
Abstract
Mowat-Wilson syndrome is a rare, autosomal dominant neurodevelopmental disorder characterized by distinctive facial gestalt and intellectual disability that is often associated with microcephaly, seizures and multiple congenital anomalies, mainly heart defects. More than 350 patients and 180 genetic variants in the ZEB2 gene, have been reported with an estimated frequency of 1 per 70,000 births. Here we report a Colombian female patient with facial gestalt, intellectual disability, microcephaly, congenital heart defects, hypothyroidism and middle ear defect associated with the nonsense pathogenic variant c.2761C>T (p.Arg921Ter) in the ZEB2 gene. This case contributes to the understanding of the clinical complications and the natural history of this complex and clinically heterogeneous disorder but also to the awareness that patients with heart congenital defects and dysmorphic facies may present an underlying genetic disorder.
Introduction
Mowat-Wilson syndrome (MWS, OMIM #235730) is a rare, complex and autosomal dominant genetic developmental disorder characterized by distinctive facial gestalt, mild-to-moderate intellectual disability, severe neurodevelopmental impairment and multiple congenital anomalies in several organs and tissues such as genital anomalies, congenital heart disease, partial or total corpus callosum agenesis, anomalies of the digestive tract, musculoskeletal anomalies, pulmonary stenosis, eye and visual defects and impaired speech (no speech or limited to few words).Citation1,Citation2 Common findings include seizures (78% of cases), microcephaly (77%), congenital heart defects (58%), short stature (46%), Hirschsprung disease (44%), genital anomalies (43%) with hypospadias (59%) and cryptorchidism (41%) in males being the most frequent, and renal anomalies (25%).Citation3 The distinctive facial features of MWS comprise uplifted earlobes, rounded nasal tip, prominent columella, broad nasal bridge, hypertelorism, open mouth, fine hair, microcephaly, high forehead, deep-set eyes and so on.Citation3 Currently, more than 350 cases have been described in the literatureCitation4 with an estimated incidence of at least 1 per 70,000 births.Citation5
MWS is caused by de novo heterozygous variants or deletions in the Zinc Finger E-Box-binding homeobox 2 (ZEB2) gene located in chromosome 2.Citation6 Zeb2 protein, made up of 1214 amino acids, is both a positive and negative regulator of gene expression that is essential for neural tube and neural crest formation, differentiation of hippocampal neurons, gliogenesis, development of interneurons and dopaminergic neurons, development of the neocortex and also has important functions in the cerebellum, telencephalon, the derivatives of neural crest (Schwann cells, sensory neurons, enteric nervous system, melanocytes), in the ventral spinal cord (affecting visceral motor development and function), among other cells; which explain several of the clinical manifestations observed in MWS patients.Citation2,Citation7
The genetic alterations in the ZEB2 gene often lead, in the majority of MWS patients, to haploinsufficiency.Citation6,Citation8 So far, more than 180 different genetic alterations have been reported in the ZEB2 gene causing MWSCitation8 with non-sense variants (33%) and small indels (40%) being the most frequent.Citation3 Although infrequent, missense variants have also been reported but patients tend to have a mild MWS phenotype.Citation8 Herein, we report a Colombian patient with a heterozygous nonsense variant in the ZEB2 gene coursing with microcephaly, congenital heart defect, hypothyroidism, facial gestalt and developmental impairment.
Case Presentation
The proband is a seven-year-old female patient first-born to non-related parents and delivered at 41 weeks of gestation by cesarean section without complications. Prenatal ultrasounds were normal, without signs suggestive of microcephaly but with the presence of polyhydramnios. Her mother and father were aged 35 and 30 years, respectively, at the time of birth. Birth weight and height were 3350 g (44th percentile) and 51 cm (71st percentile), respectively. Her maternal grandmother was diagnosed with Parkinson’s disease at 55 years old and the paternal grandfather with adolescent-onset epilepsy. No other family history was reported.
At 7 months old, the patient was diagnosed with patent ductus arteriosus (PDA), which was surgically corrected. Mild-to-moderate pulmonary valve stenosis and patent foramen ovale were also noted. At this same age, hypothyroidism was identified in the patient and pharmacological treatment was started. At 2 years old, mild stenosis of the left branch of the pulmonary artery and mild mitral regurgitation were identified. Brain computed tomography scan (CT) and Magnetic Resonance Imaging (MRI) were normal. At the age of 3 years old, episodes of tonic-clonic seizures began and to date, are treated with valproic acid and levetiracetam. At age 5, impedanciometry was suggestive of middle ear dysfunction and laryngoscopy indicated granular pharyngitis and reflux laryngopharyngitis. At age six, the echocardiogram revealed ostium secundum atrial septal defect, dysplastic pulmonary valve with stenosis and mild regurgitation, and right ventricle with mild hypertrophy and dilatation ().
Table 1 MWS Clinical Manifestations in the Patient (Not All Listed) and Some Cases Reported in the Literature with the c.2761C>T (p.Arg921Ter) Variant. Only Cases with Detailed Data Were Included
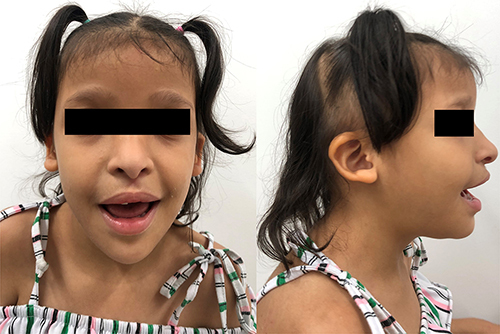
Physical examination at the current age () revealed intellectual disability, global developmental impairment, including language and motor development, wide intermammillary distance, desquamative skin rash in hands and facial gestalt comprising microcephaly (head circumference: 48.2 cm, <1st percentile), high forehead, low hairline, coarse and sparse hair, sparse eyebrows, telecanthus, low and posteriorly set ears, anteverted nares, short columella, bulbous nasal tip, high-arched palate and short neck. Weight was 29.6 kg (89th percentile) and height was 117.5 cm (23rd percentile). Holosystolic murmur II/VI was present in the upper left sternal border. RASopathies panel testing performed by next-generation sequencing was normal and G-banded karyotype was also normal (46, XX). However, the proband-only (single-based) exome sequencing, through Illumina HiSeq® and a mean depth coverage of 169X, showed the heterozygous and pathogenic variant (criteria: PVS1, PP5, PM2, according to the College of Medical Genetics and Genomics ACMGCitation9) c.2761C>T (p.Arg921Ter) in the ZEB2 gene (NM_014795.4); therefore, the diagnosis of MWS was established.
Figure 1 Colombian patient with MWS. Seven-year-old female with long face, sparse eyebrows, widely spaced eyes, downturned palpebral fissures, short columella, low-set and posteriorly rotated ears.
Discussion
Mowat-Wilson syndrome (MWS) is a rare genetic disorder with no current diagnostic criteria established and therefore, its diagnosis is based on the characteristic facial gestalt and intellectual disability with a genetic alteration in the ZEB2 gene that usually leads to haploinsufficiency. Frequent but not obligatory alterations include microcephaly, seizures, behavioral anomalies and late walking. Associated features may also be present and include short stature, eye alterations, Hirschsprung disease and congenital heart defects.Citation5 The patient described in this case, with confirmed MWS, presented typical features of this disorder (post-natal microcephaly, developmental delay, intellectual disability, congenital heart defect and epilepsy) but also middle ear dysfunction and hypothyroidism, symptoms not commonly reported as part of MWS manifestations. However, whether these last two clinical manifestations are due to the genetic alteration in the ZEB2 gene or coincidental findings is not clear.
Genetic alterations leading to MWS are de novo.Citation7 However, in rare cases, germline mosaicism in one healthy parent, with a negative genetic blood test, can be the cause of MWS in the offspringCitation10 and therefore, even if the probability of recurrence is small (1–2%),Citation5 genetic counseling and testing is necessary in all parents of MWS patients, especially when two or more siblings are affected with the same variant.Citation11 Our patient is an only child and although genetic testing of the healthy parents normally does not give useful information for MWS, given the neurological diseases of her maternal grandmother and paternal grandfather, genetic analysis of parents and grandparents could give some important information such as the presence of polymorphisms or mutations that contribute to the heterogeneity of the clinical manifestations seen in our patient (see below).
The c.2761C>T, (p.Arg921Ter) pathogenic variant detected in the ZEB2 gene of this patient affects exon 8 of the gene and has already been described in different patients. It was first described in 2005 by Zweier et alCitation10 in two MWS European patients. Currently, more than nine patients are reported in the ClinVar database, which includes patients from EuropeCitation10,Citation12–14 and Japan.Citation15 This variant seems to be a recurrent mutation leading to MWS but with a striking phenotypic variability among the patients (). Not all patients with this same variant present with microcephaly as our patient did, the same congenital heart defects or corpus callosum agenesis. The only concordant clinical features are delayed development, intellectual disability and seizures as some facial dysmorphisms can be also different. For instance, one patient had microphthalmia and some do not have microcephaly. This clinical heterogeneity with the same variants in the same gene may be caused by different mechanisms such as epigenetic modifications (environment-genome interaction), polymorphismsCitation3 or even other genetic changes not detected in the patients. However, further studies are needed to explore this hypothesis.
In the diagnostic work-up for this case, a RASopathy panel was performed due to the combination of features resembling Noonan syndrome (NS) such as short stature, intellectual disability, pulmonary valve stenosis, high forehead, and low-set ears.Citation16 This highlights the utility of a wide-scope diagnostic test such as clinical exome in the setting of patients with developmental delay/intellectual disability and multiple congenital malformations, and adds MWS to the long list of potential differential diagnoses of NS and related RASopathies.Citation17
In Colombia, as far as we know, only four cases of MWS have been published,Citation18–21 from which, only one reported the genetic variant and it was a 2q22.32-q22.3 deletion,Citation19 including the KYNU, ARHGAP15, GTDC1 and ZEB2 genes. Hence, this is the first report of the c.2761C>T, (p.Arg921Ter) genetic variant in the country. This report provides important information given the complexity of MWS and accentuates the need to understand the clinical complications and natural history of this disease.Citation5 Furthermore, early diagnosis, including the genetic diagnosis, of children with MWS is beneficial because this condition is associated with a wide spectrum of clinical manifestations and multidisciplinary follow-up is of paramount importance to improve the quality of life of MWS patients, given that there is no cure to date.Citation1 Follow-up includes evaluation by pediatricians, clinical geneticists, neurologists, nephrologists, cardiologists, ophthalmologists, immunologists, and gastroenterologists. A guideline for management and follow-up of children with MWS can be found in the work published by Ivanovski et al 2018.Citation1 Accordingly, the identification and diagnosis of this genetic disorder in our patient, led to a more informed anticipatory and multidisciplinary follow-up by different clinical specialties.
In conclusion, we report a patient with the heterozygous variant c.2761C>T, (p.Arg921Ter) in the ZEB2 gene leading to MWS. This patient presented with delayed development, intellectual disability, microcephaly, congenital heart defects, facial gestalt, hypothyroidism and middle ear defect. The last two symptoms are not commonly reported as associated with this condition; therefore, this case expands the phenotypic spectrum of the variant reported here and the overall manifestations of MWS patients. Given the complexity of the disease, early diagnosis will allow monitoring for complications that in this complex disease, with a tendency to multiple congenital defects and other severe complications, will significantly impact the life quality of the patient. In this manner, genetic analysis of patients with congenital heart defects and facial gestalt is recommended.
Abbreviations
MWS, Mowat-Wilson Syndrome; ZEB2, Zinc Finger E-Box-binding homeobox 2 gene; NS, Noonan syndrome.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Informed Consent
This study was approved by the Ethics Committee of Fundación Valle del Lili, Colombia (human study protocol #1504) and performed in accordance with the Declaration of Helsinki. Written informed consent was obtained from the parents of the patient. Information revealing the subject’s identity was not included in the manuscript. The patient was identified by number and not by his real name.
Consent for Publication
Written informed consent for publication of clinical details and images/photographs was obtained from the parents of the patient.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest in this work.
Acknowledgments
The authors would like to thank the patient and her parents for agreeing to the publication of this report. We also thank the people who have contributed to the development and execution of this study.
Additional information
Funding
References
- Evans E, Einfeld S, Mowat D, Taffe J, Tonge B, Wilson M. The behavioral phenotype of Mowat-Wilson syndrome. Am J Med Genet Part A. 2012;158(A2):358–366. doi:10.1002/ajmg.a.34405
- Cordelli DM, Di Pisa V, Fetta A, et al. Neurological phenotype of Mowat-Wilson syndrome. Genes. 2021;12(7):982. doi:10.3390/genes12070982
- Ivanovski I, Djuric O, Caraffi SG, et al. Phenotype and genotype of 87 patients with Mowat–Wilson syndrome and recommendations for care. Genet Med. 2018;20(9):965–975. doi:10.1038/gim.2017.221
- Ivanovski I, Djuric O, Broccoli S, et al. Mowat-Wilson syndrome: growth charts. Orphanet J Rare Dis. 2020;4:1–12.
- Mowat D, Wilson M. Mowat–Wilson syndromE. In: Carey JC, Battaglia A, Viskochil D, Cassidy SB, editors. Cassidy and Allanson’s Management of Genetic Syndromes. 4 ed. New York: John Wiley & Sons; 2021:597–609.
- Garavelli L, Ivanovski I, Caraffi SG, et al. Neuroimaging findings in Mowat-Wilson syndrome: a study of 54 patients. Genet Med. 2017;19(6):691–700. doi:10.1038/gim.2016.176
- Epifanova E, Babaev A, Newman AG, Tarabykin V. Role of Zeb2/Sip1 in neuronal development. Brain Res. 2019;1705:24–31. doi:10.1016/j.brainres.2018.09.034
- Ghoumid J, Drevillon L, Alavi-Naini SM, et al. ZEB2 zinc-finger missense mutations lead to hypomorphic alleles and a mild Mowat-Wilson syndrome. Hum Mol Genet. 2013;22(13):2652–2661. doi:10.1093/hmg/ddt114
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
- Zweier C, Thiel CT, Dufke A, et al. Clinical and mutational spectrum of Mowat-Wilson Syndrome. Eur J Med Genet. 2005;48(2):97–111. doi:10.1016/j.ejmg.2005.01.003
- Cecconi M, Forzano F, Garavelli L, et al. Recurrence of Mowat-Wilson syndrome in siblings with a novel mutation in the ZEB2 gene. Am J Med Genet Part A. 2008;146(23):3095–3099. doi:10.1002/ajmg.a.32568
- Pons L, Dupuis-Girod S, Cordier MP, Edery P, Rossi M. ZEB2, a new candidate gene for asplenia. Orphanet J Rare Dis. 2014;9(1):1–2. doi:10.1186/1750-1172-9-2
- Garavelli L, Zollino M, Cerruti Mainardi P, et al. Mowat-Wilson syndrome: facial phenotype changing with age: study of 19 Italian patients and review of the literature. Am J Med Genet Part A. 2009;149(3):417–426. doi:10.1002/ajmg.a.32693
- Kilic E, Cetinkaya A, Utine GE, Boduroǧlu K. A diagnosis to consider in intellectual disability: Mowat-Wilson syndrome. J Child Neurol. 2016;31(7):913–917. doi:10.1177/0883073815627884
- Yamada Y, Nomura N, Yamada K, et al. The spectrum of ZEB2 mutations causing the Mowat-Wilson syndrome in Japanese populations. Am J Med Genet Part A. 2014;164(8):1899–1908. doi:10.1002/ajmg.a.36551
- Lores J, Prada CE, Ramírez-Montaño D, Nastasi-Catanese JA, Pachajoa H. Clinical and molecular analysis of 26 individuals with Noonan syndrome in a reference institution in Colombia. Am J Med Genet Part C Semin Med Genet. 2020;184(4):1042–1051. doi:10.1002/ajmg.c.31869
- Bhoj EJ, Yu Z, Guan Q, et al. Phenotypic predictors and final diagnoses in patients referred for RASopathy testing by targeted next-generation sequencing. Genet Med. 2017;19(6):715–718. doi:10.1038/gim.2016.169
- Vaniaa Villota DA, Wilmar Saldarriaga G, Juan Fernando Gómez C. Síndrome de Mowat-Wilson: caso clínico [Mowat-Wilson’s syndrome: a case report]. Rev Chil Pediatr. 2012;83(4):371–376. doi:10.4067/S0370-41062012000400008
- María A, Johanna Z-B, Acosta-Guio C. Síndrome de Mowat – Wilson, primer caso reportado en Colombia. Importancia de un estudio ampliado por genética [Mowat-Wilson syndrome, first case reported in colombia. importance of expanded by genetic study]. Rev Salud Bosque. 2013;3(2):55–60.
- Benedetti-Isaac J, Torres-Zambrano M, Alcalá-Cerra G, Gutiérrez-Paternina J. Vagus nerve stimulation for drug-resistant epilepsy in a patient with Mowat-Wilson syndrome. Neurol India. 2013;61(3):306–307. doi:10.4103/0028-3886.115074
- Cano Sierra JD, Mestra CF, Ronderos Dumit MA. Incidental finding of pulmonary arterial sling during patent ductus arteriosus surgery in a patient with Mowat-Wilson syndrome. Cardiol Young. 2018;28(8):1074–1076. doi:10.1017/S1047951118000689