
Abstract
Background
The IQ motif and Sec7 domain ArfGEF 2 (IQSEC2), an X-linked gene that encodes the BRAG1 protein, is a guanine nucleotide exchange factor for the ADP ribosylation factor (ARF) protein family in the small guanosine triphosphate (GTP) binding protein. Mutations in this gene result in disorders such as intellectual disability (ID) and epilepsy. In this study, we analyze the clinical features of two patients with IQSEC2-mutation-related disease and discuss their possible pathogenesis.
Methods
The two patients were diagnosed with ID and epilepsy. Genetic testing was performed using whole-exome sequencing, and the three-dimensional protein structure was analyzed. UCSC Genome Browser was used to analyze the conservation of IQSEC2 in different species. We compared IQSEC2 expression in the proband families with that in a control group, as well as the expression of the postsynaptic identity protein 95 (PSD-95), synapse-associated protein 97 (SAP97), ADP ribosylation factor 6 (ARF-6), and insulin receptor substrate 53kDa (IRSP53) genes interacting with IQSEC2.
Results
We identified two semi-zygote mutations located in conserved positions in different species: an unreported de novo mutation, C.3576C>A (p. Tyr1192*), and a known mutation, c.2983C>T (p. Arg995Trp). IQSEC2 mutations resulted in significant changes in the predicted three-dimensional protein structure, while its expression in the two probands was significantly lower than that in the age-matched control group, and IQSEC2 expression in proband 1 was lower than that in his family members. The expression levels of PSD-95, ARF-6, and SAP97, IRSP 53, which interact with IQSEC2, were also significantly different from those in the family members and age-matched healthy children.
Conclusion
The clinical phenotype resulting from IQSEC2 mutations can be explained by the significant decrease in its expression, loss of function of the mutant protein, and change in the expression of related genes. Our results provide novel insights into the molecular phenotype conferred by the IQSEC2 variants.
Introduction
IQSEC2 (NCBI Reference Sequence: NM_001111125.3), located on chromosome Xp11.22, is 6011 bp in size. The transcript contains 15 exons, and BRAG1, the encoded protein, contains 1488 amino acids. IQSEC2 is named according to its two conserved regions: IQ (347–376) and the SEC7 domain (746–939). The IQ consists of approximately 30 isoleucine and glutamine amino acids and constitutes the initial domain of the IQSEC2 protein and confers calmodulin-binding ability. The SEC7 domain contains approximately 200 amino acids and is responsible for the guanine nucleotide exchange function (GEF). Other functional structures of IQSEC2 include an N-terminal coiled-coil (CC) domain (23–74), a specific PH domain (951–1085) that binds to inositol phosphate alone, and two C-terminal binding motifs that are crucial in the cell scaffold structure. A proline-rich motif (PRM) (1424–1434) and a PDZ-binding motif (1484–1488) are also present. The PDZ-binding motif is named after the initials of three homologous proteins, namely, postsynaptic density protein 95 (PSD-95), Drosophila disc large tumor suppressor (DLG1), and zonula occludens-1 protein (ZO-1).Citation1
IQSEC2 interacts with postsynaptic density protein 95 (PSD-95) through its C-terminal PDZ binding domain, forming a complex with N-methyl-D-aspartate (NMDA) receptor and allowing for Ca2+ influx, which in turn binds to the IQ domain of IQSEC2, thereby inducing a conformational change that activates its catalytic domain SEC7. IQSEC2 acts as a guanine nucleotide exchange factor (GEF) for ADP ribosylation factors (ARFs) through its Sec7 domain, promoting GTP exchange for GDP on ADP ribosylation factor 6 (ARF6) and activating it. The activated ARF6 then promotes the downregulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors through the stress-activated protein kinase JNK.Citation1,Citation2 Therefore, IQSEC2 regulates the membrane structure and synaptic function of neurons. In addition, IQSEC2 plays an important role in regulating the cell skeleton and vesicle transport density of synapses, making it a key regulator of synaptic plasticity.Citation3 All necessary for the proper development of cognition and learning. In this study, we compare and analyze the expression of PSD95, SAP97, ARF6, and IRSP 53 in two boys with IQSEC2 mutations, their family members, and a control group of healthy children of the same age. The mutations were identified as a new nonsense mutation c.3576C>A [p. Tyr1192*] and a missense mutation c.2983C>T [p. Arg995Trp]. Both cases were diagnosed with intellectual disability (ID) and epilepsy, which were consistent with the characteristics of the IQSEC2 mutations. We also evaluate their clinical features, electroencephalogram (EEG) results, laboratory tests, and molecular characteristics to elucidate the pathogenesis of IQSEC2 mutations.
Materials and Methods
Variation Detection
From February 2019 to September 2021, the peripheral venous blood of two boys and their nuclear family were collected at Shanghai Children’s Hospital. Genomic DNA was isolated from the blood samples of the patients, fragmented using Covaris Ultra Sonicator (Covaris, Inc., MA, USA), used to construct a DNA library, and evaluated for quality. Paired-end sequencing for 150-bp reads was performed on the Hiseq 2500 platform (Illumina, Inc., CA, USA) according to the manufacturer’s instructions. Subsequently, the Burrows–Wheeler alignment tool (BWA, version 0.7.15) was used to read the sequencing data and compare it with the NCBI human reference genome (GRCh37/hg19). Common mutations were filtered out based on frequencies (minor allele frequency <0.05) in the Exome Aggregation Consortium (http://exac.broadinstitute.org), the 1000 Genomes Project (http://www.1000genomes.org) database, and the Exome Sequencing Project (https://esp.gs.washington.edu) after the identified variants were filtered and interpreted using Ingenuity Variant Analysis (Qiagen Inc., CO, Germany). The DNA samples of patients and their family members were further sequenced using real-time quantitative polymerase chain reaction (qPCR) and Sanger sequencing.
RNA Extraction and cDNA Synthesis
From each participant 250 μL of blood was collected, and total RNA was extracted using a Qiagen kit (Qiagen Inc., Germany) according to the manufacturer’s instructions (Yeasen Biotechnology Co., Ltd., Shanghai, China). The purity and concentration of the total RNA (A260/280 ratio is between 2.0) were determined from their OD values measured using a nucleic acid–protein analyzer. Using the total RNA as a template, the reverse transcription cassette was reverse-transcribed using the Script Strand cDNA Synthesis Kit/RT Master Mix (Takara Shuzo Co., Ltd.) according to the manufacturer’s instructions. The reaction system was as follows: RNA template, 2 μL (≤500 ng); 5 × gDNA digester mix, 3 μL; RNase free ddH2O, 10 μL. The sample was centrifuged briefly (8000 rcf, 5 min, 4 °C) and incubated at 42 °C for 2 min. Subsequently, 5 μL of 4×Hifair®III SuperMix plus was added and heated at 25 °C for 5 min, 55 °C for 15 min, and 85 °C for 5 min to inactivate the CorYeabioIIRT Mix.
Analysis of Variance
From the GEO (GENE EXPRESSION OMNIBUS) database (https://www.ncbi.nlm.nih.gov/geo/info/datasets.html) Download and integrate the original mRNA expression data of intellectual disability and epilepsy, and analyze the expression of IQSEC2 between normal and diseased tissues. The R software package limma (version 3.40.6) was used to analyze the difference in the expression of IQSEC2. The ggplot2 package was used to generate a boxplot. The screening threshold was: log FC absolute value ≥ 1, and P<0.05.
Co-Expression Analysis
Download the Series Matrix File data file of GSE31718 from the NCBI GEO public database. The annotation file is GPL6254. Analyze the co expression of IQSEC2 in the database, and screen 455 genes significantly related to the expression of IQSEC2. The correlation coefficient is positive/negative correlation TOP25 genes heat map and co expression correlation circle. The correlation coefficient filter condition is 0.6, and the p value is 0.05. After screening the genes most significantly expressed with IQSEC2, the “corrplot” and “circle” packages were used to draw the circle diagram and heat map of IQSEC2 correlation analysis.
qPCR Analysis of the Expression of IQSEC2 and Its Interacting Genes, PSD-95, SAP97, ARF-6, and IRSP53
We extracted cDNA from the blood samples collected from the two patients and their family members and healthy children of the same age control group (4 boys and 4 girls each, aged between 3–5 years old). Review literature and select four genes significantly correlated with IQSEC2 expression from heatmaps for experimentation. Then qPCR (Takara Biomedical Technology) was then performed according to the manufacturer’s instructions to analyze the relative expression of IQSEC2 and its interacting genes, PSD-95, SAP97, ARF-6, and IRSP53, in the family members as well as in the sex- and age-matched control group. The primers used to detect the expression of IQSEC2 and PSD-95, SAP97, ARF-6, and IRSP53 are listed in . The reaction mixture (20 µL) contained cDNA (2 µL), forward primer F (10 µM; 1 µL), reverse primer R (10 µM; 1 µL), 2× SYBR Premix Ex Taq (10 µL), and sterilized water (6 µL).
Table 1 Specific Primers Used for Quantitative Real-time PCR
The reaction conditions for qPCR were as follows: pre-denaturation at 95 °C for 30s, 40 cycles of denaturation at 95 °C for 5 s, and renaturation at 60 °C for 20s, followed by a final extension at 65 °C for 15s. The relative gene expression was calculated using the 2−ΔΔCt method. The samples were analyzed in triplicate, repeat the experiment three times per group, from which the average values and their standard errors were calculated. Asterisks indicate statistically significant differences (Student’s t-test, **P <0.05, ***P <0.01).
Protein Structure Prediction
The I-TASSER server (http://zhanglab.ccmb.med~umich.edu/I-TASSER/) was used to assess missense and nonsense mutations. We predicted the structure of IQSEC2 protein, and its nonsense [c. 3576C>A (P. Tyr1192*)] and missense [c.2983C>T (p. Arg995Trp)] variants. The I-TASSER suite pipeline consisted of four general steps: threading template identification, iterative structure assembly simulation, model selection and refinement, and structure-based functional annotation.
Prediction of RNA Molecular Structure
We used the RNAfold web server (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) to predict the secondary structure of RNA molecules with missense mutation c.2983C>T (p. Arg995Trp) and wild-type IQSEC2 RNA molecules.
Conservative Sequence Analysis
We analyzed loci 1192 and 995 in the IQSEC2 protein sequence for humans, Islupus familiaris, Mus musculus, Rattus norvegicus, Bos taurus, Xenopus tropicalis, and Danio rerio to predict sequence conservation at this locus.
Genetic Pathogenicity Evaluation
The mutation prediction software SIFT (http://sift.jcvi.org), PROVEAN (http://provean.jcvi.org/index.php), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml), and Mutation-Taster (http://www.mutationtaster.org) were used to predict the pathogenicity of nonsense [c. 3576C>A (p.Tyr1192*)] and missense [c.2983C>T (p.Arg995Trp)] mutations. The pathogenicity of the two variants was also evaluated using the pathogenicity prediction guide of the American College of Medical Genetics and Genomics (ACMG).
Evaluation of Neurological Function Test Scale
The patients were unable to complete the intelligence test independently. Consequently, the parents aided the evaluation using the neurological function test scale [Warning Signs for Children’s Mental and Behavioral Development (WSC-MBD), Autism Behavior Scale (ABC), and social life ability scale (S-M)].
Statistical Analysis
Using the R Programming Language developed by Rick Becker, John Chambers and Allan Wilks exist Bell Labs evaluated the differential expression of IQSEC2 between the disease group and the control group, and the positive and negative expression of related genes. The 2−ΔΔCt method was used to analyze the expression of IQSEC2, PSD-95, SAP97, ARF-6 and IRSP53. P<0.05 is considered to indicate a statistically significant difference.
Results
Clinical Data
Proband 1
Proband 1 was a boy of 5 years and 1 month of age, a second child of unrelated parents delivered at full term by cesarean section, with a birth weight of 4100 g. His older brother was healthy, and his mother experienced a normal pregnancy. At 8 months old, the child presented with difficulties in looking up and sitting alone. Brain nuclear magnetic resonance imaging revealed the possibility of brain dysplasia, and consequently, he started rehabilitation. His parents believed that his condition improved after this treatment. When the child was 2 years and 6 months old, he experienced convulsions, which manifested with strabismus or frequent blinking of both eyes, flexing of both upper limbs, clenching of fists, rigidity and shaking of limbs, and seizures lasting 1–2 min, several times a day. The video electroencephalogram (VEEG) revealed a dominant distribution of extensive or multifocal and slow spikes and six isolated spasms. The patient was diagnosed with Infantile Spasms (IS) and ID. Oral sodium valproate (39 mg/kg/d) was prescribed; however, the treatment outcome was poor.
When the child was 3 years old, he experienced another seizure, characterized by upward eye movement and rapid, brief shaking of both upper limbs, occurring in clusters or isolated incidents, with more than 20 episodes each time.
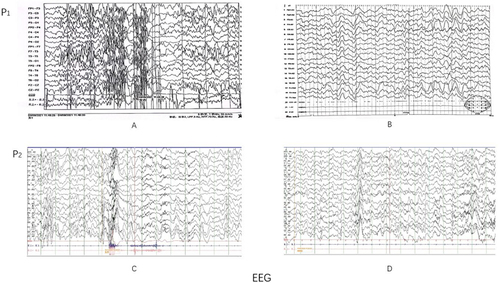
The blood ammonia and lactic acid levels were 38 µmol/L and 1.5 nmol/L, respectively, which were within the normal range. VEEG results indicated peak rhythm disorder and frequent spasms (). Topiramate (3.125 mg/kg/qn), sodium valproate (40 mL/kg/q12), clonazepam (0.015–0.03 mg/kg/qd), and prednisone acetate (0.6–1 mg/kg/d) were prescribed along with a ketogenic diet (Treatment cycle of 3 months). Reexamination of the VEEG revealed minimal discharge during sleep, and the seizures disappeared ().
Figure 1 Electrocardiograms of patients (A) Proband 1 (P1), April 2021, VEEG: spasms seizure; (B) September 2021, VEEG: the seizure disappeared, and peak rhythm disorder disappeared; (C) Proband 2 (P2), October 2019, VEEG: spasms seizure; (D) August 2021, VEEG: After sleep, the rear head showed epileptiform discharge, and the seizure disappeared.
Proband 2
Proband 2 was a boy of 4 years and 3 months of age, a fourth child of non-consanguineous parents. His mother gave birth naturally; he weighed 3350 g and has three healthy older sisters. The mother experienced a normal pregnancy. At 6 months of age, his upward gaze was unsteady; at 12 months, he sat alone, albeit unsteadily. His development lagged behind that of children of the same age. At 1 year of age (June 2019), he had convulsions without evident triggers and presented with a fever. His eyes were staring or left oblique, his lips were blue and purple, his limbs were soft, and he could not shout. Each seizure lasted approximately 1 min, and he convulsed 7–8 times a day. VEEG examination at an external hospital indicated peak rhythm disorder and an isolated spasm during sleep. He was diagnosed with Infantile Spasms (IS) and ID, and sodium valproate (43 mg/kg/d) was prescribed. A few weeks later, the number of seizures increased, and the pattern eventually changed. The seizures manifested as a series of nodding attacks, sometimes accompanied by body impact. The patient was admitted to our hospital in October 2019. The blood ammonia levels and the tandem mass spectrometry results were normal. VEEG results indicated a peak rhythm disorder, with seven spasms during waking and sleeping (). Topiramate (4 mg/kg/d) was prescribed along with adrenocorticotropic hormone ACTH (25 U/d) for three weeks, and subsequently, prednisone acetate (0.8–1.2 mg/kg/d) was administered orally. A reexamination of the VEEG in 2021 revealed intermittent epileptiform discharges during sleep; however, no seizures were detected ().
Variant Detection
Proband 1
The whole exon sequencing revealed a human genome version: Human GRCh37/hg19 (NM_001111125.3) c.3576C>A (p.Tyr1192*) hemizygote mutation in IQSEC2, which is harbored in exon 15 of the X chromosome in proband 1. This variation introduced a pretermination stop codon at amino acid 1192, resulting in the early termination of protein translation. However, Sanger sequencing results confirmed that the parents and brother of proband 1 did not carry the mutation, and thus, it was classified as de novo ().
Figure 2 (A) The sequences of genomic DNA suggest that the nonsense mutation of IQSEC2 c.3576C>A (p. Tyr1192*) was identified in proband 1 but not in his parents and brothers. (B) Missense mutation of IQSEC2 c.2983C>T (p. Arg995Trp) in proband 2 but not in his parents. (C) Secondary molecular structures of IQSEC2 RNA Wild-type. (D) missense mutant RNA.

Proband 2
The whole exon sequencing helped identify the human genome version: Human GRCh37/hg19 (NM_001111125.3) c.2983C>T (p. Arg995Trp) hemizygote variation in exon 10 of IQSEC2 in proband 2. This mutation resulted in an amino acid substitution from arginine to tryptophan at position 995. The Sanger sequencing results confirmed that the parents of proband 2 did not carry mutations, and hence were classified as de novo ().
Results of the Analysis of Variance and qPCR Analysis of the Transcription Level of IQSEC2
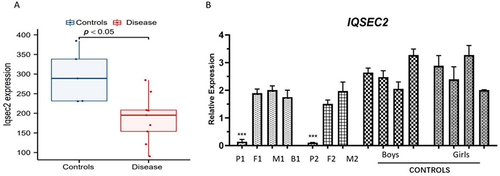
Download the Series Matrix File data file of GSE185031 from the NCBI GEO public database, and included the expression profile data of 14 patients, including 5 cases in the normal group and 9 cases in the disease group. Used for analyzing the differences between IQSEC2 and the normal group. The results showed that the expression of IQSEC2 was lower than that of the normal control group (). The expression levels of IQSEC2 in probands 1 and 2 were significantly lower than those in their core families and control group (P <0.01) ().
Figure 3 (A) Analysis of variance between IQSEC2 and the normal group. The left vertical axis represents gene expression levels, the red box plot represents gene expression levels in the disease group, and the blue box plot represents gene expression levels in the control group. The results showed that the expression of IQSEC2 was lower than that of the normal control group. (B) qPCR of IQSEC2. In proband 1 (P1) and proband 2 (P2), the expression was significantly lower than that of their core family members and normal control group.
Co-Expression Analysis
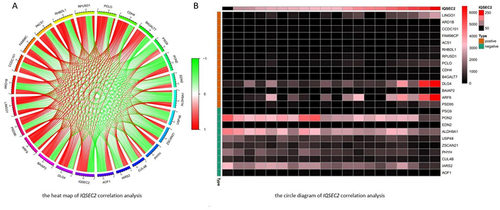
We further explored the co expression network of IQSEC2 through correlation analysis based on the expression profile of epileptic patients in GEO database, with 385 up-regulated and 70 down-regulated. The filter condition of correlation coefficient is 0.6, and the p value is 0.05. A total of 455 genes significantly related to the expression of IQSEC2 were screened (), and the heatmap of genes with positive/negative correlation coefficients with IQSEC2 () and the circular graph of co expression with IQSEC2 ().
Table 2 Genes co expressed with IQSEC2
Figure 4 (A) Analysis circle diagram of Top 25 IQSEC2 related genes, and approaching red indicates positive correlation, approaching green indicates negative correlation. (B) The heatmap of IQSEC2 correlation analysis, and brown represents positive, green represents negative.
The Transcription Level of IQSEC2 and Related Genes Expression Affected by It
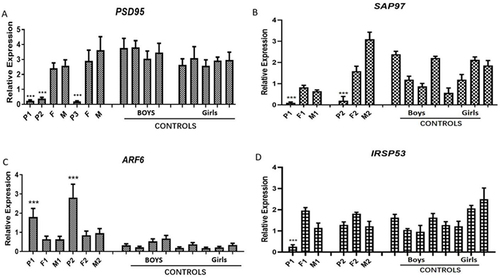
The expression levels of IQSEC2 in probands 1 and 2 were significantly lower than those in their core families and control group (P <0.01) (), and so were those of PSD-95 and SAP97, which were not observed in their parents (P <0.01) ( and ). Conversely, the expression level of ARF6 in the two probands was significantly higher than that in their parents and normal control group (). However, the expression level of IRSP53 in proband 1 was significantly lower than that in the parents and control group, while there was no significant difference in the expression level of IRSP53 in proband 2 compared with their parents and control group (P <0.01) ().
Figure 5 (A) The expression levels of PSD-95 in probands 1 and 2 were significantly lower than normal levels, which were not observed in their parents. (B) The expression levels of SAP97 in probands 1 and 2 were significantly lower than normal levels, which were not observed in their parents. (C) The expression levels of ARF6 in probands 1 and 2 were significantly higher than those in their parents and normal control group. (D) The expression level of IRSP53 in proband 1 was significantly lower than that in the parents and control group, while there was no significant difference in the expression level of IRSP53 in proband 2 compared with their parents and control group.
Changes in the Protein Structure and Function
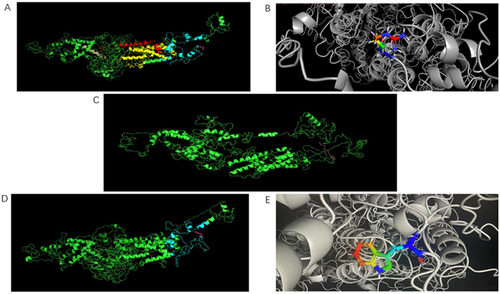
We compared the three-dimensional structures of the IQSEC2 wild-type ( and B) and the nonsense () and missense ( and ) variants. The three-dimensional structures of the two mutants differed from those of the wild-type. We speculate that these differences may change the structure of the protein, thus affecting its function and stability.
Figure 6 (A) Three-dimensional structure of IQSEC2 wild-type protein. (B) Fine structure of wild-type amino acid at position 1192. (C) Nonsense mutation c.3576C>A (p. Tyr1192*). (D) Missense mutation c.2983C>T (p. Arg995Trp). (E) Fine structure of missense mutation at position 995.
Prediction of RNA Molecular Structure
We compared the RNA secondary structure of wild-type IQSEC2 with that of missense mutation. The secondary structure of wild-type RNA () was different from that of missense mutant RNA (). We speculate that the change in the structure of the missense mutant RNA may lead to its instability and degradation before it is translated into protein. Therefore, the expression of IQSEC2 of missense mutation in proband 2 was reduced.
Conservation of the Missense and Nonsense Mutations
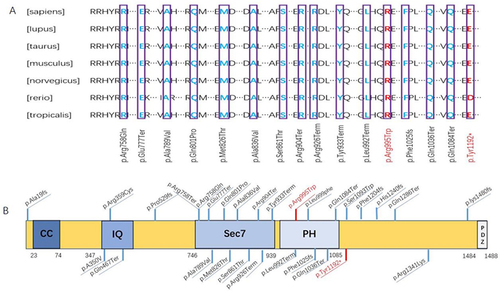
Conservation studies were conducted for c.3576C>A (p. Tyr1192*) and c.2983C>T (p. Arg995Trp) in humans, Canis lupus familiaris, Mus musculus, Rattus norvegicus, Bos taurus, Xenopus tropicalis, and Danio rerio (). These analyses suggest that the mutations are relatively stable.
Figure 7 (A) The IQSEC2 protein sequence. Conservative studies were conducted for c.3576C>A (p. Tyr1192*) and c.2983C>T (p. Arg995Trp) in humans, Canis lupus familiaris, Mus musculus, Rattus norvegicus, Bos Taurus, Xenopus tropicalis, and Danio rerio. (B) Primary structure of IQSEC2 and the common mutation sites. Coiled-coil domain (CC): 23–74; IQ motif (IQ): 347–376; SEC7 domain (SEC7):746–939; pleckstrin homology (PH): 951–1085; PDZ-binding motif (PDZ):1484–1488.
Pathogenicity of the Genetic Variants
We evaluated the pathogenicity of the identified nonsense [c. 3576C > A (p.Tyr1192*)] and missense [c.2983C>T (p.Arg995Trp)] mutations using various pathogenicity prediction programs. The results indicate that the two mutants were potentially harmful (SIFT score <0.05; PROVEAN score <-2.5; PolyPhen-2 score = 1; Mutation-Taster: Phylop > 1, Phastcons = 1). According to ACMG standard guidelines, the two variants were classified as likely pathogenic.
Neurological Function Test Scale Results
For proband 1, the WSC-MBD was positive, indicating developmental deviation. The ABC results were as follows: perception ability 30, communication ability 34, sports ability 38, language ability 31, and self-care ability 25, resulting in a total score of 158 (standard value = 53). Meanwhile, for proband 2, the WSC-MBD was positive, indicating developmental deviation. The ABC results were as follows: perception ability 30, communication ability 32, sports ability 35, language ability 30, and self-care ability 25, resulting in a total score of 152 (standard value = 53). For both probands, as the values obtained indicated the possibility of autism, the S-M score was determined. For both, the score was 5 points, lower than the standard value of 10 points. These scores indicate that the social life skills of both patients were lower than those corresponding to the normal value.
Discussion
Herein, we reported two mutants of the IQSEC2 in two male children, one novel nonsense mutation [c. 3576C>A (p.Tyr1192*)] (de novo) and one missense mutation [c.2983C>T p.Arg995Trp], which was previously detected in a female patient,Citation4 has now been identified for the first time in a male patient. The clinical manifestations in the two children were consistent with the disease characteristics of IQSEC2 mutations. Both mutants were hemizygous, and none are included in the normal population database gnome AD.
We speculate that the new nonsense mutation c.3576C>A (p. Tyr1192*) found in proband 1 may lead to the clinical features such as intellectual development disorder and epilepsy. There are several reasons: 1. The nonsense mutation identified in proband 1 will cause terminated the translation of its encoded protein at Tyr1192, resulting in the formation of truncated protein. Compared with the wild-type protein, the mutated protein will miss 297 amino acids at the C-terminus (). From the domain of IQSEC2, we found that the missing amino acids caused IQSEC2 to lose its PDZ binding motif. IQSEC2 is located in the PSD of excitatory synapses through the interaction between its C-terminal PDZ-binding motif and PSD-95.Citation5 The PDZ-binding motif of IQSEC2 maintains basal synaptic transmission as well as participates in AMPAR activity-dependent clearance.Citation2 However, mutations in the PDZ domain of IQSEC2 destroy the regulation and neurotransmission of glutamate receptors in hippocampal culturesCitation2 and alter the localization of IQSEC2 on dendritic spines.Citation6 This suggests that the nonsense mutation lacking PDZ binding motif (the key functional domain at the C-terminal) will seriously affect the function and structure of IQSEC2 protein. 2. We predicted the three-dimensional protein structure of mutant and wild-type. Research has found that truncated proteins are significantly different from proteins in the wild-type (). 3. We conducted further research on the mutants, after conducting RT qPCR analysis on their cDNA, the results showed that the significantly reduced expression level of IQSEC2 in patients compared with that in their parents, brothers (). We speculate that owing to the early termination codon, the mRNA most likely undergoes nonsense mediated RNA decay (NMD), thereby avoiding generation of a truncated protein that may be harmful to cells.Citation7
The missense mutation c.2983C>T (p.Arg995Trp) identified in proband 2, which results in an arginine-to-tryptophan substitution at amino acid 995, Although the missense mutation found in proband 2 has been reported, no relevant experiments have been conducted.Citation4 In this report, we conducted some research and found that the mutant may be the pathogenic gene of the patient, for the following reasons: 1. Conservative research has found that this site is in a conserved position in biological evolution, and mutations may alter its function (). 2. Additionally, by predicting the three-dimensional structure of the missense mutation, we observed that the protein structure of the mutant significantly differed from that of the wild-type ( and ). The mutation site is located in the pleckstrin homology domain (), which is the primary membrane-bounding domain in the human protein. It combines with phosphoinositide (PI) at different affinities and specificities and plays a vital role in membrane transport and membrane localization.Citation8,Citation9 ARF-GTPase-activating protein (ARF-GAP) is a key regulator of intracellular membrane transport, which is activated by Arf-GEF. Conversely, IQSEC2, which belongs to the GEF protein family and has a complete PH domain, can enhance the exchange between GTP and GDP, thus activating ARFs more effectively.Citation10 Therefore, we speculated that the mutation site in the PH domain might affect the coupling of IQSEC2 Arf-GEF with membrane binding function,Citation9 thereby affecting the IQSEC2 signaling pathway. 3. Real time quantitative PCR research has found a decrease in its expression level, indicating that it may lead to insufficient translation proteins, thereby affecting the stability of RNA. Therefore, the software (RNA fold web server) was used to predict the structure of wild-type IQSEC2 RNA molecules and mutant RNA molecules; structural differences between the two were found. The change in the molecular structure of missense mutant RNA may make RNA unstable before it is translated into protein, thus degrading it.
We found a decrease in gene expression in both patients, which may be the cause of the disease. We suggest that if a mutation similar to this gene is found in the future, conducting expression testing may help determine whether the mutation is pathogenic.
Approximately 2% of patients with ID and epilepsy exhibit IQSEC2 mutations.Citation11 The Human Gene Mutation Database (HGMD) includes 89 reported cases of IQSEC2 mutations across 113 mutation sites. Among these, the most common is the frameshift mutation, which has been reported in 27 cases. The most common mutations among these are C.804 del C del 1 bp codon 268, which has been reported in 5 cases,Citation12–16 and c.2052 _ 2053 del CG del 2bp Codon 684 and c.4039dupG ins 1 bp codon 1347, which have been reported in 2 cases.Citation12,Citation17 There are 27 nonsense mutations, among which the most common are [c.3163C>T(p.Arg1055*)] and [c.2563C>T(p.Arg855*)],Citation14,Citation16,Citation18 each of which has been reported in 3 cases and [c.2776C>T(p.Arg926*)] and [c.2317C>T(p.Gln773*)] and [c.895C>T(p. Gln299Term)], each of which has been reported in 2 cases.Citation14–16 There are 25 missense mutations, among which the most common are [[c.2312G>A (p.Gly771Asp)], [c.1049C>T(p.Ala350Val)], and [c.2582G>C(p.Ser861Thr)] which have been reported in 3 cases,Citation17,Citation19–21 and [c.2507C>T(p.Ala836Val)] and [c.3206G>C(p.Arg1069Pro)] which have been reported in 2 cases.Citation15,Citation22,Citation23 There are 5 cases of splice, and intraframe mutations occurred in 3 casesCitation16,Citation24,Citation25 ( and ).We found that 61% (31/52) of these missense mutations and nonsense mutations were distributed in the SEC7 and PH region, and that these mutations were located in conserved sequences and important domains of the protein ( and ), suggesting that the SEC7 and PH region plays an important role in the function of the IQSEC2 protein. And out of 113 reported mutation cases, 39% (44/113) were accompanied by epilepsy.
Table 3 IQSEC2 mutations previously reported
Table 4 Most common IQSEC2 mutations and related phenotypes reported for research
Mutations in IQSEC2, an X-linked gene, are associated with intellectual disability (ID), epilepsy and autism.Citation20,Citation26 Four missense mutants of IQSEC2 were identified in families diagnosed with X chromosome-linked intellectual disability (XLID).Citation27 Common clinical features and signs related to IQSEC2 include language regression, stereotypic hand movements, hypotonia, ataxia, microcephaly, and seizures.Citation28 The common types of epilepsy associated with IQSEC2 mutations include muscle tone disorders, myoclonus, and spasmodic epilepsy, and most of them are generalized epileptic seizures. However, in patients with combined ID, other symptoms include developmental delay, speech impairment, walking delay, and autism. Other rare symptoms may also include limited hand movement, strabismus, and low intraocular pressure.Citation20 In addition, research reports have found that differences were observed between males and females in patients with missense functional IQSEC2 variants, with males demonstrating mild to severe non-syndromic ID accompanied by seizures and females with missense functional IQSEC2 mutations being generally either asymptomatic carriers or mildly affected.Citation29 The research has shown that the majority of pathogenic variants are predicted to lead to premature termination that is subject to the RNA surveillance process of nonsense-mediated decay, leading to a complete loss of mutant IQSEC2 protein, male patients with loss of function variants invariably present with severe ID, seizures.Citation17 The two variants we reported highly conform to the clinical phenotype of IQSEC2 pathogenesis. In previous reports, Subtelomere FISH analysis was performed on peripheral blood lymphocytes to investigate the pathogenicity of the X chromosome homozygous deletion 4q35.2 in IQSEC2.Citation30 However, research on IQSEC2 A350V is mostly focused on mouse models, which investigate the pathogenicity of mutations by observing the behavior of mice and the expression of iqsec2 in the brain.Citation31–33 Our report is the first to explore the pathogenic mechanism of IQSEC2 by examining its expression in peripheral blood cells of patients.
Through the analysis of the co expression of IQSEC2 and the IQSEC2 signaling pathway, four genes with significant expression of IQSEC2 were screened IQSEC2: PSD-95, SAP-97, ARF-6 and IRSP 53. To investigate whether the mutation of this gene affects the function of its downstream genes, we used quantitative PCR to further investigate the expression of IQSEC2 related genes in patient blood samples. IQSEC2 encodes IQSEC2 protein, which is an upstream regulatory factor of PSD-95. It can regulate the transcription and translation of PSD-95 by interacting with PSD-95 through PDZ binding motif. A lack of PSD-95 results in impaired cognition and learning.Citation34 PSD-95 exists in the 1.5 MDa NMDA complex, which also includes IQSEC2.In mice lacking PSD-95, IQSEC2 could not form the above complex.Citation35 We observed that the expression of PSD-95 in probands 1 and 2 was significantly lower than in the control group and the parents of proband 1 (). We speculate that the mutation of IQSEC2 may affect the expression and function of IQSEC2 protein, leading to changes in the expression level of PSD-95.Citation36
Petersen et al also found that IQSEC2 also interacts with synapse associated protein 97 (SAP97) through PDZ. Research showed that deletion of the SAP97 may lead to disrupted glutamate neurotransmitter transmission and impaired function of glutamate receptors.Citation37 We also found that IQSEC2 is a membrane protein that participates in vesicular transportCitation3 and binds with SAP97 to participate in vesicle formation and play an important role in vesicular transport.Citation38 Therefore, we speculate that the decreased expression of IQSEC2 in the proband 1 and proband 2 may affect the localization and stability of SAP97, resulting in a decrease in SAP97 expression (). This impedes vesicle formation and transport, leading to the development of the disease.
Arf-6 protein is the substrate of IQSEC2-GEF. Although most mammals have six kinds of ARF proteins, only arf-6 is related to the transport of cell membrane substances and the regulation of neurotransmitters. AFR6 is one of the downstream target genes of IQSEC2. Consequently, arf-6 is involved in the IQSEC2 signal transduction pathway.Citation5 IQSEC2 promotes the exchange of GDP and GTP on the arf-6 protein, leading to its activation. arf-6 activation subsequently activates a downstream effector, which leads to changes in cell membrane transport and actin dynamics. These physiological processes are closely related to the development of cortical neurons, formation of dendritic spines, and promotion of prominent plasticityCitation39 speculated that the over-expression of ARF-6 delays neuronal migration. ARF6 mRNA expression in probands 1 and 2 was significantly higher than that in their parents and control group (). The results indicate that the overexpression of ARF6 caused by IQSEC2 mutations may be related to the pathogenesis of epilepsy and ID. Unfortunately, we did not obtain sufficient sample size. Our experiment had some shortcomings, such as the activity changes of ARF6-GEF. We intend to further verify the role of ARF6 activity changes caused by IQSEC2 mutations in ID and epilepsy in future studies.
Previous studies have demonstrated that the interaction between IQSEC2 and IRSp53 is mediated by a proline-rich sequence (aa 1424–1434) located in the C-terminus.Citation33 IRSp53 has been shown to regulate spine formation downstream of the Rac1 pathway.Citation40 Meanwhile, ARF6 can regulate dendritic and spine formation upstream of Rac1.Citation41,Citation42 The interaction between IQSEC2 and IRSp53 activates ARF6 at the optimal position, and the activated ARF6 can enhance Rac1 near its effector IRSp53. Therefore, the reduction of IRSp53 caused by IQSEC2 mutation may hinder the formation and maintenance of dendrites and spines. Our experiment showed that the expression level of IRSP53 in proband 1 was lower than that of the parents and control group, but there was no change in the expression level of proband 2 (). This result suggests that the nonsense mutation in proband 1 resulted in the loss of function of the truncated protein, which affected the expression level of IRSP53; while the missense mutation in proband 2 led to RNA instability, with the translated product may have retained some function, enabling it to interact with IRSP53, hence the expression level of IRSP53 did not decrease.
The epilepsy caused by IQSEC2 mutation has different clinical phenotypes.Citation9,Citation20,Citation43,Citation44 The two patients in this study were diagnosed with IS, and the initial VEEG indicated peak rhythm disorders accompanied by frequent spasms. The EEG of infantile spasticity showed that high potential slow waves and spike slow complex waves are widely distributed, and low voltage activities are involved, which appear repeatedly and periodically. It is reported that periodic discharges will occur in the cortex, such as the inhibitory burst mode of nonketotic hyperglycinemia, and excessive N-methyl-D-aspartate (NMDA) transmission may cause these discharges.Citation45 The specific bidirectional modulation of IQSEC2 on synaptic transmission is specific in the modulation activity dependent signaling process through NMDA.Citation2 Therefore, Patients with pathogenic variation of IQSEC2 may have reduced synaptic transmission, so they show enhanced transmission, which is the basis of these characteristic periodic discharges on EEG. However, according to reports,Citation46 changes in glutamate receptors in brain regions should be considered a potential factor in the pathogenesis of infantile spasms. We speculate that the two variants we reported may have strong potential to affect NMDA receptors. This preliminary speculation needs further research and demonstration.
Conclusions
Our findings suggest that the two IQSEC2 variants, including the de novo mutation, are likely to be highly pathogenic, and therefore provide novel insights into IQSEC2 variant spectrum. In practice, clinical doctors can conduct early genetic testing and routine follow-up for children with growth retardation and epilepsy. And we also need to prevent heart and kidney diseases caused by mutations in this gene. We believe that a better understanding of the diseases associated with this gene mutation may lead to the development of novel diagnostics and mutation-specific personalized pharmacotherapy. In the future, we intend to obtain stem cells from affected patients to further explore the possible mutation-specific functional alterations to clarify the function of IQSEC2.
Study Limitations
Given that children are the most vulnerable population and their diseases are complex and diverse. The most important limitations of this study are the lack of new variants or the same variant from many infants, as well as the absence of other tissue samples for validation. More tissue samples and mutants will be able to explore the pathogenic mechanisms of variants from different perspectives.
Ethics Approval and Consent to Participate
The study was approved by the Ethics Committee of Shanghai Children’s Hospital (Approval number: 2019R071-F03) and complies with the ethical principles of the Helsinki Declaration. And the authors have obtained informed consent from the patient’s guardian regarding the publication of the article.The parents/guardians of both patients have agreed to publish case details and institutional approval was not required to publish case details.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
Acknowledgments
We would like to express our gratitude to the patients and their parents for their cooperation. We would also like to thank Chao Wang for his help during this study.
Additional information
Funding
References
- Levy NS, Umanah GKE, Rogers EJ, Jada R, Lache O, Levy AP. IQSEC2-Associated Intellectual Disability and Autism. Int J Mol Sci. 2019;20(12):12. doi:10.3390/ijms20123038
- Brown JC, Petersen A, Zhong L, et al. Bidirectional regulation of synaptic transmission by BRAG1/IQSEC2 and its requirement in long-term depression. Nat Commun. 2016;7:11080. doi:10.1038/ncomms11080
- Ewans LJ, Field M, Zhu Y, et al. Gonadal mosaicism of a novel IQSEC2 variant causing female limited intellectual disability and epilepsy. Eur J Hum Genet. 2017;25(6):763–767. doi:10.1038/ejhg.2017.29
- Srivastava S, Desai S, Cohen J, et al. Monogenic disorders that mimic the phenotype of Rett syndrome. Neurogenetics. 2018;19(1):41–47. doi:10.1007/s10048-017-0535-3
- Sakagami H, Sanda M, Fukaya M, et al. IQ-ArfGEF/BRAG1 is a guanine nucleotide exchange factor for Arf6 that interacts with PSD-95 at postsynaptic density of excitatory synapses. Neurosci Res. 2008;60(2):199–212. doi:10.1016/j.neures.2007.10.013
- Kang J, Park H, Kim E. IRSp53/BAIAP2 in dendritic spine development, NMDA receptor regulation, and psychiatric disorders. Neuropharmacology. 2016;100:27–39. doi:10.1016/j.neuropharm.2015.06.019
- Oren YS, Pranke IM, Kerem B, Sermet-Gaudelus I. The suppression of premature termination codons and the repair of splicing mutations in CFTR. Curr Opin Pharmacol. 2017;34:125–131. doi:10.1016/j.coph.2017.09.017
- Feng J, He L, Li Y, Xiao F, Hu G. Modeling of PH Domains and Phosphoinositides Interactions and Beyond. Protein Reviews. 2018;19–32.
- Shoubridge C, Dudding-Byth T, Pasquier L, Goel H, Yap P, McConnell V. IQSEC2-related encephalopathy in males due to missense variants in the pleckstrin homology domain. Clin Genet. 2022;102(1):72–77. doi:10.1111/cge.14136
- Aizel K, Biou V, Navaza J, et al. Integrated conformational and lipid-sensing regulation of endosomal ArfGEF BRAG2. PLoS Biol. 2013;11(9):e1001652. doi:10.1371/journal.pbio.1001652
- Heyne HO, Singh T, Stamberger H, et al. De novo variants in neurodevelopmental disorders with epilepsy. Nat Genet. 2018;50(7):1048–1053. doi:10.1038/s41588-018-0143-7
- Helbig KL, Farwell Hagman KD, Shinde DN, et al. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet Med. 2016;18(9):898–905. doi:10.1038/gim.2015.186
- Farwell KD, Shahmirzadi L, El-Khechen D, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med. 2015;17(7):578–586. doi:10.1038/gim.2014.154
- Hamdan FF, Myers CT, Cossette P, et al. High Rate of Recurrent De Novo Mutations in Developmental and Epileptic Encephalopathies. Am J Hum Genet. 2017;101(5):664–685. doi:10.1016/j.ajhg.2017.09.008
- Helm BM, Powis Z, Prada CE, et al. The role of IQSEC2 in syndromic intellectual disability: narrowing the diagnostic odyssey. Am J Med Genet A. 2017;173(10):2814–2820. doi:10.1002/ajmg.a.38404
- Mignot C, McMahon AC, Bar C, et al. IQSEC2-related encephalopathy in males and females: a comparative study including 37 novel patients. Genet Med. 2019;21(4):837–849. doi:10.1038/s41436-018-0268-1
- Gandomi SK, Farwell Gonzalez KD, Parra M, et al. Diagnostic exome sequencing identifies two novel IQSEC2 mutations associated with X-linked intellectual disability with seizures: implications for genetic counseling and clinical diagnosis. J Genet Couns. 2014;23(3):289–298. doi:10.1007/s10897-013-9671-6
- Tzschach A, Grasshoff U, Beck-Woedl S, et al. Next-generation sequencing in X-linked intellectual disability. Eur J Hum Genet. 2015;23(11):1513–1518. doi:10.1038/ejhg.2015.5
- de Kovel CG, Brilstra EH, van Kempen MJ, et al. Targeted sequencing of 351 candidate genes for epileptic encephalopathy in a large cohort of patients. Mol Genet Genomic Med. 2016;4(5):568–580. doi:10.1002/mgg3.235
- Zerem A, Haginoya K, Lev D, et al. The molecular and phenotypic spectrum of IQSEC2-related epilepsy. Epilepsia. 2016;57(11):1858–1869. doi:10.1111/epi.13560
- Hynynen J, Pokka T, Komulainen-Ebrahim J, et al. Variants p.Q1236H and p.E1143G in mitochondrial DNA polymerase gamma POLG1 are not associated with increased risk for valproate-induced hepatotoxicity or pancreatic toxicity: a retrospective cohort study of patients with epilepsy. Epilepsia. 2018;59(11):2125–2136. doi:10.1111/epi.14568
- Staněk D, Laššuthová P, Štěrbová K, et al. Detection rate of causal variants in severe childhood epilepsy is highest in patients with seizure onset within the first four weeks of life. Orphanet J Rare Dis. 2018;13(1):71. doi:10.1186/s13023-018-0812-8
- Radley JA, O’Sullivan RBG, Turton SE, et al. Deep phenotyping of 14 new patients with IQSEC2 variants, including monozygotic twins of discordant phenotype. Clin Genet. 2019;95(4):496–506. doi:10.1111/cge.13507
- Zhang Y, Kong W, Gao Y, et al. Gene Mutation Analysis in 253 Chinese Children with Unexplained Epilepsy and Intellectual/Developmental Disabilities. PLoS One. 2015;10(11):e0141782. doi:10.1371/journal.pone.0141782
- Sajan SA, Jhangiani SN, Muzny DM, et al. Enrichment of mutations in chromatin regulators in people with Rett syndrome lacking mutations in MECP2. Genet Med. 2017;19(1):13–19. doi:10.1038/gim.2016.42
- Shoubridge C, Harvey RJ, Dudding-Byth T. IQSEC2 mutation update and review of the female-specific phenotype spectrum including intellectual disability and epilepsy. Hum Mutat. 2019;40(1):5–24. doi:10.1002/humu.23670
- Shoubridge C, Tarpey PS, Abidi F, et al. Mutations in the guanine nucleotide exchange factor gene IQSEC2 cause nonsyndromic intellectual disability. Nat Genet. 2010;42(6):486–488. doi:10.1038/ng.588
- Alexander-Bloch AF, McDougle CJ, Ullman Z, Sweetser DA. IQSEC2 and X-linked syndromal intellectual disability. Psychiatr Genet. 2016;26(3):101–108. doi:10.1097/ypg.0000000000000128
- Brant B, Stern T, Shekhidem HA, et al. IQSEC2 mutation associated with epilepsy, intellectual disability, and autism results in hyperexcitability of patient-derived neurons and deficient synaptic transmission. Mol Psychiatry. 2021;26(12):7498–7508. doi:10.1038/s41380-021-01281-0
- Karaman Mercan T, Altiok Clark O, Erkal O, et al. Coexistence of a Homozygous Chromosome 4q35.2 Deletion and Hidden IQSEC2 Pathogenic Variant in a Child with Intellectual Disability. Cytogenet Genome Res. 2021;161(3–4):153–159. doi:10.1159/000515368
- Lichtman D, Bergmann E, Kavushansky A, et al. Structural and functional brain-wide alterations in A350V Iqsec2 mutant mice displaying autistic-like behavior. Transl Psychiatry. 2021;11(1):181. doi:10.1038/s41398-021-01289-8
- Myers KR, Wang G, Sheng Y, Conger KK, Casanova JE, Zhu JJ. Arf6-GEF BRAG1 regulates JNK-mediated synaptic removal of GluA1-containing AMPA receptors: a new mechanism for nonsyndromic X-linked mental disorder. J Neurosci. 2012;32(34):11716–11726. doi:10.1523/JNEUROSCI.1942-12.2012
- Sanda M, Kamata A, Katsumata O, et al. The postsynaptic density protein, IQ-ArfGEF/BRAG1, can interact with IRSp53 through its proline-rich sequence. Brain Res. 2009;1251:7–15. doi:10.1016/j.brainres.2008.11.061
- Coley AA, Gao WJ. PSD95: a synaptic protein implicated in schizophrenia or autism? Prog Neuropsychopharmacol Biol Psychiatry. 2018;82:187–194. doi:10.1016/j.pnpbp.2017.11.016
- Frank RAW, Zhu F, Komiyama NH, Grant SGN. Hierarchical organization and genetically separable subfamilies of PSD95 postsynaptic supercomplexes. J Neurochem. 2017;142(4):504–511. doi:10.1111/jnc.14056
- Petersen A, Brown JC, Gerges NZ. BRAG1/IQSEC2 as a regulator of small GTPase-dependent trafficking. Small GTPases. 2020;11(1):1–7. doi:10.1080/21541248.2017.1361898
- Sato J, Shimazu D, Yamamoto N, Nishikawa T. An association analysis of synapse-associated protein 97 (SAP97) gene in schizophrenia. J Neural Transm. 2008;115(9):1355–1365. doi:10.1007/s00702-008-0085-9
- Zheng CY, Seabold GK, Horak M, Petralia RS. MAGUKs, synaptic development, and synaptic plasticity. Neuroscientist. 2011;17(5):493–512. doi:10.1177/1073858410386384
- Falace A, Buhler E, Fadda M, et al. TBC1D24 regulates neuronal migration and maturation through modulation of the ARF6-dependent pathway. Proc Natl Acad Sci U S A. 2014;111(6):2337–2342. doi:10.1073/pnas.1316294111
- Choi J, Ko J, Racz B, et al. Regulation of dendritic spine morphogenesis by insulin receptor substrate 53, a downstream effector of Rac1 and Cdc42 small GTPases. J Neurosci. 2005;25(4):869–879. doi:10.1523/jneurosci.3212-04.2005
- Choi S, Ko J, Lee JR, et al. ARF6 and EFA6A regulate the development and maintenance of dendritic spines. J Neurosci. 2006;26(18):4811–4819. doi:10.1523/jneurosci.4182-05.2006
- Hernández-Deviez DJ, Casanova JE, Wilson JM. Regulation of dendritic development by the ARF exchange factor ARNO. Nat Neurosci. 2002;5(7):623–624. doi:10.1038/nn865
- Izumi T, Aihara Y, Kikuchi A, Kure S. Electroencephalographic findings and genetic characterization of two brothers with IQSEC2 pathogenic variant. Brain Dev. 2021;43(5):652–656. doi:10.1016/j.braindev.2020.12.020
- Choi MH, Yang JO, Min JS, et al. A Novel X-Linked Variant of IQSEC2 is Associated with Lennox-Gastaut Syndrome and Mild Intellectual Disability in Three Generations of a Korean Family. Genet Test Mol Biomarkers. 2020;24(1):54–58. doi:10.1089/gtmb.2019.0177
- Dulac O. Epileptic encephalopathy with suppression-bursts and nonketotic hyperglycinemia. Handb Clin Neurol. 2013;113:1785–1797. doi:10.1016/b978-0-444-59565-2.00048-4
- Riikonen R. Biochemical mechanisms in pathogenesis of infantile epileptic spasm syndrome. Seizure. 2023;105:1–9. doi:10.1016/j.seizure.2023.01.004