
Abstract
Primary ciliary dyskinesia (PCD) is a rare genetic disease caused by mutations of genes coding motile-cilia-related proteins. CCDC40 variants can cause PCD via disrupting the assembling of inner dynein and dynein regulating complex in cilia and flagella, but none has been reported associated with multiple morphological abnormalities of the sperm flagella (MMAF). We identified and validated the disease-causing variants in our patient via whole-exome and Sanger sequencing. We used high-speed video microscopy analysis (HSVA) and immunofluorescence to analyze the functional and structural deficiency of respiratory cilia. Papanicolaou staining and scanning electron microscope was applied to analyze the morphological sperm defects resulted from the PCD associated variants. We identified novel compound variants (c.901C>T, p.(Arg301*); c.2065_2068dup, p.(Ala690Glyfs*67)) in CCDC40 in a male patient with male infertility. HSVA revealed the rigid and stiff ciliary beating pattern. Immunofluorescence indicated loss of inner dynein arm protein DNAH2 both in cilia and the sperms of the patient. Diagnosis of MMAF was confirmed through sperm Papanicolaou staining and scanning electron microscope. We first describe a patient with a combination of PCD and MMAF associated with novel compound heterozygous variants in CCDC40. Our results present initial evidence that CCDC40 associated with MMAF, which expands the genetic spectrum of PCD and MMAF and provides precise clinical genetic counseling to this family.
Keywords:
Introduction
Primary ciliary dyskinesia (PCD) is a rare autosomal recessive disease caused by mutations of genes coding motile-cilia-related proteins and characterized by chronic respiratory infections due to decreased airway clearance.Citation1,Citation2 Up to now, no diagnostic test can be relied on solely to confirm this disease.Citation3 Mutations in over 50 genes of PCD have been reported.Citation4 However, very few of them are related to multiple morphological abnormalities of the sperm flagella (MMAF) because details about male infertility and the results of sperm morphological analysis are rarely reported in most of the PCD patients.Citation5
MMAF, first described in 2014,Citation6 is one kind of severe sperm malformations characterized by short, absent, bent, coiled or irregular sperm flagella, caused by flagellum assembly and organization defect, resulting in decreased male fertility. ARMC4, CCDC39, DRC1, SPEF2, CFAP74, and BRWD1 are the few PCD-associated genes reported to be related to MMAF phenotype.Citation7–Citation12
CCDC40 is a gene encoding a highly-preserved protein that controls ciliary beating via regulating the assembling of inner dynein arm and dynein regulating complex.Citation13 Mutation in CCDC40 has been reported in a male PCD patient with infertility, but it has not been reported to be associated with MMAF.Citation14 In this study, we first report variants in CCDC40 associated with PCD and MMAF.
Materials and Methods
Ethical Compliance
The Review Board of the Second Xiangya Hospital of Central South University in China approved this study (no.2020082). Written informed consent was obtained from the patient and the healthy control.
Whole Exome Sequencing and Variants Analysis
We collected peripheral blood sample from the patient and his parents and extracted genomic DNA with the QIAamp DNA Blood MiniKit (250) (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. Whole-exome sequencing was performed as previously described in another study.Citation9 The sequencing reads were aligned to the human reference genome (GRCh37/hg19, http://genome.ucsc.edu/) by using the Burrows Wheeler Aligner,Citation15 and the variants were detected by HaplotypeCaller in GATK4.0 with default parameters and annotated with ANNOVAR.Citation16,Citation17 To identify the disease-causing variants, we selected the rare variants (minor allele frequency <0.01) from the 1000 Genomes Project data set (http://browser.1000genomes.org), NHLBI Exome Sequencing Project Exome Variant Server (http://evs.gs.washington.edu/EVS), Genome Aggregation Database (all datasets and East Asian population datasets of gnomAD genome database, http://gnomad.broadinstitute.org/) and in-house database of Novogene. Noncoding, intronic, and synonymous missense variants were then filtered. Sanger sequencing was used to detect and validate the variants in the patient and his parents. The sequences of the primers are listed as following: Mutation 1, forward primer: 5’-GGAGGGTAACCAGAAAGGTAAC-3’, reverse primer: 5’-CTGCTGCACCTCATAGAGATT-3’; Mutation 2, forward primer: 5’-ACATCTGGGTTCCAACAAGTAG-3’, reverse primer: 5’-TTTGTCTGTAAAGCAGGTAGGG-3’.
Reverse Transcription and Quantitative Real-Time Polymerase Chain Reaction (RT-qPCR)
We collected peripheral blood sample from the patient and a healthy control and extracted total RNA using a Whole Blood RNA Purification Kit (B0006, EZ Bioscience) according to the manufacturer’s instructions. Then, we used 4×EZscript Reverse Transcription Mix II (with gDNA Remover, EZB-RT2GQ, EZBioscience) to synthesize cDNA based on the manufacturer’s recommendations. Subsequently, we performed the quantitative real-time polymerase chain reaction (qPCR) process in a Thermo ABI 7500 Fast Real-time PCR system using PowerUp SYBR Green Master Mix (LT-02241, Thermo Fisher Scientific). The qPCR conditions were set according to manufacturer’s instructions. The melting curve was analyzed using the default setting. The reaction system used a 10ul reaction system, and the sample amount of cDNA was 50ng per reaction. GAPDH was used as an internal control,Citation6 and fold changes in mRNA levels were determined using the 2−ΔΔCT method. The sequences of the primers are listed as following: GAPDH, forward primer: 5’-AATCCCATCACCATCTTCCAG-3’, reverse primer’: 5’-AAATGAGCCCCAGCCTTC-3’; CCDC40, forward primer: 5’-CGCCTAGCAACGGGAAAT-3’; reverse primer: 5’-CATCATCCTTCTCTGGTGGTG-3’.
High-Speed Video Microscopy Analysis
Nasal brush biopsy samples were imaged using an upright Olympus BX53 microscope (Olympus, Tokyo, Japan) and scientific complementary metal oxide semiconductor camera (Prime BSI, Teledyne Photometrics Inc., USA) as described.Citation9
Sperm Morphological Analysis
Semen samples were collected from the patient after at least five days of sexual abstinence. The flagellum morphology was evaluated according to the World Health Organization guideline.Citation18 The abnormal flagella of the sperms were classified as absent, short, bent, coiled or irregular after Papanicolaou staining.Citation6 One spermatozoon was classified to only one morphological category according to its major flagellar abnormality.Citation19,Citation20
Scanning Electron Microscope of Sperms
Sperms were fixed in 2.5% glutaraldehyde; then sample was washed in 0.1 mol/L phosphate buffer for 30 min and post-fixed in osmic acid. Next, the sperms were washed again in 0.1 mol/L phosphate buffer for 30 min; thereafter, sample was progressively dehydrated with ethanol and isoamyl acetate, and was dried with a CO2 critical-point dryer (Eiko HCP-2, Hitachi, Tokyo, Japan) were conducted. Subsequently, the sample was mounted on aluminum stubs, sputter-coated with an ionic sprayer meter (Eiko E-1020, Hitachi, Tokyo, Japan), and analyzed via scanning electron microscopy (SEM) (NOVA NANOSEM 450, America) under an accelerating voltage of 20 kV.
Immunofluorescence
Respiratory epithelial tissues and sperms were fixed in 4% paraformaldehyde. Respiratory epithelial tissues were incubated overnight at 4°C and then embedded in paraffin, processed, sectioned, and performed immunofluorescence. The outer-dynein arm, inner-dynein arm, and the cilia axoneme were stained with primary antibodies DNAH5 (HPA037470, 1:100, Sigma-Aldrich, Missouri, USA), DNAH2 (HPA067103, 1:50, Sigma-Aldrich, Missouri, USA), and anti-acetylated tubulin monoclonal antibody (T7451, 1:500, Sigma-Aldrich, Missouri, USA). Sperms were coated on the slides and then incubated overnight at 4°C with anti-acetylated tubulin monoclonal antibody (T7451, 1:100, Sigma-Aldrich, Missouri, USA) and DNAH2 (HPA067103, 1:25, Sigma-Aldrich, Missouri, USA). Antibody binding was detected using Alexa Fluor 488 anti-mouse IgG (A-21121, 1:200, Invitrogen, Carlsbad, CA, USA) and Alexa Fluor 555 anti-rabbit IgG (A31572, 1:400, Invitrogen, Carlsbad, CA, USA). After incubation for 1.5 h at 37°C, all the slides were stained with 2-(4-amidinophenyl)-1H-indole-6-carboxamidine (DAPI) for 5 min at 25°C. Fluorescence signals were recorded using an Olympus BX53 microscope (Olympus, Tokyo, Japan) and scientific complementary metal oxide semiconductor (sCMOS) camera (Prime BSI, Teledyne Photometrics Inc, USA).
Results
Case Presentation
The proband (II-2) is a 20-year-old unmarried male, having non-consanguineous parents and a healthy sister (). He had suffered from productive cough and shortness of breath for 6 years. He exhibited recurrent cough when he was about 1 year old and had chronic otitis media and sinusitis. Respiratory symptoms did not get better despite multiple anti-infection treatment courses with macrolide and diagnostic anti-tuberculosis treatment.
Figure 1 (A) Pedigree of patient’s family with inherited CCDC40 pathogenic variants. Black arrow, proband. Half-colored symbol, heterozygous CCDC40 variant carrier. Solid symbol, patient (affected). (B) Lung high-resolution computed tomography of the patient showed diffuse nodules among small airway and bronchiectasis in right middle lung and both lower lungs. (C) High-resolution computed tomography of the patient showed sinusitis.
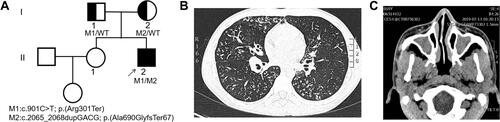
Lung high-resolution computed tomography (HRCT) showed diffuse nodules among small airway and bronchiectasis in right middle lung and both lower lungs (). Sinus HRCT showed sinusitis and bilateral inferior turbinate hypertrophy (). Pulmonary function test showed predicted forced expiratory volume during the first second (FEV1) was 64.2%. Cold agglutinin test was negative. Fiberoptic bronchoscope examination demonstrated suppurative inflammation of bronchus. Echocardiography was normal. The nasal nitric oxide level was 9.0 nL/min. He was first diagnosed with diffuse panbronchiolitis, but regular treatments of macrolides were not effective, thus we suspected that he had PCD.
Identification of the CCDC40 Variants
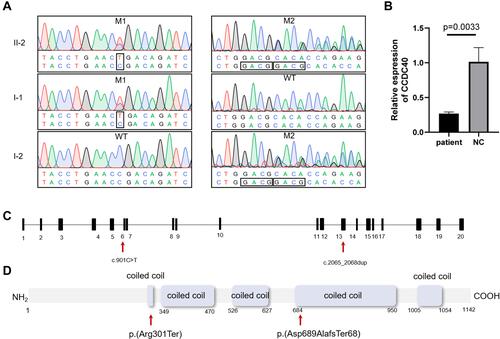
We performed whole-exome sequencing (WES) of peripheral-blood DNA sample from the patient, and detected two novel compound heterozygous mutations in CCDC40. Then we validated the variants from the patient and his parents using Sanger sequencing, and we found that each parent carried different variant and passed it on to the patient. The patient’s father carried mutation 1 (NM_017950.3:c.901C>T, p.(Arg301*)) which was expected to be pathogenic (PVS1 + PM2 + PM3) according to the American College of Medical Genetics and Genomics guidelines.Citation21 The patient’s mother carried mutation 2 (NM_017950.3:c.2065_2068dup, p.(Ala690Glyfs*67)), a pathogenic variant (PVS1 + PM2 + PM3). These variants may lead to nonsense-mediated decay of the truncate mRNA. The patient carried both variants ( and ). To confirm that the variants lead to nonsense-mediated decay of mRNA, we used RT-qPCR to analyze the expression levels of CCDC40 in the patient. As we predicted above, CCDC40 expression level was significantly reduced in the peripheral blood from the patient compared with the healthy control (). The locations of the variants are shown in and .
Figure 2 (A) Sanger-sequencing chromatograms and co-segregation analysis for patient’s family. Mutation 1 (NM_017950.3:c.901C>T, p.(Arg301*)) and mutation 2 (NM_017950.3:c.2065_2068dup, p. (Ala690Glyfs*67)) were identified in patient’s family. (B) Expression levels of CCDC40 are verified using RT-qPCR. A significantly reduced expression of CCDC40 is observed in the peripheral blood from the patient. (C and D) Location of CCDC40 mutations identified in this study are shown with arrows in the gene and protein. Predicted protein domains (coiled coil domains) are indicated by rectangles.
Analysis of Respiratory Cilia and Sperm Flagella
High speed video analysis of ciliated nasal brush biopsies showed that the ciliary beating pattern was typically stiff and rigid, and it also showed a decrease in ciliary beating amplitude and capacity of axonemal bending ( and Supplementary Video 1) compared to that in healthy control (Supplementary Video 2).
Figure 3 (A) Ciliary beating patterns of healthy cilia and our patient with CCDC40 mutations. (B) Papanicolaou staining showing morphology of the sperms from the patient demonstrated short, bent, coiled, and irregular flagella and other MMAF phenotypes. Scale bar, 10μm. (C) Scanning electron microscope of the sperms from the patient, showing bent, short, coiled and irregular flagella. Scale bar, 5μm.
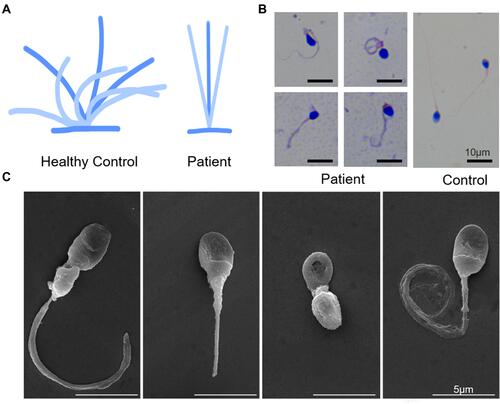
The computer-aided sperm analysis showed the sperms were completely immotile. Papanicolaou staining and scanning electron microscope demonstrated the abnormal morphology of the sperms from the patient demonstrated short, curled, and irregular flagella ( and ), in comparison with that from the normal control. We conducted semen analysis according to the WHO guidelines () and concluded that the patient had asthenoteratospermia given the immotility and the abnormal morphology of the sperm.
Table 1 Semen Parameters and Sperm Flagella Morphology in the Patient Carrying CCDC40 Variants and the Normal Control
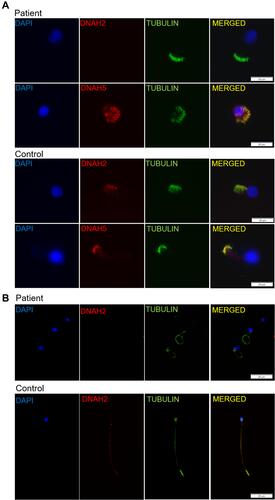
Immunofluorescence analysis of nasal ciliated cells showed absence of inner dynein arm (IDA) protein DNAH2, while the outer dynein arm (ODA) protein DNAH5 was intact (). We also performed immunofluorescence of sperms using antibodies targeting DNAH2, which was characterized as a marker of MMAF. As is shown in , the flagellum of the sperm from the patient lacked DNAH2. These results together confirmed the MMAF phenotype in the patient.
Figure 4 (A) Immunofluorescence analysis of nasal cilia cell of the patient and healthy control, showing presence of outer dynein arm protein DNAH5 and absence of the inner dynein arm protein DNAH2 in the patient. Scale bar, 20μm. (B) Immunofluorescence staining of sperms from the patient and the healthy control with α-tubulin antibodies (green) and DNAH2 (red), a marker for MMAF phenotype. DNAH2 is absent in the sperms from the patient. Scale bar, 20μm.
Hence, the patient was finally diagnosed PCD (caused by CCDC40 compound heterozygous mutations) and MMAF.
Discussion
We report a patient with novel compound heterozygous variants in CCDC40, exhibiting PCD and MMAF. To our knowledge, it is also the first report of CCDC40 mutation presenting MMAF phenotype.
CCDC40, located on chromosome 17 with 20 exons, encodes a 1142-residue protein. CCDC40 was first identified by Becker-Heck to possibly play an important role in assembly of dynein regulatory complex and inner dynein arm (IDA) complex which regulated ciliary beat.Citation13 Dynein proteins form the IDA and outer dynein arms (ODA). Both IDA and ODA are crucial components of the 9+2 structure of the axoneme, the core structure of the motile cilia and sperm flagella.Citation4 Becker-Heck’s discovery was emphasized by their following identification of 13 mutant alleles in CCDC40 from 8 PCD families. These variants were nonsense, frameshift or splicing mutations, thus deprived the structure of the protein, and led to its function loss.Citation24,Citation25 In our case, similar to prior reported cases, both of the two variants are in the exons encoding coiled-coil domains, and are predicted to lead to nonsense-mediated decay of mRNA.
Pathogenic variants of CCDC40 result in inner dynein arm absence, abnormal central apparatus and microtubular disorganization.Citation26 In addition, CCDC40 is expressed in the embryonic node and midline, and it controls left-right patterning.Citation13 Pereira et al reported a splicing (c.1989+1G>A) and a frameshift insertion (c.2824_2825insCTGT) variant in CCDC40 which resulted in Kartagener Syndrome in a female child.Citation27 However, about half of the patients with PCD do not exhibit situs inversus,Citation28 as in our case, the patient does not present situs inversus, which makes it difficult to confirm the diagnosis relying only on the clinical characteristics.
PCD is caused by mutations of genes coding motile-cilia-related proteins, and it is characterized by chronic respiratory infections due to decreased airway clearance, with or without situs inversus and infertility.Citation1,Citation2 Up to now, no diagnostic test can be relied on solely to confirm this disease.Citation3 To confirm PCD in a patient with typical clinical features, a hallmark transmission electron microscope defect of ciliary or biallelic causative variants in PCD-associated genes should be established.Citation29 We confirmed PCD in our case due to identification of the compound heterozygous variants in CCDC40, and the patient’s symptoms of chronic sinusitis, bronchiectasis, reduced nasal nitric oxide, which fit classic PCD phenotype.
CCDC40 pathogenic variants associate with poor and early-presented pulmonary functions, but significant heterogeneity of pulmonary involvement exists among different patients.Citation30 Ghandourah et al reported a female PCD neonate suffered from severe respiratory distress two hours after birth. Genetic test of the patient identified homozygous frameshift in CCDC40 (c.1416delG).Citation31 Emiralioğlu et al’s study reported CCDC40 mutant patients had lower FEV1 predicted values (median 53%, min 46%, max 96%) than those with other mutants.Citation23 In a Chinese study conducted by Guo et al involving 50 children with PCD, children with CCDC40 pathogenic variants present with mild lung disease.Citation30 In our report, FEV1 was 64.2% of the predicted value after salbutamol inhalation, consistent with Emiralioğlu et al’s findings.Citation23 High speed video analysis of respiratory cilia showed that the ciliary beating pattern was typically stiff and rigid, and it showed a decrease in ciliary beating amplitude and capacity of axonemal bending, in accordance with Raidt et al’s study.Citation32
The sperm flagellum and motile cilia of the airways share evolutionarily-highly-conserved structural elements, thus it is understandable that male infertility has been reported in some patients with PCD.Citation5 However, most studies did not report MMAF in PCD for several reasons. Firstly, most of the previously reported PCD patients were children whose semen was unavailable for collection and analysis. Secondly, the majority of PCD cases were identified in respiratory units and the symptoms of concern were predominantly respiratory, thus MMAF may be underdiagnosed. Thirdly, the concept of MMAF was introduced decades later than PCD. It was since 2014 when MMAF was first reported in 7 subjects carrying DNAH1 variants that it was widely used to describe the abnormal morphology of sperm flagella.Citation6 However, these subjects with DNAH1 variants presented with MMAF but without typical PCD symptoms.Citation6 It was not until Tu et al reported the SPEF2 mutation case series in 2020 that MMAF was first associated with PCD.Citation10 Up to now, ARMC4, CCDC39, DRC1, CFAP74 and BRWD1 are the other few PCD-associated genes reported to be related to MMAF phenotype.Citation7–Citation9,Citation11,Citation12 CCDC40 protein functions properly with CCDC39, forming a molecular ruler to determine the 96-nm repeat length and arrangements of components in cilia and flagella, thus CCDC40 was also assumed a good candidate for MMAF-associated genes.Citation5,Citation33 Sylvain et al reported a CCDC40 mutant cohort, two males of which presented sperm defects of asthenozoospermia.Citation25 Recently, Liu et al reported 2 novel mutations (c.1259delA and EX17_20 deletion) in CCDC40 in a male PCD patient and his family, resulting in sperm immobility and infertility.Citation22 However, these patients were not reported to have MMAF. In our report, the patient carrying CCDC40 variants was confirmed to have MMAF phenotype, verifying the previous assumption.
Conclusion
In conclusion, we identified novel compound homozygous variants of CCDC40 in a patient with PCD and MMAF. We first describe the case of a patient with a combination of PCD and MMAF associated with CCDC40 variants. Our study provides initial evidence that CCDC40 is associated with MMAF, which expands the genetic spectrum of PCD and MMAF, and provides precise clinical genetic counseling to this family.
Data Sharing Statement
The original contributions presented in the study are included in the article, and further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Review Board of the Second Xiangya Hospital of Central South University in China (approval number 2020082). Written informed consent was obtained from the patient for the publication of any potentially identifiable images or data included in this article.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Acknowledgments
The authors wish to thank the patient and medical staff of the department of Pulmonary and Critical Care Medicine for their help and collaborations.
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Funding
References
- Zariwala MA, Knowles MR, Omran H. Genetic defects in ciliary structure and function. Annu Rev Physiol. 2007;69:423–450. doi:10.1146/annurev.physiol.69.040705.141301
- Escudier E, Duquesnoy P, Papon JF, Amselem S. Ciliary defects and genetics of primary ciliary dyskinesia. Paediatr Respir Rev. 2009;10(2):51–54. doi:10.1016/j.prrv.2009.02.001
- Shapiro AJ, Zariwala MA, Ferkol T, et al. Diagnosis, monitoring, and treatment of primary ciliary dyskinesia: PCD foundation consensus recommendations based on state of the art review. Pediatr Pulmonol. 2016;51(2):115–132. doi:10.1002/ppul.23304
- Wallmeier J, Nielsen KG, Kuehni CE, et al. Motile ciliopathies. Nat Rev Dis Primers. 2020;6(1):77. doi:10.1038/s41572-020-0209-6
- Coutton C, Escoffier J, Martinez G, Arnoult C, Ray PF. Teratozoospermia: spotlight on the main genetic actors in the human. Hum Reprod Update. 2015;21(4):455–485. doi:10.1093/humupd/dmv020
- Ben Khelifa M, Coutton C, Zouari R, et al. Mutations in DNAH1, which encodes an inner arm heavy chain dynein, lead to male infertility from multiple morphological abnormalities of the sperm flagella. Am J Hum Genet. 2014;94(1):95–104. doi:10.1016/j.ajhg.2013.11.017
- Gao Y, Xu C, Tan Q, et al. Case report: novel biallelic mutations in ARMC4 cause primary ciliary dyskinesia and male infertility in a Chinese family. Front Genet. 2021;12:715339. doi:10.3389/fgene.2021.715339
- Chen D, Liang Y, Li J, et al. A novel CCDC39 mutation causes multiple morphological abnormalities of the flagella in a primary ciliary dyskinesia patient. Reprod Biomed Online. 2021;43:920–930. doi:10.1016/j.rbmo.2021.07.005
- Lei C, Yang D, Wang R, et al. DRC1 deficiency caused primary ciliary dyskinesia and MMAF in a Chinese patient. J Hum Genet. 2021;67:197–201. doi:10.1038/s10038-021-00985-z
- Tu C, Nie H, Meng L, et al. Novel mutations in SPEF2 causing different defects between flagella and cilia bridge: the phenotypic link between MMAF and PCD. Hum Genet. 2020;139(2):257–271. doi:10.1007/s00439-020-02110-0
- Sha Y, Wei X, Ding L, et al. Biallelic mutations of CFAP74 may cause human primary ciliary dyskinesia and MMAF phenotype. J Hum Genet. 2020;65(11):961–969. doi:10.1038/s10038-020-0790-2
- Guo T, Tu CF, Yang DH, et al. Bi-allelic BRWD1 variants cause male infertility with asthenoteratozoospermia and likely primary ciliary dyskinesia. Hum Genet. 2021;140(5):761–773. doi:10.1007/s00439-020-02241-4
- Becker-Heck A, Zohn IE, Okabe N, et al. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat Genet. 2011;43(1):79–84. doi:10.1038/ng.727
- Sui W, Hou X, Che W, et al. CCDC40 mutation as a cause of primary ciliary dyskinesia: a case report and review of literature. Clin Respir J. 2016;10(5):614–621. doi:10.1111/crj.12268
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–595. doi:10.1093/bioinformatics/btp698
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi:10.1093/nar/gkq603
- Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:111011–111033.
- Cooper TG, Noonan E, von Eckardstein S, et al. World Health Organization reference values for human semen characteristics. Hum Reprod Update. 2010;16(3):231–245. doi:10.1093/humupd/dmp048
- Yang SM, Li HB, Wang JX, et al. Morphological characteristics and initial genetic study of multiple morphological anomalies of the flagella in China. Asian J Androl. 2015;17(3):513–515. doi:10.4103/1008-682X.146100
- Tang S, Wang X, Li W, et al. Biallelic mutations in CFAP43 and CFAP44 cause male infertility with multiple morphological abnormalities of the sperm flagella. Am J Human Gene. 2017;100(6):854–864. doi:10.1016/j.ajhg.2017.04.012
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
- Liu L, Zhou K, Song Y, Liu X. CCDC40 mutation as a cause of infertility in a Chinese family with primary ciliary dyskinesia. Medicine. 2021;100(51):e28275. doi:10.1097/MD.0000000000028275
- Emiralioglu N, Taskiran EZ, Kosukcu C, et al. Genotype and phenotype evaluation of patients with primary ciliary dyskinesia: first results from Turkey. Pediatr Pulmonol. 2020;55(2):383–393. doi:10.1002/ppul.24583
- Antony D, Becker-Heck A, Zariwala MA, et al. Mutations in CCDC39 and CCDC40 are the major cause of primary ciliary dyskinesia with axonemal disorganization and absent inner dynein arms. Hum Mutat. 2013;34(3):462–472. doi:10.1002/humu.22261
- Blanchon S, Legendre M, Copin B, et al. Delineation of CCDC39/CCDC40 mutation spectrum and associated phenotypes in primary ciliary dyskinesia. J Med Genet. 2012;49(6):410–416. doi:10.1136/jmedgenet-2012-100867
- Davis SD, Ferkol TW, Rosenfeld M, et al. Clinical features of childhood primary ciliary dyskinesia by genotype and ultrastructural phenotype. Am J Respir Crit Care Med. 2015;191(3):316–324. doi:10.1164/rccm.201409-1672OC
- Pereira R, Barbosa T, Gales L, et al. Clinical and genetic analysis of children with kartagener syndrome. Cells. 2019;8(8):900. doi:10.3390/cells8080900
- Lucas JS, Davis SD, Omran H, Shoemark A. Primary ciliary dyskinesia in the genomics age. Lancet Respir Med. 2020;8(2):202–216. doi:10.1016/S2213-2600(19)30374-1
- Shapiro AJ, Davis SD, Polineni D, et al. Diagnosis of primary ciliary dyskinesia. An official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;197(12):e24–e39. doi:10.1164/rccm.201805-0819ST
- Guo Z, Chen W, Wang L, Qian L. Clinical and genetic spectrum of children with primary ciliary dyskinesia in China. J Pediatr. 2020;225(157–165):e155. doi:10.1016/j.jpeds.2020.05.052
- Ghandourah H, Dell SD. Severe disease due to CCDC40 gene variants and the perils of late diagnosis in primary ciliary dyskinesia. BMJ Case Rep. 2018;2018. doi:10.1136/bcr-2018-224964
- Raidt J, Wallmeier J, Hjeij R, et al. Ciliary beat pattern and frequency in genetic variants of primary ciliary dyskinesia. Eur Respir J. 2014;44(6):1579–1588. doi:10.1183/09031936.00052014
- Oda T, Yanagisawa H, Kamiya R, Kikkawa M. A molecular ruler determines the repeat length in eukaryotic cilia and flagella. Science. 2014;346(6211):857–860. doi:10.1126/science.1260214
- Auger J, Jouannet P, Eustache F. Another look at human sperm morphology. Hum Reprod. 2016;31(1):10–23. doi:10.1093/humrep/dev251