ME3277 by prodrug formation is ascribed to the intestinal efflux mediated by breast cancer resistant protein (BCRP/ABCG2)Pharm Res20052261361815846469
- MerinoGJonkerJWWagenaarEThe breast cancer resistance protein (BCRP/ABCG2) affects pharmacokinetics, hepatobiliary excretion, and milk secretion of the antibiotic nitrofurantoinMol Pharmacol2005671758176415709111
- HiranoMMaedaKMatsushimaSInvolvement of BCRP (ABCG2) in the biliary excretion of pitavastatinMol Pharmacol20056880080715955871
- IidaASaitoSSekineACatalog of 605 single-nucleotide polymorphisms (SNPs) among 13 genes encoding human ATP-binding cassette transporters: ABCA4, ABCA7, ABCA8, ABCD1, ABCD3, ABCD4, ABCE1, ABCF1, ABCG1, ABCG2, ABCG4, ABCG5, and ABCG8J Hum Genet20024728531012111378
- ImaiYNakaneMKageKC421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low-level drug resistanceMol Cancer Ther2002161161612479221
- ZamberCPLambaJKYasudaKNatural allelic variants of breast cancer resistance protein (BCRP) and their relationship to BCRP expression in human intestinePharmacogenetics200313192812544509
- IshikawaTTamuraASaitoHPharmacogenomics of the human ABC transporter ABCG2: from functional evaluation to drug molecular designNaturwissenschaften20059245146316160819
- MitomoHKatoRItoAA functional study on polymorphism of the ATP-binding cassette transporter ABCG2: critical role of arginine-482 in methotrexate transportBiochem J200337376777412741957
- KobayashiDIeiriIHirotaTFunctional assessment of ABCG2 (BCRP) gene polymorphisms to protein expression in human placentaDrug Metab Dispos2005339410115475413
- LepperERNooterKVerweijJMechanisms of resistance to anticancer drugs: the role of the polymorphic ABC transporters ABCB1 and ABCG2Pharmacogenomics2005611513815882131
- SparreboomAGelderblomHMarshSDiflomotecan pharmacokinetics in relation to ABCG2 421C>A genotypeClin Pharmacol Ther200476384415229462
- LiJCusatisGBrahmerJAssociation of variant ABCG2 and the pharmacokinetics of epidermal growth factor receptor tyrosine kinase inhibitors in cancer patientsCancer Biol Ther2007643243817312388
- CusatisGGregorcVLiJPharmacogenetics of ABCG2 and adverse reactions to gefitinibJ Natl Cancer Inst2006981739174217148776
- NiemiMRole of OATP transporters in the disposition of drugsPharmacogenomics2007878780218240907
- Meyer zu SchwabedissenHEKimRBHepatic OATP1B transporters and nuclear receptors PXR and CAR: interplay, regulation of drug disposition genes, and single nucleotide polymorphismsMol Pharm200961644166119558188
- KameyamaYYamashitaKKobayashiKFunctional characterization of SLCO1B1 (OATP-C) variants, SLCO1B1*5, SLCO1B1*15 and SLCO1B1*15+C1007G, by using transient expression systems of HeLa and HEK293 cellsPharmacogenet Genomics20051551352215970799
- NozawaTMinamiHSugiuraSRole of organic anion transporter OATP1B1 (OATP-C) in hepatic uptake of irinotecan and its active metabolite, 7-ethyl-10-hydroxycamptothecin: in vitro evidence and effect of single nucleotide polymorphismsDrug Metab Dispos20053343443915608127
- TironaRGLeakeBFMerinoGPolymorphisms in OATP-C: identification of multiple allelic variants associated with altered transport activity among European- and African-AmericansJ Biol Chem2001276356693567511477075
- NiemiMPasanenMKNeuvonenPJSLCO1B1 polymorphism and sex affect the pharmacokinetics of pravastatin but not fluvastatinClin Pharmacol Ther20068035636617015053
- NiemiMBackmanJTKajosaariLIPolymorphic organic anion transporting polypeptide 1B1 is a major determinant of repaglinide pharmacokineticsClin Pharmacol Ther20057746847815961978
- NishizatoYIeiriISuzukiHPolymorphisms of OATP-C (SLC21A6) and OAT3 (SLC22A8) genes: consequences for pravastatin pharmacokineticsClin Pharmacol Ther20037355456512811365
- XiangXJadaSRLiHHPharmacogenetics of SLCO1B1 gene and the impact of *1b and *15 haplotypes on irinotecan disposition in Asian cancer patientsPharmacogenet Genomics20061668369116906022
- HanJYLimHSShinESInfluence of the organic anion-transporting polypeptide 1B1 (OATP1B1) polymorphisms on irinotecan-pharmacokinetics and clinical outcome of patients with advanced non-small cell lung cancerLung Cancer200859697517766002
- LinkEParishSArmitageJSLCO1B1 variants and statin-induced myopathym – a genomewide studyN Engl J Med200835978979918650507
- FrankeRMScherkenbachLASparreboomAPharmacogenetics of the organic anion transporting polypeptide 1A2Pharmacogenomics20091033934419290786
- LetschertKKepplerDKonigJMutations in the SLCO1B3 gene affecting the substrate specificity of the hepatocellular uptake transporter OATP1B3 (OATP8)Pharmacogenetics20041444145215226676
- NozawaTNakajimaMTamaiIGenetic polymorphisms of human organic anion transporters OATP-C (SLC21A6) and OATP-B (SLC21A9): allele frequencies in the Japanese population and functional analysisJ Pharmacol Exp Ther200230280481312130747
- Hayer-ZillgenMBrussMBonischHExpression and pharmacological profile of the human organic cation transporters hOCT1, hOCT2 and hOCT3Br J Pharmacol200213682983612110607
- ZhangSLovejoyKSShimaJEOrganic cation transporters are determinants of oxaliplatin cytotoxicityCancer Res2006668847885716951202
- ChoiMKSongISOrganic cation transporters and their pharmacokinetic and pharmacodynamic consequencesDrug Metab Pharmacokinet20082324325318762711
- PentikainenPJNeuvonenPJPenttilaAPharmacokinetics of metformin after intravenous and oral administration to manEur J Clin Pharmacol197916195202499320
- SongISShinHJShimEJGenetic variants of the organic cation transporter 2 influence the disposition of metforminClin Pharmacol Ther20088455956218401339
- WangZJYinOQTomlinsonBOCT2 polymorphisms and in-vivo renal functional consequence: studies with metformin and cimetidinePharmacogenet Genomics20081863764518551044
- TzvetkovMVVormfeldeSVBalenDThe effects of genetic polymorphisms in the organic cation transporters OCT1, OCT2, and OCT3 on the renal clearance of metforminClin Pharmacol Ther20098629930619536068
- ShuYSheardownSABrownCEffect of genetic variation in the organic cation transporter 1 (OCT1) on metformin actionJ Clin Invest20071171422143117476361
- XuGBhatnagarVWenGAnalyses of coding region polymorphisms in apical and basolateral human organic anion transporter (OAT) genes [OAT1 (NKT), OAT2, OAT3, OAT4, URAT (RST)]Kidney Int2005681491149916164626
- BhatnagarVXuGHamiltonBAAnalyses of 5’ regulatory region polymorphisms in human SLC22A6 (OAT1) and SLC22A8 (OAT3)J Hum Genet20065157558016648942
- HeidelbergerCChaudhuriNKDannebergPFluorinated pyrimidines, a new class of tumour-inhibitory compoundsNature195717966366613418758
- OmuraKClinical implications of dihydropyrimidine dehydrogenase (DPD) activity in 5-FU-based chemotherapy: mutations in the DPD gene, and DPD inhibitory fluoropyrimidinesInt J Clin Oncol2003813213812851836
- SoongRShahNSalto-TellezMPrognostic significance of thymidylate synthase, dihydropyrimidine dehydrogenase and thymidine phosphorylase protein expression in colorectal cancer patients treated with or without 5-fluorouracil-based chemotherapyAnn Oncol20081991591918245778
- SalongaDDanenbergKDJohnsonMColorectal tumors responding to 5-fluorouracil have low gene expression levels of dihydropyrimidine dehydrogenase, thymidylate synthase, and thymidine phosphorylaseClin Cancer Res200061322132710778957
- LuZZhangRDiasioRBDihydropyrimidine dehydrogenase activity in human peripheral blood mononuclear cells and liver: population characteristics, newly identified deficient patients, and clinical implication in 5-fluorouracil chemotherapyCancer Res199353543354388221682
- MorelABoisdron-CelleMFeyLClinical relevance of different dihydropyrimidine dehydrogenase gene single nucleotide polymorphisms on 5-fluorouracil toleranceMol Cancer Ther200652895290417121937
- VrekenPVan KuilenburgABMeinsmaRA point mutation in an invariant splice donor site leads to exon skipping in two unrelated Dutch patients with dihydropyrimidine dehydrogenase deficiencyJ Inherit Metab Dis1996196456548892022
- WeiXMcLeodHLMcMurroughJMolecular basis of the human dihydropyrimidine dehydrogenase deficiency and 5-fluorouracil toxicityJ Clin Invest1996986106158698850
- Van KuilenburgABVrekenPBeexLVHeterozygosity for a point mutation in an invariant splice donor site of dihydropyrimidine dehydrogenase and severe 5-fluorouracil related toxicityEur J Cancer199733225822649470816
- IchikawaWUetakeHShirotaYBoth gene expression for orotate phosphoribosyltransferase and its ratio to dihydropyrimidine dehydrogenase influence outcome following fluoropyrimidine-based chemotherapy for metastatic colorectal cancerBr J Cancer2003891486149214562021
- IchikawaWTakahashiTSutoKOrotate phosphoribosyltransferase gene polymorphism predicts toxicity in patients treated with bolus 5-fluorouracil regimenClin Cancer Res2006123928393416818689
- PullarkatSTStoehlmacherJGhaderiVThymidylate synthase gene polymorphism determines response and toxicity of 5-FU chemotherapyPharmacogenomics J20011657011913730
- KawakamiKOmuraKKanehiraEPolymorphic tandem repeats in the thymidylate synthase gene is associated with its protein expression in human gastrointestinal cancersAnticancer Res1999193249325210652619
- MandolaMVStoehlmacherJMuller-WeeksSA novel single nucleotide polymorphism within the 5’ tandem repeat polymorphism of the thymidylate synthase gene abolishes USF-1 binding and alters transcriptional activityCancer Res2003632898290412782596
- MandolaMVStoehlmacherJZhangWA 6 bp polymorphism in the thymidylate synthase gene causes message instability and is associated with decreased intratumoral TS mRNA levelsPharmacogenetics20041431932715115918
- LurjeGManegoldPCNingYPohlAZhangWLenzHJThymidylate synthase gene variations: predictive and prognostic markersMol Cancer Ther2009851000100719383851
- SaltzLBCoxJVBlankeCIrinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. Irinotecan Study GroupN Engl J Med200034390591411006366
- SlatterJGSuPSamsJPBioactivation of the anticancer agent CPT-11 to SN-38 by human hepatic microsomal carboxylesterases and the in vitro assessment of potential drug interactionsDrug Metab Dispos199725115711649321519
- RivoryLPRiouJFHaazMCIdentification and properties of a major plasma metabolite of irinotecan (CPT-11) isolated from the plasma of patientsCancer Res199656368936948706009
- DoddsHMHaazMCRiouJFIdentification of a new metabolite of CPT-11 (irinotecan): pharmacological properties and activation to SN-38J Pharmacol Exp Ther19982865785839655905
- LuoFRParanjpePVGuoAIntestinal transport of irinotecan in Caco-2 cells and MDCK II cells overexpressing efflux transporters Pgp, cMOAT, and MRP1Drug Metab Dispos20023076377012065434
- SchellensJHMaliepaardMScheperRJTransport of topoisomerase I inhibitors by the breast cancer resistance protein. Potential clinical implicationsAnn NY Acad Sci200092218819411193894
- MathijssenRHvan AlphenRJVerweijJClinical pharmacokinetics and metabolism of irinotecan (CPT-11)Clin Cancer Res200172182219411489791
- TukeyRHStrassburgCPMackenziePIPharmacogenomics of human UDP-glucuronosyltransferases and irinotecan toxicityMol Pharmacol20026244645012181419
- IyerLDasSJanischLUGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicityPharmacogenomics J20022434711990381
- InnocentiFUndeviaSDIyerLGenetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecanJ Clin Oncol2004221382138815007088
- PremawardhenaAFisherCALiuYTThe global distribution of length polymorphisms of the promoters of the glucuronosyltransferase 1 gene (UGT1A1): hematologic and evolutionary implicationsBlood Cells Mol Dis2003319810112850492
- HanJYLimHSShinESComprehensive analysis of UGT1A polymorphisms predictive for pharmacokinetics and treatment outcome in patients with non-small-cell lung cancer treated with irinotecan and cisplatinJ Clin Oncol2006242237224416636344
- KitagawaCAndoMAndoYGenetic polymorphism in the phenobarbital-responsive enhancer module of the UDP-glucuronosyltransferase 1A1 gene and irinotecan toxicityPharmacogenet Genomics200515354115864124
- De JongFAde JongeMJVerweijJRole of pharmacogenetics in irinotecan therapyCancer Lett20062349010616343744
- ZhouQSparreboomATanEHPharmacogenetic profiling across the irinotecan pathway in Asian patients with cancerBr J Clin Pharmacol20055941542415801936
- MathijssenRHMarshSKarlssonMOIrinotecan pathway genotype analysis to predict pharmacokineticsClin Cancer Res200393246325312960109
- SaiKKaniwaNItodaMHaplotype analysis of ABCB1/MDR1 blocks in a Japanese population reveals genotype-dependent renal clearance of irinotecanPharmacogenetics20031374175714646693
- KimRBMDR1 single nucleotide polymorphisms: multiplicity of haplotypes and functional consequencesPharmacogenetics20021242542712172210
- HoskinsJMMarcuelloEAltesAIrinotecan pharmacogenetics: influence of pharmacodynamic genesClin Cancer Res2008141788179618347181
- DehalSSKupferDCYP2D6 catalyzes tamoxifen 4-hydroxylation in human liverCancer Res199757340234069270005
- DestaZWardBASoukhovaNVComprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: prominent roles for CYP3A and CYP2D6J Pharmacol Exp Ther20043101062107515159443
- PartePKupferDOxidation of tamoxifen by human flavin-containing monooxygenase (FMO) 1 and FMO3 to tamoxifen-N-oxide and its novel reduction back to tamoxifen by human cytochromes P450 and hemoglobinDrug Metab Dispos2005331446145215987777
- CreweHKNotleyLMWunschRMMetabolism of tamoxifen by recombinant human cytochrome P450 enzymes: formation of the 4-hydroxy, 4′-hydroxy and N-desmethyl metabolites and isomerization of trans-4-hydroxytamoxifenDrug Metab Dispos20023086987412124303
- StearnsVJohnsonMDRaeJMActive tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetineJ Natl Cancer Inst2003951758176414652237
- JinYDestaZStearnsVCYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatmentJ Natl Cancer Inst200597303915632378
- LimHSJu LeeHSeok LeeKClinical implications of CYP2D6 genotypes predictive of tamoxifen pharmacokinetics in metastatic breast cancerJ Clin Oncol2007253837384517761971
- HoskinsJMCareyLAMcLeodHLCYP2D6 and tamoxifen: DNA matters in breast cancerNat Rev Cancer2009957658619629072
- HigginsMJStearnsVCYP2D6 polymorphisms and tamoxifen metabolism: clinical relevanceCurr Oncol Rep20101271520425602
- SchrothWGoetzMPHamannUAssociation between CYP2D6 polymorphisms and outcomes among women with early stage breast cancer treated with tamoxifenJAMA20093021429143619809024
- BonanniBMacisDMaisonneuvePPolymorphism in the CYP2D6 tamoxifen-metabolizing gene influences clinical effect but not hot flashes: data from the Italian Tamoxifen TrialJ Clin Oncol20062437083709 author reply370916877740
- NowellSAAhnJRaeJMAssociation of genetic variation in tamoxifen-metabolizing enzymes with overall survival and recurrence of disease in breast cancer patientsBreast Cancer Res Treat20059124925815952058
- NowellSSweeneyCWintersMAssociation between sulfotransferase 1A1 genotype and survival of breast cancer patients receiving tamoxifen therapyJ Natl Cancer Inst2002941635164012419790
- DavisWVenittSPhillipsDHThe metabolic activation of tamoxifen and alpha-hydroxytamoxifen to DNA-binding species in rat hepatocytes proceeds via sulphationCarcinogenesis1998198618669635875
- DasaradhiLShibutaniSIdentification of tamoxifen-DNA adducts formed by alpha-sulfate tamoxifen and alpha-acetoxytamoxifenChem Res Toxicol1997101891969049430
- JinYHayesDFLiLEstrogen receptor genotypes influence hot flash prevalence and composite score before and after tamoxifen therapyJ Clin Oncol2008265849585419018086
- ZhangQXBorgAWolfDMAn estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancerCancer Res199757124412499102207
- SolusJFAriettaBJHarrisJRGenetic variation in eleven phase I drug metabolism genes in an ethnically diverse populationPharmacogenomics2004589593115469410
- MizutaniTPM frequencies of major CYPs in Asians and CaucasiansDrug Metab Rev2003359910612959412
- XieHGKimRBWoodAJMolecular basis of ethnic differences in drug disposition and responseAnnu Rev Pharmacol Toxicol20014181585011264478
- Suarez-KurtzGPharmacogenomics in admixed populationsTrends Pharmacol Sci20052619620115808344
- SistonenJSajantilaALaoOCYP2D6 worldwide genetic variation shows high frequency of altered activity variants and no continental structurePharmacogenet Genomics2007179310117301689
- RoyJNLajoieJZijenahLSCYP3A5 genetic polymorphisms in different ethnic populationsDrug Metab Dispos20053388488715833928
- BozinaNBradamanteVLovricMGenetic polymorphism of metabolic enzymes P450 (CYP) as a susceptibility factor for drug response, toxicity, and cancer riskArh Hig Rada Toksikol20096021724219581216
- Ingelman-SundbergMPharmacogenetics of cytochrome P450 and its applications in drug therapy: the past, present and futureTrends Pharmacol Sci20042519320015063083
- AgundezJAPolymorphisms of human N-acetyltransferases and cancer riskCurr Drug Metab2008952053118680472
- HamdySIHiratsukaMNaraharaKGenotype and allele frequencies of TPMT, NAT2, GST, SULT1A1 and MDR-1 in the Egyptian populationBr J Clin Pharmacol20035556056912814450
- FungKLGottesmanMMA synonymous polymorphism in a common MDR1 (ABCB1) haplotype shapes protein functionBiochim Biophys Acta2009179486087119285158
- LeslieEMLetourneauIJDeeleyRGFunctional and structural consequences of cysteine substitutions in the NH2 proximal region of the human multidrug resistance protein 1 (MRP1/ABCC1)Biochemistry2003425214522412731862
- ConradSKauffmannHMItoKA naturally occurring mutation in MRP1 results in a selective decrease in organic anion transport and in increased doxorubicin resistancePharmacogenetics20021232133012042670
- HulotJSVillardEMaguyAA mutation in the drug transporter gene ABCC2 associated with impaired methotrexate eliminationPharmacogenet Genomics20051527728515864128
- MizuaraiSAozasaNKotaniHSingle nucleotide polymorphisms result in impaired membrane localization and reduced atpase activity in multidrug transporter ABCG2Int J Cancer200410923824614750175
- TamuraAWakabayashiKOnishiYRe-evaluation and functional classification of non-synonymous single nucleotide polymorphisms of the human ATP-binding cassette transporter ABCG2Cancer Sci20079823123917297656
- TamuraAWatanabeMSaitoHFunctional validation of the genetic polymorphisms of human ATP-binding cassette (ABC) transporter ABCG2: identification of alleles that are defective in porphyrin transportMol Pharmacol20067028729616608919
- KonigJSeithelAGradhandUPharmacogenomics of human OATP transportersNaunyn Schmiedebergs Arch Pharmacol200637243244316525793
- ZairZMElorantaJJStiegerBPharmacogenetics of OATP (SLC21/SLCO), OAT and OCT (SLC22) and PEPT (SLC15) transporters in the intestine, liver and kidneyPharmacogenomics2008959762418466105
- MarzoliniCTironaRGKimRBPharmacogenomics of the OATP and OAT familiesPharmacogenomics2004527328215102542
- ZhouFYouGMolecular insights into the structure-function relationship of organic anion transporters OATsPharm Res200724283617103332
- WaltherAJohnstoneESwantonCMidgleyRTomlinsonIKerrDGenetic prognostic and predictive markers in colorectal cancerNat Rev Cancer2009948949919536109
- van ErpNPBakerSDZhaoMEffect of milk thistle (Silybum marianum) on the pharmacokinetics of irinotecanClin Cancer Res2005117800780616278402