Abstract
Targeted therapies for cancer bring the hope of specific treatment, providing high efficacy and in some cases lower toxicity than conventional treatment. Although targeted therapeutics have helped immensely in the treatment of several cancers, like chronic myelogenous leukemia, colon cancer, and breast cancer, the benefit of these agents in the treatment of lung cancer remains limited, in part due to the development of drug resistance. In this review, we discuss the mechanisms of drug resistance and the current strategies used to treat lung cancer. A better understanding of these drug-resistance mechanisms could potentially benefit from the development of a more robust personalized medicine approach for the treatment of lung cancer.
Introduction
Lung cancer is the leading cause of cancer-related deaths in the US, with a mortality rate that is nearly twice as large as its closest follower in both men (prostate) and women (breast).Citation1 Based on histology, lung cancers are classified into two major classes: small-cell lung cancer (SCLC), comprising 15% of cases, and non-small-cell lung cancer (NSCLC), which comprise 85% of cases. While cigarette smokers constitute the main population at risk for developing lung cancer, the fastest-growing demographic currently is in nonsmoking women between the ages of 30 and 50.Citation2,Citation3 Unfortunately, due to the unavailability of early diagnostic tools, disease in two-thirds of these patients is not diagnosed until a later stage, leaving surgery as a nonviable course of action. Despite decades of research, the treatment options for lung cancer patients remain insufficient, either to provide a cure or even an appreciable survival benefit. The average 5-year survival rate has not improved greatly over the last 40 years, being cited currently at a mere 17%,Citation4 and highlighting the need for improved or novel therapeutic options.
The first major advancement in the treatment of lung cancer, however, came with the introduction of platinum-based chemotherapeutics, specifically cisplatin and carboplatin. The therapeutic use of platinum-based chemotherapies together with other agents such as gemcitabine, docetaxel, vinorelbine, and pemetrexed increased the 5-year survival rate from 5% to 14%.Citation5 However, even with various combinations of these drugs, it soon became clear that the usefulness of chemotherapy in the treatment of lung cancer had reached its limit. Despite this shortcoming, chemotherapy was still the best course of action until the approach to cancer treatment changed drastically with the observation of “oncogene addiction.” This phenomenon describes when the loss of even a single mutated protein of which the cells have come to rely on can induce massive cell death and prevent disease progression.Citation6 This new idea of targeting specific proteins opened the possibility of honing treatments, specific to the diseased cells, lessening the deleterious side effects of traditional treatments. This new therapeutic direction, initially championed with the development of imatinib,Citation7 an Abelson murine leukemia viral oncogene homolog 1 kinase inhibitor, created a new frontier in the battle against many types of cancer. To date, a few of these targetable mutations have been identified in lung cancer. Some therapies designed to exploit these mutations have shown promise, both as single-line treatments and in combination with the standard platinum-based chemotherapies. In this context, receptor tyrosine kinase inhibitors (TKIs), which target epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK), have shown great promise in tailoring treatments to common kinase mutations found in NSCLC.
Unfortunately, despite the advances provided by these drugs, even the “addicted” cancers have a high incidence of relapse due to the development of resistance, limiting the “success” of these drugs in prolonging the median survival times by only a few months. Therefore, the need for focused research to identify new drugs and/or testing the existing drug combinations to mitigate drug-resistance mechanisms is critical to any future success in the field of lung cancer therapy. In this review, we highlight the current targeted therapies in use, as well as those under development for the treatment of NSCLC. In addition, we describe the mechanisms by which these therapies work, as well as why they also frequently fail.
Current strategies used to treat lung cancer
Receptor tyrosine kinase inhibitors in current clinical use
Epidermal growth-factor receptor
EGFR is a receptor tyrosine kinase that is expressed in 60% of NSCLC. The binding of growth-factor ligands to EGFR initiates cell-signaling events activating the Phosphatidylinositide 3-kinase (PI3K)/Akt (involved in survival signaling) and the mitogen-activated protein kinases (MAPKs/ERK, involved in proliferation) pathways.Citation8 The desire to inhibit the proliferative activity of EGFR in cancers led to the development of TKIs specific to EGFR. These TKIs function by binding to the adenosine triphosphate (ATP) pocket of the kinase, thereby preventing the receptor from activating its downstream-signaling cascades. In the case of NSCLC, two such drugs gained US Food and Drug Administration approval in 2004: gefitinibCitation9 and erlotinib.Citation10 However, in trials performed just after being put on the market, gefitinib showed no survival advantage, showing a median survival of 5.6 months for gefitinib and 5.1 months on placebo (with a hazard ratio [HR] of 0.89).Citation11 By comparison, erlotinib only improved median progression-free survival by 0.4 months compared to placebo (HR = 0.61), and overall survival only improved by 2 months.Citation12 In the following years, response to gefitinib was found to occur in only about 10% of cases in North American and European unselected populations (with slightly higher response rates in female Asian nonsmokers with adenocarcinoma), which corresponded with the presence of an activating mutation, but not with expression levels of EGFR. Thus far, the mutations correlating with TKI sensitivity are encoded within the tyrosine kinase domain of EGFR: in exons 18, 19, or 21, and often close to the ATP-binding pocket, gefitinib’s target site. An extremely common mutation is on lysine 858, which is usually mutated to either arginine (41% of EGFR-activating mutationsCitation13), or more rarely to a methionine. Other identified mutations include the G719C substitution and several other deletion mutations, occurring between codons 746–750, 747–749, 747–751, 747–753, and 752–759, all of which were observed to be heterozygous.Citation14–Citation17 Erlotinib is believed to have similar efficacy to gefitinib, though this might be due to a lack of direct comparison in trials to highlight differences.Citation18 Additionally, it has been shown that the type of mutation, in many cases, dictates the degree of sensitivity to treatment.Citation19 Unfortunately, despite any initial response in slowing disease progression, patients with EGFR-mutant NSCLC will develop progression of disease on TKI therapy after 10–16 months, resulting in these drugs only improving the median survival time by several weeks.Citation20–Citation22 This is the major drawback with existing therapy, which has driven research to further explore the mechanisms of acquired resistance.
Resistance to EGFR TKIs
The molecular mechanisms in acquiring resistance to EGFR TKIs are numerous. Often a secondary mutation, substituting threonine with a methionine at position 790 results in an increased affinity for ATP.Citation23 This increases competition for binding, rendering the drugs ineffective.Citation24 This mutation is seen in about 50% of cases with acquired resistance.Citation25,Citation26 Several other similar mutations in the kinase domain have been shown to cause resistance, including D761Y, L747S, and T854A.Citation27 Another mechanism is via the amplification of another receptor that signals to similar downstream mediators like that of EGFR. A fairly common example is the upregulation of Met, which can also activate the downstream PI3K/Akt pathway without requiring EGFR.Citation28–Citation30 Along similar lines, the upregulation of the ligand hepatocyte growth factor can increase Met activity. While these mechanisms are common, a number of cases have been observed where resistance appears through alternative means. These include the downregulation of repressors, such as phosphatase and tensin homologue, or even activation of completely different pathways, such as turning on the nuclear factor kappa-light-chain enhancer of activated B cells transcription factor.Citation27,Citation31 An additional mechanism for the development of resistance is via the epithelial–mesenchymal transition (EMT).Citation32 This describes alterations in a cell where it loses the adherence-junction protein E-cadherin, becomes more mesenchymal in morphology, and with increased motility. While EMT is a natural event during development, its aberrant occurrence makes it an important mediator of cell invasion and metastasis. Additionally, cells that have undergone EMT also show development of drug resistance to TKIs.Citation32 Specifically, a more recent work has identified an E-cadherin repressor, Slug, as a critical mediator of EMT and as a mediator of resistance to gefitinib in NSCLC cell lines.Citation33,Citation34 Therefore, inhibition of Slug might as well represent a possible mechanism for resensitization of TKI resistant EGFR-mediated cancers.
While these mechanisms describe acquired resistance, cancer cells can also develop resistance to a drug without ever being exposed to it. Recently it has been observed that a polymorphism in BCL2-like 11, a proapoptotic protein, inhibits normal cell-death mechanisms and provides an intrinsic resistance to TKI therapy even in the presence of an activating mutation.Citation35 Another typical example of de novo resistance is the mutation of Kirsten rat sarcoma viral oncogene homolog (KRAS). As previously stated, EGFR signaling could take two paths: the first being the PI3K/Akt pathway that supports survival, and the other being the MAPK/ERK pathway, which supports proliferation. KRAS, a key component of both of these pathways, is mutated in ~30% of NSCLC. Furthermore, cancers with this mutation do not respond to either gefitinib or erlotinib.Citation36 In fact, the utility of KRAS mutations as a biomarker for predicting resistance to TKI therapy was also investigated.Citation37 It has been shown that mutations in EGFR and KRAS are usually mutually exclusive,Citation25,Citation38–Citation40 making KRAS an independent therapeutic target. While direct attempts of drugging KRAS via farnesyl transferase inhibitors failed to show efficacy in NSCLC,Citation41 treatment of adenocarcinomas, which are also KRAS-driven in mouse models, with the combination of a PI3K inhibitor (NVP-BEZ235) and an MAPK kinase inhibitor (ARRY-142886) showed some promise.Citation42 Indeed, a very recent study using docetaxel in combination with either placebo or selumetinib (AZD6244, ARRY-142866) in patients with a KRAS mutation showed a 3.2 month increase in progression free survival (HR = 0.58).Citation43 This study provides some hope that targeting the MAPK pathway may also be an effective treatment for KRAS-mutant lung cancer patients in the future.
While TKIs inactivate receptor activity by binding to the intracellular kinase domain, another drug, cetuximab, a monoclonal antibody, inactivates EGFR activity by binding to the extracellular domain of EGFR. Cetuximab has been commonly used in the treatment of metastatic colorectal cancer,Citation44 and while it is not currently FDA-approved for single-line treatment in lung cancer, early phase II trials showed a 1-month improvement of median survival time when combined with cisplatin and/or vinorelbine in NSCLC (HR = 0.71).Citation45 In a phase III trial, cetuximab was able to increase overall survival by 1.2 months (HR = 0.871).Citation46 However, a recent study combining cetuximab, carboplatin, and paclitaxel closed due to excessive high-grade toxicity events.Citation47 Another trial compared the effects of cetuximab and radiotherapy in combination with either cisplatin and vinorelbine, or cisplatin and etoposide, and found a progression-free survival rate at 57% and 43% for each arm, respectively.Citation48 Despite the fact that the median survival time in several studies only increased by a few months,Citation49 the addition of cetuximab to certain combinations of conventional chemotherapy has been generally well tolerated, and continues to be investigated for use as a first-line treatment option for advanced cases of NSCLC.Citation50
Anaplastic lymphoma kinase
Identified as a transforming mutation in 2007,Citation51 the ALK fusion protein is a result of a chromosomal inversion, which commonly results in a fusion with echinoderm microtubule-associated protein-like 4 (EML4). Initially observed in 6.7% of Japanese patients, this mutation appears even less frequently in Koreans, African Americans, and North Americans (a mere 3%). Early studies found that in a fraction of the cases with mutations in ALK4, ALK inhibitors may serve as a potential therapy.Citation52 An early phase I trial of crizotinib not only showed extremely mild toxicity in treated patients but also a response rate of 57% and 6-month progression-free survival in 72% of their patients.Citation53 Indeed, the ALK/Met inhibitor crizotinib was conceived, tested, and finally approved by the FDA for use in NSCLC in only 4 years, and continues to show impressive results in clinical trials.Citation54–Citation56 This success story is extremely encouraging, especially compared with the early studies showing lack of efficacy with the EGFR TKIs. It is also important to highlight that in the case of gefitinib and erlotinib, the early studies that often failed to show response were evaluated in unselected populations. The phase I trial described for crizotinib showing exceedingly impressive results, and receiving partial credit for the extremely fast approval of the drug, specifically screened on their test population. Starting with 1500 patients, they narrowed down their cohort to the 82 people who actually possessed an ALK rearrangement, which resulted in a real demonstration of the drug’s efficacy, and highlights the need for analysis of each and every patient.
In addition to fusing with ELM4, ALK has been shown to fuse with KIF5B,Citation57 KLC1, and is suspected to fuse with TFG, though the TFG-TRK-fused gene fusion partner still requires confirmation.Citation58 While the effects of different fusion partners remain to be identified, all fusions result in the activation of ALK kinase activity.
Resistance to ALK TKIs
Similar to the cancers with EGFR mutations, acquired resistance (as well as an unfortunate side effect of hypogonadism and slight risk of hepatotoxicityCitation59,Citation60) is also a problem for patients with ALK4 mutations treated with crizotinib. A wide range of secondary mutations in the kinase domain, similar to the T790M mutations in EGFR, has been identified.Citation61 These include mutations such as L1196M, S1206Y, G1202R, G1269A, D1203N, C1156Y, and L1152R.Citation62–Citation64 It is hypothesized that due to the fact that ALK kinase is activated by a translocation and not by a mutation within the gene, the larger range of mutations identified in the kinase domain might not affect its function.Citation64 Alternative mechanisms of resistance include an increase in copy number and the activation of different pathways via receptor tyrosine kinase activation, such as Kit and EGFR.Citation62,Citation63
There are a few strategies being employed to counteract resistance. Inhibitors directed against heat-shock protein 90 (HSP90), which forms a chaperone complex with EML4–ALK, are effective in interrupting the fusion protein’s function. Early in vitro work and clinical trials have shown promise for HSP90 inhibition in treating lung cancers with this genotype.Citation65,Citation66 Very recently, a phase II trial was conducted for the HSP90 inhibitor AUY922 in several different mutation classes of NSCLC. Though the trial is ongoing, 25% of the ALK mutated patients have shown a partial response to treatment, though responders seem to be restricted to the patients with no prior treatment with crizotinib.Citation67 However, its efficacy in cases that have already developed resistance to targeted ALK inhibitors remains to be conclusively shown.Citation64,Citation68 Also, there are several different kinds of ALK fusion proteins, some of which have de novo resistance to crizotinib. This, in addition to the prospect of regaining sensitivity after the development of crizotinib resistance, has sparked the development of several other ALK inhibitors targeting different sites on the kinase. Early results show that in some cases (for example the L1196M, F1174L, or G1269S mutations) an alternative ALK inhibitor may able to produce/reproduce sensitivity in NSCLC.Citation64,Citation69
Strategies in development for lung cancer treatment
In the search for more effective treatment for lung cancer, several genes and cellular processes were probed to identify drug targets. These include EGFR and ALK signaling, vascular endothelial growth factor (VEGF) receptors (ie, receptor tyrosine kinases involved in angiogenesis), matrix metalloproteases (which are implicated in metastasis), and insulin-like growth-factor receptors (IGFRs).Citation41 In the following section, we describe the mechanisms and their therapeutic targets that are still under investigation (summarized in ).
Table 1 Summary of the current therapies, their targets, and their function
Angiogenesis inhibitors
The ability of a cancer to develop its own blood supply through angiogenesis is an incredibly important process, and designated as one of the “hallmarks” of cancer.Citation70 Without angiogenesis, a tumor could not grow larger than a few millimeters, making it an attractive target for therapeutic gain.Citation71 Several angiogenesis inhibitors, including SU5416 and thalidomide, have already failed in clinical trials, due to an unacceptable risk or pure lack of benefit.Citation41,Citation72,Citation73 However, a recombinant humanized monoclonal antibody against the VEGF receptor, called bevacizumab, showed some promise, and has been included in phase II and III trials for various metastatic cancers in the last decade,Citation58,Citation74,Citation75 specifically for metastatic NSCLC since 2005.Citation76,Citation77 Several studies have seen increased overall survival times when combined with chemotherapy, though these studies did not account for any additional lines of therapy that may have contributed to the health of the patients.Citation78 While high toxicity occurred in squamous carcinomas, adenocarcinomas tended to respond with minimal adverse side affects.Citation78 A recent phase II single-arm trial found that a combination of bevacizumab and platinum-based chemotherapy followed by maintenance therapy was able to extend progression-free survival to 7.1 months, and median survival time to 17.1 months in 85% of the patients.Citation79 While this is still an improvement over the existing therapies, a major disadvantage is that this therapy does not seem to render any benefit to patients over the age of 65 years, which is the main demographic group for lung cancer.Citation1,Citation80,Citation81 However, recent studies have shown that in a small cohort of non-squamous NSCLC selected patients (at an average age of 65 years), bevacizumab in addition to docetaxel as a second-line or above therapy was able to produce a median progression-free survival time of 7.9 months and control disease in 95% of the patients.Citation82 This warrants further investigation as to the usefulness of bevacizumab in this age-group. Along a parallel path, several other TKIs directed against angiogenesis, such as sunitinib, sorafenib, and linifanib, are also in clinical trials. While they are intended to inhibit VEGF, they are nonspecific, and therefore could also inhibit other kinases, such as platelet-derived growth factor receptor and Kit.Citation83 Early phase II trials showed some positive activity against recurrent NSCLC, though the benefit gained by these agents was only in the order of months.Citation84,Citation85 A recent phase II trial tested the addition of linifanib or placebo to a combination of carboplatin and paclitaxel therapy for non-squamous NSCLC. At lower doses of linifanib, progression-free survival was improved by 2.9 months (HR = 0.51); however, overall survival was not greatly improved.Citation86 This is contradictory to a phase III trial of sorafenib, which seemed to show no benefit.Citation87 However, a recurrent explanation provided in the literature for the failure of the trials is due to the lack of patient selection by biomarkers that would respond well to anti-VEGF agents.Citation88,Citation89
Histone deacetylase inhibitor
Recently there has been some investigation into drugs that can inhibit histone deacetylases (HDACs) as therapeutics. HDACs are responsible for removing acetyl groups from histones. This helps to condense chromatin, and in the case of cancer often shuts down crucial genes involved in proliferative regulation.Citation90,Citation91 In a phase I trial, treating a small cohort (n = 19) with an HDAC inhibitor, vorinostat, produced response in 53% of patients, compared to the 20%–30% response rate of chemotherapy. Following up with in vitro testing of the effects of vorinostat with carboplatin and paclitaxel showed a synergistic benefit.Citation91 Another in vitro study found that the effects of vorinostat on cancer cells could be due in part to its effects on telomerases, a protein required to maintain the immortality of cells.Citation92 While it is still too early to tell how quickly resistance might be acquired, and the mechanisms thereof, the results of future phase II and III trials are eagerly awaited.
Insulin-like growth-factor 1 receptor inhibitor (IGF-1R)
IGF-1R is yet another TKI that can initiate proliferative as well as antiapoptotic signaling in the cell. The finding that IGF-1R is overexpressed in a wide range of cancers made it an interesting target.Citation93 A drug designed to inhibit IGF-IR, IMC-A12 (later called cixutumumab [CIX]), is a human monoclonal antibody, which was identified to have tumor-suppressive potential.Citation94 It is thought that IGF-1R is centrally placed in several different survival signaling mechanisms, which leads to increased resistance to cytotoxic agents. These include the Akt/PI3K pathway, and activation of p38 and Rad51, which increase DNA-damage repair.Citation94 CIX began early clinical trials by being combined with chemotherapy as a method to regain sensitivity to chemotherapy.Citation95 Of these trials, those specifically aimed at NSCLC were phase I or II trials using CIX in various combinations with erlotinib, cetuximab, and chemotherapy. However, no reports were found to have noteworthy success. A recent phase I/II trial also suggested that the combination of CIX with erlotinib in nonselected patients had high toxicity and little benefit.Citation96
Despite the seeming failure of CIX, a slew of other IGF-1R inhibitors have been developed.Citation97 Another IGF-1R monoclonal antibody – CP-751871, better known as figitumumab – was able to increase objective response by 12% and progression-free survival by 1 month (HR = 0.46) when combined with carboplatin and paclitaxel in a recent phase II study. Of note, it seemed that those with squamous cell lung cancer were particularly responsive.Citation98 Despite early optimism, phase III trials were discontinued due to lack of benefit. However, the search for biomarkers to identify the appropriate patient population began. Currently, it is thought that circulating levels of IGF-1 might be useful in identifying possible beneficiaries of figitumumab treatment.Citation99
Cyclooxygenase-2 inhibitor
Cyclooxygenase 2 (COX-2) is a member of the COX family of enzymes that are involved in the production of prostaglandin H2 (PGH2). In NSCLC, the expression of COX-2 has been highly correlated with increased survivin, an antiapoptotic factor,Citation100 and COX-2 has been found to be overexpressed in ~90% of NSCLC patient samples.Citation101 Once COX-2 produces PGH2, PGH2 can then be converted to PGE2. Increased PGE2 levels lead to increased EGFR signaling, which in turn promotes survival and proliferation. Elevated PGE2 levels have been correlated with neoplastic lung tissues,Citation102 making COX-2 and its signaling pathway an attractive target for therapy. Early phase II studies found that in unselected patients, the use of celecoxib, a COX-2 inhibitor, did not render any benefit; however, a possible biomarker for response was identified.Citation103 Unfortunately, there does not seem to be a large body of evidence that celecoxib could be useful in the treatment of NSCLC, but studies have been conducted looking at its use as a cancer preventative, rather than a cure. One study found that celecoxib given to former smokers not only showed a decrease in biomarkers of early carcinogenesis but also showed a reduction in lung nodules.Citation104
While the inhibition of PGH2 is desired in order to decrease PHE2 levels, PGH2 can alternatively be made into prostacyclin (PGI2), if PGI2 synthase is present. Unlike PGE2, PGI2 has actually been shown to have a tumor-suppressive function. PGI2 synthase, however, is downregulated in many NSCLCs, resulting in low levels of PGI2.Citation105 Recent studies have investigated the possible use of PGI2 as a chemoprevention strategy, finding that in mouse models increasing PGI2 (alone and with gefitinib in moderate doses) prevented tumorigenesis.Citation106,Citation107 Additionally, studies in mouse models found that the activation of peroxisome proliferator-activated receptor gamma (PPARγ) was in part responsible for the tumor-suppressive phenotype of PGI2.Citation108 PPARγ is a potent transcription factor, which has roles in suppressing inflammatory and immune responses,Citation109 as well as directing the differentiation of adipocytes. Low PPARγ in NSCLC has also been correlated with poor prognosis.Citation110 Additionally, a large trial looking at patients across ten Veterans Affairs medical centers compared patients using thiazolidinediones (which are ligands for PPARγ activation) to those who were not on the drug. The investigators found that those individuals taking thiazolidinediones had a 33% reduced risk for lung cancer, highlighting PPARγ as a possible target warranting further investigation for clinical application.Citation111
As previously mentioned, the prevalence of the EGFR and ALK mutations that are currently targetable by therapy can account for only a small percentage of NSCLC patients, and current treatments are only useful for a short period of time due to resistance. The concept of reversing resistance (sensitization) has driven an appreciable amount of research in this area. For example, there have been studies showing that a combination of erlotinib with cetuximab can induce apoptosis in formerly TKI-resistant cells in vitro.Citation112
In general, the idea of combinatorial therapeutics is becoming more popular. Though EGFR treatments, as previously discussed, are looking at combinatorial-targeted inhibition, Src, the first discovered oncogene, and its downstream effects also closely mirror that of many of the receptor tyrosine kinases. To that end, a recent study attempting to concomitantly target Src and EGFR has shown some promising initial results. In a small trial, the inhibition of Src with different doses of dasatinib (all combined with erlotinib) in patients with lung cancer (50% adenocarcinoma, 21% squamous cell histology, 29% NSCLC) was seen to have a favorable response in 29 of 34 patients treated, as evinced by a disease-control rate (partial response plus stable disease) of 62% of patients, with a median progression-free survival time of 2.7 months and an overall survival time of 5.6 months (ranging from 4.9 to 12.2 months). This trial also determined that dasatinib may be combined safely with erlotinib for treatment.Citation113
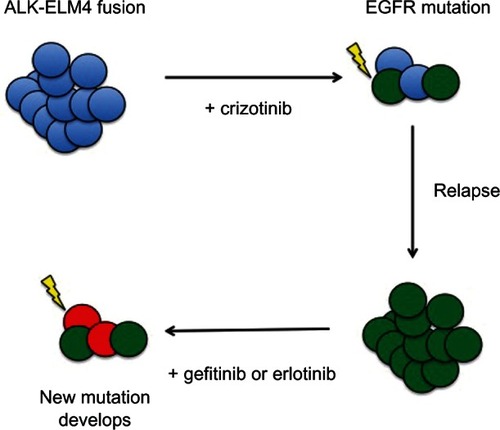
However, even combinatorial treatments might not be effective for all cases. Given the propensity of cancer to develop resistant cells, the next logical step in preventing relapse would be to target multiple pathways, or rather attacking cancer as a heterogeneous body. Perhaps targeting the primary and common resistance mechanisms simultaneously could improve the treatments. A recent study did just this, adding a c-Met inhibitor, tivantinib, to erlotinib. Though it was a small phase I trial, and further studies are being conducted, all 13 of their patients were able to achieve either a partial response or stable disease.Citation114 Several examples in the literature describe cases where two distinct driving mechanisms have been found in clonal populations within the same cancer, also suggesting that combination therapy might be the best approach to treatment.Citation52,Citation64 Combination of erlotinib and the VEGF inhibitor sorafenib has shown early promise in phase II trials.Citation115 Conversely, combination of erlotinib with another VEGF inhibitor, sunitinib, did not have any effect in a phase III trial.Citation116 In several of these cases, the clonal populations of cells with different driving mutations, and those with new mutations, gained resistance, and tended to fall into categories for which targeted therapies already exist in NSCLC (). For example, a patient who develops lung cancer with an ALK fusion protein and treated with crizotinib may have their lung tumors shrink, but subsequently develop a resistant population of cells, via EGFR mutations. In that case, since crizotinib would no longer be effective for this patient, treatment with gefitinib or erlotinib might be beneficial. Another example would be a case where the lung cancer driven by an EGFR mutation develops resistance to therapy by the upregulation of Met. In that case, the cancer may now be sensitive to a subsequent treatment with crizotinib.Citation28,Citation29,Citation64 Given the advances in personalized medicine via genetic and biomarker screening, we may be much more effective at treating these various scenarios.
Figure 1 A hypothetical line of treatment based on the availability of a range of targeted therapies.
Abbreviations: ALK, anaplastic lymphoma kinase; ELM4, echinoderm microtubule-associated protein-like 4; EGFR, epidermal growth factor receptor.
Future directions for the treatment of lung cancer
While the kinase pathways have been the major focus of therapeutic development, recently a small body of literature is suggestive of an important role for the Wnt-signaling pathway in lung cancer. The common conversation about Wnt signaling references the canonical pathway, where binding of a Wnt ligand to a Frizzled receptor results in stabilization of β-catenin. Normally important as a structural protein, when β-catenin is allowed to persist in the cytosol, it translocates into the nucleus, where it stimulates not only the genes associated with increased proliferation and motility but also factors that are native to stem cells. While this transcriptional program is very important during development, improper activation in adult tissues contributes to oncogenesis.Citation117–Citation119 Therefore, attempts to use this pathway as a target for therapeutic gain have all been focused on inhibiting canonical signaling.Citation120,Citation121 Interestingly, recent studies have identified a possible benefit in NSCLC, not from inhibiting Wnt signaling, but from restoring its function.
Specifically, Wnt 7a, when paired with the receptor Frizzled 9, activates a β-catenin independent (noncanonical) signaling pathway. This leads to a cascade of events including the activation of c-Jun N-terminal kinase signaling,Citation122 Sprouty-4 (a TKI),Citation122,Citation123 and the stabilization of PPARγ.Citation117,Citation123 Intriguingly, Wnt 7a is lost in a high percentage of NSCLC. In fact, a staggering 85% of patient tissue samples and 88% of dysplastic lesions have lost Wnt 7a, possibly via DNA methylation-mediated gene silencing.Citation124 Moreover, it was also observed that the reconstitution of this noncanonical Wnt-signaling pathway displayed significant tumor-suppressive effects.Citation122 These factors make this pathway an exciting prospect not only for its possible efficacy in treatment but also for its widespread applicability.
However, it is a challenge to purify recombinant Wnt 7a that can be used in therapy. Interestingly, it has been shown that a PGI2 analogueCitation125 can mimic many of the effects of Wnt 7a in the context of Frizzled 9. Recent studies have also shown that the addition of prostacyclin to lung cancer cell lines that have retained Wnt 7a receptor Frizzled 9 results in decreased proliferation, inhibition of anchorage-independent growth,Citation126 and significantly decreases lung cancer growth in vitro.123,126
While the Wnt pathway presents an exciting new path to investigate, we must also continue to find new and different targets in the treatment of lung cancer. New methods are currently in development to assist in the screening of individual patients for important mutations, which may help identify which therapy would be effective for them.Citation127,Citation128 Additionally, steps have been taken towards identifying novel mutations that still require therapeutic development via whole-genome analysis methods. Recent studies in SCLC have analyzed the genomes, transcriptomes, and exomes of SCLC samples, either alone or paired to normal tissues. By looking at the entire cancer rather than just the mutations that have been previously reported, several trends emerged and pointed towards new targets.Citation129,Citation130 This method has also been applied to NCSLCs, specifically of the squamous histology type. Of the 178 lung squamous cell carcinomas that were characterized, eleven genes among hundreds of alterations in gene copy number, gene rearrangements, and mutations were recurrently mutated. While these include some of the obvious suspects, such as TP53, alternate pathways were also found to be commonly mutated, including proteins important for cellular differentiation and oxidative stress.Citation131 If the goal of achieving truly personalized medicine requires that we screen each and every patient in order to ascertain their therapeutic needs, we then also need to prepare therapies for any number of possible mutant genotypes so that once the problem is identified, we can be equipped to treat it.
Discussion
Lung cancer remains the leading cause of cancer deaths in the United States. While there have been a number of drugs approved for use, they have suffered from limited applicability and an incredibly high rate of acquired resistance. At the current rate, real advancement in the treatment of lung cancer may still be years or even decades away. That being said, several themes have emerged in the literature. First, there is a significant need for the advancement of personalized medicine. The power of targeted therapies is clear when applied to those individuals harboring the specific mutations for which the drug was designed to treat. However, the success of many of these targeted therapies has often been confounded by the design of the trials. The low prevalence of mutations that are specifically targeted by drugs result in trials that mask efficacy in large and unselected populations of patients. This highlights the need for a more personalized approach to therapy. The identification of biomarkers is progressing, but will do little good if the information is not translated to the clinical trial arena, allowing drugs to be tested against their intended population. The perfect example of this was previously described in the cases of gefitinib and crizotinib. The selection of patients via the use of biomarkers and individualized analysis can mean the difference between a null result and the clear demonstration of a medicine’s potential.
Secondly, there is no magic bullet for any of these subsets of cancers. Where one mutation may dominate, other populations will rise or adapt upon selection by drugs. While there may not be a single cure for lung cancer, there could be a way to either cycle treatment or combine drugs in such a way as to keep the cancer cells under constant submission. This also requires that attention be paid to the patient on an individual basis, as the mechanisms of resistance are many, and proper treatment relies on the ability to determine which resistance mechanism has been employed. Based on the adaptive nature of lung cancer, one must constantly be vigilant in looking for alternative mechanisms to target this disease, and the need to develop more robust personalized approaches for the treatment of lung cancer has never been greater.
Acknowledgments
This study was supported by a merit award from the US Department of Veterans Affairs, and National Institute of Health grants R01CA1385282522717 and 5R21CA153268-02 to Robert A Winn.
Disclosure
The authors have no conflicts of interest to disclose.
References
- SiegelRNaishadhamDJemalACancer statistics, 2012CA Cancer J Clin201262102922237781
- OkuboKMarkEJFliederDBronchoalveolar carcinoma: clinical, radiologic, and pathologic factors and survivalJ Thorac Cardiovasc Surg199911870270910504637
- RazDJHeBRosellRJablonsDMCurrent concepts in bronchioloal-veolar carcinoma biologyClin Cancer Res2006123698370416778095
- SiegelRDeSantisCVirgoKCancer treatment and survivorship statistics, 2012CA Cancer J Clin20126222024122700443
- KhuriFRHerbstRSFossellaFVEmerging therapies in non-small-cell lung cancerAnn Oncol20011273974411484947
- WeinsteinIBCancer. Addiction to oncogenes – the Achilles heal of cancerScience2002297636412098689
- HabeckMFDA licences imatinib mesylate for CMLLancet Oncol20023611905608
- CarpenterGCohenSEpidermal growth factorJ Biol Chem1990265770977122186024
- CohenMHWilliamsGASridharaRChenGPazdurRFDA drug approval summary: gefitinib (ZD1839) (Iressa) tabletsOncologist2003830330612897327
- CohenMHJohnsonJRChenYFSridharaRPazdurRFDA drug approval summary: erlotinib (Tarceva) tabletsOncologist20051046146616079312
- ThatcherNChangAParikhPGefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer)Lancet20053661527153716257339
- ShepherdFARodrigues PereiraJCiuleanuTErlotinib in previously treated non-small-cell lung cancerN Engl J Med200535312313216014882
- GazdarAFActivating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitorsOncogene200928Suppl 1S24S3119680293
- LynchTJBellDWSordellaRActivating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinibN Engl J Med20043502129213915118073
- MarchettiAMartellaCFelicioniLEGFR mutations in non-small-cell lung cancer: analysis of a large series of cases and development of a rapid and sensitive method for diagnostic screening with potential implications on pharmacologic treatmentJ Clin Oncol20052385786515681531
- PaezJGJännePALeeJCEGFR mutations in lung cancer: correlation with clinical response to gefitinib therapyScience20043041497150015118125
- WuYLZhongWZLiLYEpidermal growth factor receptor mutations and their correlation with gefitinib therapy in patients with non-small cell lung cancer: a meta-analysis based on updated individual patient data from six medical centers in mainland ChinaJ Thorac Oncol2007243043917473659
- ShaoYYLinCCYangCHGefitinib or erlotinib in the treatment of advanced non-small cell lung cancerDiscov Med2010953854520587343
- CostaECTaronMQueraltCDifferential progression-free survival (PFS) to erlotinib according to EGFR exon 19 deletion type in non-small cell lung cancer (NSCLC) patients (p) in the EURTAC studyJ Clin Oncol201230Suppl7540
- MokTSWuYLThongprasertSGefitinib or carboplatin-paclitaxel in pulmonary adenocarcinomaN Engl J Med200936194795719692680
- OxnardGRArcilaMESimaCSAcquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutationClin Cancer Res2011171616162221135146
- RosellRMoranTQueraltCScreening for epidermal growth factor receptor mutations in lung cancerN Engl J Med200936195896719692684
- YunCHMengwasserKETomsAVThe T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATPProc Natl Acad Sci USA20081052070207518227510
- PaoWMillerVAPolitiKAAcquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domainPLoS Med20052e7315737014
- KosakaTYatabeYEndohHAnalysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinibClin Cancer Res2006125764576917020982
- HammermanPSJannePAJohnsonBEResistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancerClin Cancer Res2009157502750920008850
- SudaKMizuuchiHMaeharaYMitsudomiTAcquired resistance mechanisms to tyrosine kinase inhibitors in lung cancer with activating epidermal growth factor receptor mutation-diversity, ductility, and destinyCancer Metastasis Rev20123180781422736441
- EngelmanJAZejnullahuKMitsudomiTMET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signalingScience20073161039104317463250
- CappuzzoFJännePASkokanMMET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patientsAnn Oncol20092029830418836087
- KosakaTYamakiEMogiAKuwanoHMechanisms of resistance to EGFR TKIs and development of a new generation of drugs in non-small-cell lung cancerJ Biomed Biotechnol2011201116521421687596
- SosMLKokerMWeirBAPTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFRCancer Res2009693256326119351834
- ThomsonSBuckEPettiFEpithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibitionCancer Res2005659455946216230409
- ChangTHTsaiMFSuKYSlug confers resistance to the epidermal growth factor receptor tyrosine kinase inhibitorAm J Respir Crit Care Med20111831071107921037017
- WinnRATuderRM“Un-slugging” resistance to epidermal growth factor receptor inhibition. A positive outlook for the future of lung cancer therapyAm J Respir Crit Care Med201118397097221498821
- NgKPHillmerAMChuahCTA common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancerNat Med20121852152822426421
- PaoWWangTYRielyGJKRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinibPLoS Med20052e1715696205
- MassarelliEVarella-GarciaMTangXKRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancerClin Cancer Res2007132890289617504988
- SchmidKOehlNWrbaFPirkerRPirkerCFilipitsMEGFR/ KRAS/BRAF mutations in primary lung adenocarcinomas and corresponding locoregional lymph node metastasesClin Cancer Res2009154554456019584155
- ShigematsuHLinLTakahashiTClinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancersJ Natl Cancer Inst20059733934615741570
- KrisMGJohnsonBEKwiatkowskiIdentification of driver mutations in tumor specimens from 1,000 patients with lung adenocarcinoma: the NCI’s Lung Cancer Mutation Consortium (LCMC)J Clin Oncol201129SupplCRA7506
- BrokerLEGiacconeGThe role of new agents in the treatment of non-small cell lung cancerEur J Cancer2002382347236112460778
- EngelmanJAChenLTanXEffective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancersNat Med2008141351135619029981
- JännePAShawATPereiraJREfficacy and patient (pt)-reported outcomes (PROs) with selumetinib (AZD6244, ARRY-142866; SEL) + docetaxel (Doc) in KRAS-mutant advanced non-small cell lung cancer (NSCLC): a randomized, phase II trialAnn Oncol201223Suppl 91233PD
- RamalingamSBelaniCPRecent advances in targeted therapy for non-small cell lung cancerExpert Opin Ther Targets20071124525717227238
- RosellRRobinetGSzczesnaARandomized phase II study of cetuximab plus cisplatin/vinorelbine compared with cisplatin/ vinorelbine alone as first-line therapy in EGFR-expressing advanced non-small-cell lung cancerAnn Oncol20081936236917947225
- PirkerRPereiraJRSzczesnaACetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX): an open-label randomised phase III trialLancet20093731525153119410716
- HannaNHDahlbergSEKolesarJThree-arm randomized phase II study of carboplatin (C) and paclitaxel (P) in combination with cetuximab (CET), IMC-A12, or both for advanced non-small cell lung cancer (NSCLC) patients who will not receive bevacizumab-based therapy: an Eastern Cooperative Oncology Group (ECOG) study (E4508)J Clin Oncol201230Suppl7516
- GreillierLMartel-LafayIArpinDA randomised phase II study of cetuximab (c) in combination with two cisplatin-based concurrent chemoradiotherapy regimens in patients (pts) with stage III non-small cell lung cancer (NSCLC). Final results of the GFPC 08-03 trialPaper presented at the 37th European Society for Medical Oncology (ESMO) CongressSeptember 28–October 2, 2012Vienna, Austria
- GridelliCMaionePFerraraMLRossiACetuximab and other anti-epidermal growth factor receptor monoclonal antibodies in the treatment of non-small cell lung cancerOncologist20091460161119482958
- MaPCPersonalized targeted therapy in advanced non-small cell lung cancerCleve Clin J Med201279Suppl 1eS56eS6022614968
- SodaMChoiYLEnomotoMIdentification of the transforming EML4-ALK fusion gene in non-small-cell lung cancerNature200744856156617625570
- KoivunenJPMermelCZejnullahuKEML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancerClin Cancer Res2008144275428318594010
- KwakELBangYJCamidgeDRAnaplastic lymphoma kinase inhibition in non-small-cell lung cancerN Engl J Med20103631693170320979469
- FordePMRudinCMCrizotinib in the treatment of non-small-cell lung cancerExpert Opin Pharmacother2012131195120122594847
- GalettaDRossiAPiscontiSColucciGThe emerging role of ALK inhibitors in the treatment of advanced non-small cell lung cancerExpert Opin Ther Targets201216Suppl 2S45S5422443113
- KimuraHALK fusion gene positive lung cancer and 3 cases treated with an inhibitor for ALK kinase activityLung Cancer201275667221757253
- TakeuchiKChoiYLTogashiYKIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancerClin Cancer Res2009153143314919383809
- TogashiYSodaMSakataSKLC1-ALK: a novel fusion in lung cancer identified using a formalin-fixed paraffin-embedded tissue onlyPLoS One20127e3132322347464
- WeickhardtAJRothmanMSSalian-MehtaSRapid-onset hypogonadism secondary to crizotinib use in men with metastatic nonsmall cell lung cancerCancer20121185302530922488744
- SchnellPSaffermanAZBartlettCHTangYWilnerKDClinical presentation of hepatotoxicity-associated crizotinib in ALK-positive (ALK+) advanced non-small cell lung cancer (NSCLC)J Clin Oncol201230Suppl7598
- ZhangSWangFKeatsJCrizotinib-resistant mutants of EML4-ALK identified through an accelerated mutagenesis screenChem Biol Drug Des201178999100522034911
- DoebeleRCPillingABAisnerDLMechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancerClin Cancer Res2012181472148222235099
- KatayamaRShawATKhanTMMechanisms of acquired crizotinib resistance in ALK-rearranged lung cancersSci Transl Med20124120ra17
- CamidgeDRDoebeleRCTreating ALK-positive lung cancer – early successes and future challengesNat Rev Clin Oncol2012926827722473102
- ChenZSasakiTTanXInhibition of ALK, PI3K/MEK, and HSP90 in murine lung adenocarcinoma induced by EML4-ALK fusion oncogeneCancer Res2010709827983620952506
- KatayamaRKhanTMBenesCTherapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALKProc Natl Acad Sci USA20111087535754021502504
- GaronEBMoranTBarlesiFPhase II study of the HSP90 inhibitor AUY922 in patients with previously treated, advanced non-small cell lung cancer (NSCLC)J Clin Oncol201230Suppl7543
- SasakiTJannePANew strategies for treatment of ALK-rearranged non-small cell lung cancersClin Cancer Res2011177213721822010214
- HeuckmannJMHölzelMSosMLALK mutations conferring differential resistance to structurally diverse ALK inhibitorsClin Cancer Res2011177394740121948233
- HanahanDWeinbergRAThe hallmarks of cancerCell2000100577010647931
- FolkmanJWhat is the evidence that tumors are angiogenesis dependent?J Natl Cancer Inst199082461688381
- ReckMGatzemeierUTargeted therapies: thalidomide in lung cancer therapy – what have we learned?Nat Rev Clin Oncol2010713413520190796
- HoangTDahlbergSESchillerJHRandomized phase III study of thoracic radiation in combination with paclitaxel and carboplatin with or without thalidomide in patients with stage III non-small-cell lung cancer: the ECOG 3598 studyJ Clin Oncol20123061662222271472
- YangJCHaworthLSherryRMA randomized trial of bevacizumab, n anti-vascular endothelial growth factor antibody, for metastatic renal cancerN Engl J Med200334942743412890841
- KindlerHLShulmanKLMetastatic colorectal cancerCurr Treat Options Oncol2001245947112057092
- AzimHAJrGantiAKTargeted therapy in advanced non-small cell lung cancer (NSCLC): where do we stand?Cancer Treat Rev20063263063617034953
- LaskinJJSandlerABFirst-line treatment for advanced non-small-cell lung cancerOncology (Williston Park)20051916711676 discussion1678168016425521
- DempkeWCSutoTReckMTargeted therapies for non-small cell lung cancerLung Cancer20106725727419914732
- StevensonJPLangerCJSomerRAPhase 2 trial of maintenance bevacizumab alone after bevacizumab plus pemetrexed and carboplatin in advanced, nonsquamous nonsmall cell lung cancerCancer20121185580558722544579
- RoehrBBevacizumab as adjuvant therapy for lung cancer does not help patients over 65BMJ2012344e285522517975
- DasanuCABevacizumab in lung cancer: lackluster performance and unjustified expense?J Oncol Pharm Pract20121838138222777997
- OhyanagiFTanimotoASakataniTPhase II trial of bevacizumab plus docetaxel in patients with previously treated nonsquamous non-small cell lung cancerJ Clin Oncol201230Supple18004
- RamalingamSSOwonikokoTKKhuriFRLung cancer: new biological insights and recent therapeutic advancesCA Cancer J Clin2011619111221303969
- GridelliCMaionePDel GaizoFSorafenib and sunitinib in the treatment of advanced non-small cell lung cancerOncologist20071219120017296815
- TanEHGossGDSalgiaRPhase 2 trial of Linifanib (ABT-869) in patients with advanced non-small cell lung cancerJ Thorac Oncol201161418142521597387
- RamalingamSSShtivelbandMSooRARandomized phase II study of carboplatin and paclitaxel with either linifanib or placebo for advanced nonsquamous NSCLCJ Clin Oncol201230Suppl7512
- Paz-AresLGBiesmaBHeigenerDPhase III, randomized, double-blind, placebo-controlled trial of gemcitabine/cisplatin alone or with sorafenib for the first-line treatment of advanced, nonsquamous non-small-cell lung cancerJ Clin Oncol2012303084309222851564
- BriaEPilottoSTortoraGSorafenib for lung cancer: is the “Battle” still open?Expert Opin Investig Drugs20122114451448
- ZhangJGoldKAKimESorafenib in non-small cell lung cancerExpert Opin Investig Drugs20122114171426
- NealJWSequistLVExciting new targets in lung cancer therapy: ALK, IGF-1R, HDAC, and HhCurr Treat Options Oncol201011364420676809
- OwonikokoTKRamalingamSSKanterewiczBBaliusTEBelaniCPHershbergerPAVorinostat increases carboplatin and paclitaxel activity in non-small-cell lung cancer cellsInt J Cancer201012674375519621389
- LiCTHsiaoYMWuTCLinYWYehKTKoJLVorinostat, SAHA, represses telomerase activity via epigenetic regulation of telomerase reverse transcriptase in non-small cell lung cancer cellsJ Cell Biochem20111123044305321678477
- OubanAMuracaPYeatmanTCoppolaDExpression and distribution of insulin-like growth factor-1 receptor in human carcinomasHum Pathol20033480380814506643
- RowinskyEKYoussoufianHTonraJRSolomonPBurtrumDLudwigDLIMC-A12, a human IgG1 monoclonal antibody to the insulin-like growth factor I receptorClin Cancer Res2007135549s5555s17875788
- McKianKPHaluskaPCixutumumabExpert Opin Investig Drugs20091810251033
- WeickhardtADoebeleROtonAA phase I/II study of erlotinib in combination with the anti-insulin-like growth factor-1 receptor monoclonal antibody IMC-A12 (cixutumumab) in patients with advanced non-small cell lung cancerJ Thorac Oncol2012741942622237261
- ScagliottiGVNovelloSThe role of the insulin-like growth factor signaling pathway in non-small cell lung cancer and other solid tumorsCancer Treat Rev20123829230221907495
- KarpDDPaz-AresLGNovelloSPhase II study of the anti-insulin-like growth factor type 1 receptor antibody CP-751,871 in combination with paclitaxel and carboplatin in previously untreated, locally advanced, or metastatic non-small-cell lung cancerJ Clin Oncol2009272516252219380445
- GualbertoAHixonMLKarpDDPre-treatment levels of circulating free IGF-1 identify NSCLC patients who derive clinical benefit from figitumumabBr J Cancer2011104687421102589
- KrysanKMerchantFHZhuLCOX-2-dependent stabilization of survivin in non-small cell lung cancerFASEB J20041820620814597555
- LiFLiuYChenHEGFR and COX-2 protein expression in non-small cell lung cancer and the correlation with clinical featuresJ Exp Clin Cancer Res2011302721385353
- TennisMAVanscoykMKeithRLWinnRAThe role of prostacyclin in lung cancerTransl Res2010155576120129485
- MutterRLuBCarboneDPA phase II study of celecoxib in combination with paclitaxel, carboplatin, and radiotherapy for patients with inoperable stage IIIA/B non-small cell lung cancerClin Cancer Res2009152158216519276291
- MaoJTRothMDFishbeinMCLung cancer chemoprevention with celecoxib in former smokersCancer Prev Res (Phila)2011498499321733822
- CathcartMCGraySGBairdAMProstacyclin synthase expression and epigenetic regulation in nonsmall cell lung cancerCancer20111175121513221523772
- KeithRLKaroorVMozerABHudishTMLeMMillerYEChemoprevention of murine lung cancer by gefitinib in combination with prostacyclin synthase overexpressionLung Cancer201070374220116128
- KeithRLMillerYEHoshikawaYManipulation of pulmonary prostacyclin synthase expression prevents murine lung cancerCancer Res20026273474011830527
- NemenoffRMeyerAMHudishTMProstacyclin prevents murine lung cancer independent of the membrane receptor by activation of peroxisomal proliferator – activated receptor gammaCancer Prev Res (Phila)2008134935619138979
- HazraSPeeblesKASharmaSMaoJTDubinettSMThe role of PPARgamma in the cyclooxygenase pathway in lung cancerPPAR Res2008200879056818769553
- SasakiHTanahashiMYukiueHDecreased perioxisome proliferator-activated receptor gamma gene expression was correlated with poor prognosis in patients with lung cancerLung Cancer200236717611891036
- GovindarajanRRatnasingheLSimmonsDLThiazolidinediones and the risk of lung, prostate, and colon cancer in patients with diabetesJ Clin Oncol2007251476148117442990
- WangMZhaoJZhangLMCombined erlotinib and cetuximab overcome the acquired resistance to epidermal growth factor receptors tyrosine kinase inhibitor in non-small-cell lung cancerJ Cancer Res Clin Oncol20121382069207722821179
- HauraEBTanvetyanonTChiapporiAPhase I/II study of the Src inhibitor dasatinib in combination with erlotinib in advanced non-small-cell lung cancerJ Clin Oncol2010281387139420142592
- YamamotoNMurakamiHTakahashiTFinal results of a Japanese phase 1 trial evaluating a c-met inhibitor tivantinib in combination with an EGFR inhibitor erlotinib in advanced/metastatic non-small cell lung cancer (ARQ 197-003/005 study)Ann Oncol201223Suppl 9ix424
- GridelliCMorgilloFFavarettoASorafenib in combination with erlotinib or with gemcitabine in elderly patients with advanced non-small-cell lung cancer: a randomized phase II studyAnn Oncol2011221528153421212155
- ScagliottiGVKrzakowskiMSzczesnaASunitinib plus erlotinib versus placebo plus erlotinib in patients with previously treated advanced non-small-cell lung cancer: a phase III trialJ Clin Oncol2012302070207822564989
- Van ScoykMRandallJSergewAWilliamsLMTennisMWinnRAWnt signaling pathway and lung diseaseTransl Res200815117518018355764
- TennisMVan ScoykMWinnRARole of the wnt signaling pathway and lung cancerJ Thorac Oncol2007288989217909349
- Pacheco-PinedoECDurhamACStewartKMWnt/β-catenin signaling accelerates mouse lung tumorigenesis by imposing an embryonic distal progenitor phenotype on lung epitheliumJ Clin Invest20111211935194521490395
- KimJYouLXuZWnt inhibitory factor inhibits lung cancer cell growthJ Thorac Cardiovasc Surg200713373373717320573
- YaoHAshiharaEStrovelJWAV-65, a novel Wnt/beta-catenin signal inhibitor, successfully suppresses progression of multiple myeloma in a mouse modelBlood Cancer J20111e4322829079
- WinnRAMarekLHanSYRestoration of Wnt-7a expression reverses non-small cell lung cancer cellular transformation through frizzled-9-mediated growth inhibition and promotion of cell differentiationJ Biol Chem2005280196251963415705594
- TennisMAVan ScoykMMFreemanSVVandervestKMNemenoffRAWinnRASprouty-4 inhibits transformed cell growth, migration and invasion, and epithelial-mesenchymal transition, and is regulated by Wnt7A through PPARgamma in non-small cell lung cancerMol Cancer Res2010883384320501643
- TennisMAVanscoykMMWilsonLAKelleyNWinnRAMethylation of Wnt7a is modulated by DNMT1 and cigarette smoke condensate in non-small cell lung cancerPLoS One20127e3292122403725
- KaukinenSYlitaloPPessiTVapaataloHHemodynamic effects of iloprost, a prostacyclin analogClin Pharmacol Ther1984364644696206978
- TennisMAVan ScoykMHeasleyLEProstacyclin inhibits non-small cell lung cancer growth by a frizzled 9-dependent pathway that is blocked by secreted frizzled-related protein 1Neoplasia20101224425320234818
- ZhangSZhangHYangXDetection of EGFR and KRAS somatic mutations in tumor tissue and peripheral blood by a liquidchip technology for patients with advanced non-small cell lung cancerJ Clin Oncol201230Supple18142
- WangKStephensPYelenskyRFrequency of actionable genomic alterations in early-stage lung adenocarcinoma (LA) detected by next-generation sequencing (NGS)J Clin Oncol201230Supple17541
- PeiferMFernández-CuestaLSosMLIntegrative genome analyses identify key somatic driver mutations of small-cell lung cancerNat Genet2012441104111022941188
- RudinCMDurinckSStawiskiEWComprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancerNat Genet2012441111111622941189
- Cancer Genome Atlas Research NetworkComprehensive genomic characterization of squamous cell lung cancersNature201248951952522960745