Abstract
Neonatal diabetes mellitus (NDM) is a monogenic disorder caused by mutations in genes involved in regulation of insulin secretion from pancreatic β-cells. Mutations in the KCNJ11 and ABCC8 genes, encoding the adenosine triphosphate (ATP)-sensitive potassium (KATP) channel Kir6.2 and SUR1 subunits, respectively, are found in ∼50% of NDM patients. In the pancreatic β-cell, KATP channel activity couples glucose metabolism to insulin secretion via cellular excitability and mutations in either KCNJ11 or ABCC8 genes alter KATP channel activity, leading to faulty insulin secretion. Inactivation mutations decrease KATP channel activity and stimulate excessive insulin secretion, leading to hyperinsulinism of infancy. In direct contrast, activation mutations increase KATP channel activity, resulting in impaired insulin secretion, NDM, and in severe cases, developmental delay and epilepsy. Many NDM patients with KCNJ11 and ABCC8 mutations can be successfully treated with sulfonylureas (SUs) that inhibit the KATP channel, thus replacing the need for daily insulin injections. There is also strong evidence indicating that SU therapy ameliorates some of the neurological defects observed in patients with more severe forms of NDM. This review focuses on the molecular and cellular mechanisms of mutations in the KATP channel that underlie NDM. SU pharmacogenomics is also discussed with respect to evaluating whether patients with certain KATP channel activation mutations can be successfully switched to SU therapy.
Introduction
Neonatal diabetes mellitus (NDM), either transient or permanent, is characterized by the occurrence of insulin-requiring diabetes in the first 6 months of life. The incidence of NDM is estimated to be 1 in ∼200,000 live births.Citation1,Citation2 The diabetes in 50%–60% of NDM is transient in nature, resolving within 18 months of birth and is thus termed TNDM.Citation3 The remaining 40%–50% of NDM cases are permanent (PNDM) and require insulin treatment throughout life.Citation3 In the most severe cases of NDM, the diabetes may be accompanied by marked developmental delay, muscle weakness, and epilepsy, termed DEND (developmental delay, epilepsy, and neonatal diabetes) syndrome.Citation4 A form of NDM, between PNDM and DEND in severity, is known as intermediate DEND (iDEND), in which patients with PNDM show developmental delay or muscle weakness but not epilepsy.Citation4
The evidence to date indicates that NDM is a monogenic disorder. Although mutations in multiple genes can cause NDM, such as INS (insulin gene) mutationsCitation5,Citation6 and GCK (glucokinase gene) mutations,Citation7,Citation8 much attention has focused on the most common forms of NDM caused by heterozygous activation mutations in the KCNJ11Citation9–Citation12 and ABCC8Citation13–Citation15 genes that encode the two subunits Kir6.2 and SUR1, respectively, of the adenosine triphosphate (ATP)-sensitive potassium (KATP) channel that couples cellular metabolism to cellular excitability.Citation16 KATP channels composed of Kir6.2 and SUR1 subunits are predominately expressed in endocrine tissues such as the pancreatic islet and nervous system. Therefore, the diabetic phenotype of NDM is believed to arise from KATP channel activation mutations in pancreatic β-cells,Citation10,Citation16 whereas neurological features associated with the more severe iDEND/DEND syndromes are likely the result of KATP channel activation mutations deleteriously affecting the nervous system.Citation4,Citation17
The physiological role of KATP channels in pancreatic β-cells
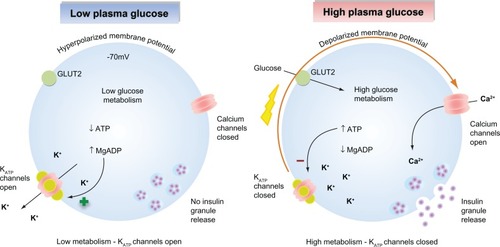
KATP channels sense changes in the cytosolic ATP/ADP ratio as a result of cellular metabolism and are a major regulator of the β-cell membrane potential. As glucose-stimulated insulin secretion is primarily controlled by the β-cell membrane potential, KATP channels serve to couple glucose metabolism to insulin secretion.Citation16,Citation18 When plasma glucose levels are low, the cytosolic ATP/ADP ratio is reduced, leading to a basal efflux of potassium ions from the cell via KATP channel activity that maintains the membrane potential of the β-cell at approximately −70 mV. This polarized membrane potential prevents calcium entry through voltage-gated calcium channels. As elevations in cytosolic calcium are the primary trigger for insulin granule exocytosis, insulin secretion is suppressed when plasma glucose levels are low ().Citation19,Citation20 When plasma glucose levels rise, glucose enters the β-cells via the glucose transporter 2. Subsequent glucose metabolism leads to an increase in the ratio of cytosolic ATP/ADP ratio, promoting KATP channel closure. The resultant decrease in potassium ion efflux depolarizes the β-cell membrane potential, leading to activation of voltage-gated calcium channels, calcium influx, and calcium-stimulated insulin granule exocytosis ().Citation21 Graded increases in plasma glucose and subsequent metabolism lead to proportional decreases in KATP channel activity, resulting in an appropriate insulin secretory response that is tightly coupled to the plasma glucose concentration.
Figure 1 Glucose-stimulated insulin secretion in pancreatic β-cells. (Left) When plasma glucose is low, the decreased ratio of ATP/Mg-ADP will increase KATP channel opening. Consequently, the cell membrane is hyperpolarized, preventing voltage-gated calcium channel opening, Ca2+ influx, and insulin secretion. (Right) When plasma glucose is high, glucose is transported into the cell via GLUT2. Glucose metabolism leads to an increased ratio of ATP/Mg-ADP, resulting in KATP channel closure, membrane depolarization, opening of voltage-gated calcium channels, Ca2+ influx, and insulin secretion.
As the electrical resistance of β-cell is high,Citation22 only small changes in KATP channel activity are required to change β-cell excitability (and hence insulin secretion) via alterations in the β-cell membrane potential.Citation23 Mutations within the KATP channel complex that change their intrinsic activity and/or ability to sense changes in either ATP or ADP will result in altered KATP channel activity that is correlated to the specific effects of the individual mutation on KATP channel activity.
KATP channels encoded by the KCNJ11 and ABCC8 genes are also expressed in other excitable tissues such as the nervous system. As KATP channels are involved in the control of neuronal excitability, mutations may also cause neuronal abnormalities, again dependent on the severity of the individual mutation.Citation24–Citation27
Molecular structure of pancreatic KATP channels
The KATP channel is a hetero-octameric membrane protein complexCitation28,Citation29 composed of four pore-forming inwardly rectifying potassium channel (Kir6.x) subunits and four regulatory sulfonylurea receptor (SURx) subunits ().Citation30 There are two isoforms of the Kir6.x subunit, Kir6.1 and Kir6.2. Kir6.2 is more widely expressed than Kir6.1, which is predominately expressed in vascular smooth muscle.Citation31,Citation32 There are two isoforms of the SUR subunit (SUR1 and SUR2), and the subunit composition of KATP channel differs between tissue types.Citation33 In pancreatic β-cells and neurons, KATP channels are assembled from Kir6.2 and SUR1 subunits.Citation34 In cardiac tissue and skeletal muscle, KATP channels are composed of Kir6.2 and the SUR2A splice variant subunits,Citation35 whereas in smooth muscle, KATP channels contain Kir6.1/Kir6.2 and SUR2A/SUR2B splice variant subunits.Citation36,Citation37
Figure 2 Molecular make-up of the KATP channel complex. (Lower left) KATP channel is a hetero-octameric complex composed of four pore Kir6.2 subunits and four regulatory SUR1 subunits. (Right) Membrane topology of SUR1 and Kir6.2 subunits of the KATP channel. ATP binds to the Kir6.2 subunit, inhibiting KATP channels. Hydrolysis of MgATP within the SUR1 subunit nucleotide-binding domains (NBDs) leads to generation of stimulatory MgADP. The A and B sites for sulfonylurea drug binding on both subunits are labeled as indicated.
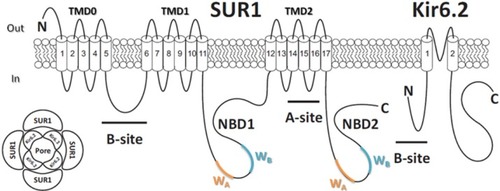
The Kir6.2 subunit contains ∼390 amino acid and is encoded by KCNJ11 gene, while ∼1,580 amino acid SUR1 subunits are encoded by ABCC8 gene. Both KCNJ11 and ABCC8 genes are located at the same chromosomal locus (11p15.1) and are only 4.5 kb apart.Citation30 Each Kir6.2 subunit consists of two transmembrane (TM) helices connected by a pore-forming loop that confers potassium selectivity to the channel.Citation38 The α-helix linking TM helix 1 (TM1) and intracellular N-terminus, termed as the “slide helix,” plays an important role in channel gating.Citation39 Extensive interactions are found between the cytosolic N- and C-termini of adjacent Kir6.2 subunits that contribute to the formation of binding pocket for the inhibitory ATP molecule.Citation40 Each SUR1 subunit consists of three TM domains (TMD) with a total of 17 TM segments.Citation41 Each SUR1 subunit contains two nucleotide-binding domains (NBD1 and NBD2) that dimerize to form catalytic sites for the intrinsic Mg-ATPase activity of the channel complex, regulating channel activity through binding and hydrolysis of magnesium-bound ATP and the formation of stimulatory Mg-ADP.Citation42,Citation43 Each NBD contains two amino acid sequence nucleotide hydrolysis “Walker A” and “Walker B” motifs ().Citation44,Citation45 TMD0 and the cytosolic loop linking TMD0 and TMD1 of the SUR1 subunit are responsible for the interaction between Kir6.2 subunit.Citation46 The Kir6.2 and SUR1 subunits each possess an endoplasmic reticulum retention motif that requires masking via subunit co-assembly to enable correct trafficking of the assembled hetero-octameric channel complex to the cell membrane.Citation47
KATP channels are inhibited by ATP binding to the Kir6.2 subunits but are activated by the binding and hydrolysis of Mg-ATP in the NBD1/NBD2 dimers on SUR1 subunit, the resulting Mg-ADP generated antagonizes the inhibitory action of ATP on the Kir6.2 subunits. Therefore, the overall activity of the KATP channel complex, and hence the excitability of pancreatic β-cells, is primarily governed by the ratio of cytosolic ATP/ADPCitation45,Citation48 in the close vicinity of the KATP channel complex.
Mutations in either subunit that alter 1) the correct ATP/ADP-sensing machinery within the KATP channel complex, 2) subunit assembly, or 3) trafficking to the cell membrane may adversely affect the appropriate insulin secretion in response to plasma glucose. Inactivation mutations in the KATP channel complex decrease channel activity, causing over-secretion of insulin that is poorly coupled to plasma glucose levels. Indeed, mutations in SUR1 subunit that 1) reduce the stimulatory effect of Mg-ADP or 2) prevent correct trafficking of the channel complex to the cell membrane cause persistent hyperinsulinemia that presents as hypoglycemia in infancy (HI).Citation49,Citation50
Conversely, activation mutations in the KATP channel complex lead to increased channel opening, resulting in a suppression of insulin secretion and subsequent hyperglycemia. Consistent with the cellular regulation of KATP channel activity are the findings that mutations 1) in the Kir6.2 subunit that reduce sensitivity to inhibitory ATP and 2) in the SUR1 subunit that enhance the stimulatory effects of Mg-ADP may precipitate DEND,Citation17 iDEND,Citation4 PNDM,Citation51 TNDM,Citation52 MODY (maturity onset diabetes of the young),Citation53 and type II diabetes (T2D).Citation54,Citation55
KATP channel inactivation mutations underlie HI
Inactivation mutations in both KATP channel subunits can cause HI, which is characterized by severe hypoglycemia.Citation56 Mutant KATP channels with reduced or completely abolished channel activity lead to persistent depolarization of cell membrane, which results in continuous calcium influx and excessive insulin secretion that is uncoupled from the plasma glucose level, producing the hyperinsulinemic hypoglycemia phenotype.Citation12,Citation57,Citation58 Compared to inactivation mutations in the KCNJ11 gene (Kir6.2 subunit), more inactivation mutations have been reported in the ABCC8 gene encoding the SUR1 subunit. lists the reported inactivation mutations causing HI in both KCNJ11 and ABCC8 genes and their corresponding locations on each subunit.Citation59,Citation60 Inactivation mutations in the KATP channel complex can be divided into two functional classes: class I, a reduced number of functional KATP channels inserted into the cell membrane, and class II, mutant KATP channels that are correctly inserted but remain refractory to opening regardless of the cellular metabolic state of the cell.Citation22 Class I mutations in either SUR1 or Kir6.2 subunits lead to reduced surface expression of KATP channels, which may result from a total loss of protein, defective channel assembly, or faulty trafficking to the cell membrane.Citation61–Citation63 Class II mutations impair the ability of Mg-ADP to stimulate channel activity,Citation64–Citation66 such that ATP inhibition becomes dominant and the KATP channel is permanently closed even at low glucose concentrations. The majority of class II mutations are located in the NBDs of SUR1 subunit, where the binding and hydrolysis of Mg-ATP occurs. In general, class I mutations produce a more severe phenotype, often requiring near-total or total pancreatectomy, whereas a number of class II mutations result in a milder phenotype as some residual response to stimulatory Mg-ADP may remain. However, there is no strict genotype–phenotype correlation as the same mutation in different patients can produce HI with differing degrees of severity. As HI class II mutations lead to cell membrane expression of dysfunctional KATP channels, less severe forms of HI can often be treated with KATP channel opener diazoxide.Citation67
Table 1 Mutations in KATP channel genes KCNJ11 and ABCC8 causing hyperinsulinism of infancy
KATP channel activation mutations underlie NDM
Monogenic activation mutations in the KCNJ11 and ABCC8 genes can be found in all forms of NDM (DEND,Citation4,Citation17 PNDM,Citation51 TNDM,Citation52 and MODY).Citation53 Activation mutations result in a reduced coupling of channel activity to plasma glucose levels via glucose metabolism. In general, the more stimulatory the mutation, the greater the suppression of insulin secretion and the resulting level of hyperglycemia ().Citation68–Citation71 The underlying molecular mechanisms for the majority of activation mutations can be tested experimentally and correlated well with their specific locations within the KATP channel subunits as follows.
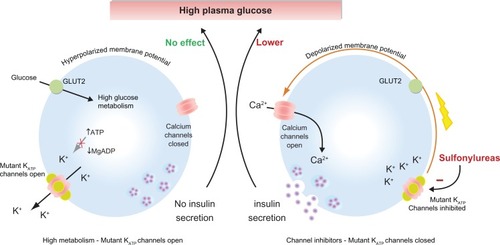
Figure 3 Sulfonylureas stimulate insulin secretion in neonatal diabetes caused by KATP channel mutations. (Left) Activation mutations in the KATP channel prevent channel closure in response to high plasma glucose. Consequently, the membrane potential remains hyperpolarized even, thereby preventing insulin secretion. (Right) Sulfonylureas bind directly to KATP channels causing channel inhibition that triggers membrane potential and insulin secretion resulting in a lowering of plasma glucose.
Activation mutations in the Kir6.2 subunit
Heterozygous activation mutations in Kir6.2 subunit have been identified in ∼50% of PNDM cases and also have been found in a large number of TNDM cases.Citation3 To date, >40 activation mutations in Kir6.2 subunit have been reported at 30 distinct residues ().Citation60 The locations of these mutations are clustered into three common regions in the Kir6.2 subunit. One cluster of mutations line the putative ATP-binding pocket (eg, R50, R201, and Y330) and reduce channel ATP inhibition by decreasing ATP-binding affin-ity.Citation69,Citation72–Citation74 Another cluster of mutations reside in subunit regions involved in channel gating such as the slide helix (eg, V59), the cytosolic mouth of the channel (eg, I296), or gating loops (eg, C166) between ATP-binding site and the slide helix. These mutations decrease ATP inhibition by stabilizing the open conformation of the channel in both the absence and the presence of ATP, leading to increases in channel activity.Citation75–Citation77 The third cluster of mutations is located at the interface between the subunits such as the interface between adjacent Kir6.2 subunits (eg, F35 and E322) and the interface between Kir6.2 and SUR1 subunits (eg, Q52 and G53). These mutations likely alter channel activity by affecting the interactions between adjacent Kir6.2 and SUR1 subunits that are important for correct channel gating.Citation78–Citation82
Table 2 Mutations in KATP channel genes KCNJ11 and ABCC8 causing diabetes in terms of DEND, PNDM, TNDM, MODY, and T2D
To directly study the ability of ATP to inhibit the KATP channel via the Kir6.2 subunit, Mg-free experimental conditions can be used to eliminate the channel stimulatory effect of Mg-ATP on the SUR1 subunit of the channel.Citation83 In Mg-free conditions, homomeric channels containing Kir6.2 activation mutations are less sensitive to ATP inhibition compared to wild-type channels. There are two major molecular mechanisms by which Kir6.2 activation mutations elicit a reduction in ATP sensitivity. 1) An increase in the maximal open probability (Po) of the channel in the absence of ATP.Citation17,Citation84 In the ATP-unbound state of channels (ATP absent), mutations in the region involved in channel gating (eg, V59G)Citation4 exhibit a higher maximal channel Po compared to wild-type channels. (In the absence of Mg2+, IC50 was 7.0 ± 1.1 μmol/L and 7.4 ± 1.5 mmol/L for wild-type channels and homomeric V59G channels, respectively; P < 0.001. Channel Po was 0.53 ± 0.02 and 0.83 ± 0.01 for wild-type channels and homomeric V59G channels, respectively; P < 0.001.)Citation4 2) A decrease in ATP-binding affinity.Citation51,Citation85,Citation86 Homomeric channels containing mutations in the ATP-binding region (eg, R201C)Citation4 display altered ATP inhibition, yet their maximal Po in the absence of ATP is not significantly different compared to wild-type channels. (In the absence of Mg2+, IC50 was 7.0 ± 1.1 and 106 ± 12 μmol/L for wild-type channels and homomeric R201C channels, respectively; P < 0.001. Channel Po was 0.53 ± 0.02 and 0.6 ± 0.03 for wild-type channels and homomeric R201C channels, respectively; P is not significant.)Citation4
Activation mutations in the SUR1 subunit
There are more than 30 individual activation mutations in SUR1 subunit that have been reported to cause NDM ().Citation60 Many of these mutations are dispersed throughout the SUR1 subunit sequence, although a large number of mutations reside in two specific regions of the SUR1 subunit. One cluster of mutations is concentrated in TMD0 and the cytosolic loop linking TMD0 and TMD1.Citation14,Citation87–Citation91 As this region is known to interact with adjacent Kir6.2 subunit, mutations in this region are believed to reduce ATP inhibition via the Kir6.2 subunit.Citation92,Citation93 The second cluster of mutations resides in the NBD2 of the SUR1 subunit, where stimulatory Mg–nucleotide diphosphates such as Mg-ADP bind.Citation94–Citation96 Therefore, NBD2 mutations are thought to either increase direct Mg-ADP stimulation or enhance MgATPase activity in NBD2, leading to increased Mg-ADP stimulation. (For example, R1380 L, Vmax of ATPase activity was 60.8 ± 1.8 and 104.3 ± 9.9 nmol/min/mg for wild-type NBD2 and R1380L NBD2, respectively; P < 0.01. Km of ATPase activity was 0.41 ± 0.04 and 0.55 ± 0.09 mmol/L for wild-type NBD2 and R1380L NBD2, respectively. P is not significant.)Citation94
Genotype–phenotype correlation in NDM caused by mutations in KATP channels
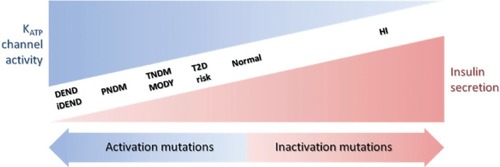
There is a wide spectrum of NDM severity associated with different degrees of insulin secretion deficiency and neuronal defects caused by activation mutations in KATP channels (). The severity of these clinical phenotypes increases in the order of T2D < MODY/TNDM < PNDM < iDEND/DEND.Citation22 In general, the greater the activation of KATP channels, the more severe the phenotype; however, several factors need to be considered when attempting to predict the clinical severity caused by a specific mutation.
Figure 4 Relationship between insulin secretion and KATP channel activity in a spectrum of clinical presentations of hypo- and hyperglycemia. The clinical severity of the disease is correlated with the extent of KATP channel activity caused by the mutations.
Heterozygosity is an important factor affecting the clinical phenotype of a mutation. NDM patients with activation mutations in either Kir6.2 or SUR1 subunits are heterozygousCitation77 for the mutation; thus, both wild-type and mutant subunits are expressed in the same cell. The assembly of Kir6.2 subunits can be used to explain the nature of heterozygosity in NDM patients. In a heterozygous NDM patient carrying one copy of normal (wild type) and one copy of mutant KCNJ11 gene,Citation28 there will be a mixed population of channels, each of which carries from 0 to 4 mutant Kir6.2 subunits ().Citation4,Citation22 Two factors determine the inhibitory ATP sensitivity of any individual channel in this population. One is the number of mutant subunits that an individual channel contains and the other is the contribution of mutant subunits to overall channel ATP sensitivity. This contribution is also linked to the molecular mechanism of each activation mutation in Kir6.2 subunit.
Figure 5 Schematic of the KATP channel Kir6.2 subunit compositions expected when wild-type (blue) and mutant (red) Kir6.2 are co-expressed in the heterozygous state. If the co-assembly wild-type (blue) and mutant (red) Kir6.2 subunits is independent and random and follows a binomial distribution, as a single KATP channel is made of 4 Kir6.2 subunits, there will be 1/16 channel with 0 mutant subunit, 4/16 channel with 1 mutant subunit, 6/16 channel with 2 mutant subunits, 4/16 channel with 3 mutant subunits, and 1/16 channel with all 4 mutant subunits.
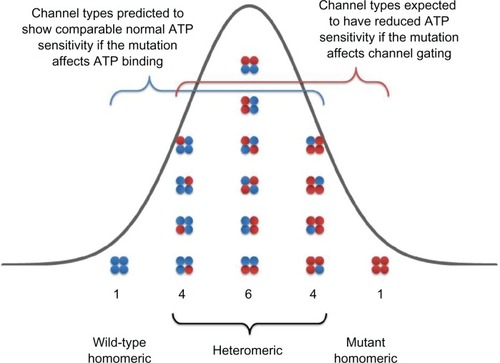
If the mutation impairs ATP-binding affinity alone, there will be only a small reduction in ATP sensitivity in heterozygous population compared to wild-type population. (For example, R201H of Kir6.2 subunit. In the absence of Mg2+, IC50 were 7.0 ± 1.1, 12.5 ± 1.1, and 298 ± 25 μmol/L for wild type, heterozygous, and homomeric R201H channels, respectively; P < 0.05 and P < 0.001 vs wild-type, respectively.)Citation4 This is because binding of a single ATP molecule to 1 of 4 ATP-binding sites is sufficient to inhibit the KATP channel.Citation97 The ATP sensitivity of the channel will only be substantially impaired when all four subunits contain the mutation; otherwise, the mutant Kir6.2 subunit’s effects will be largely compensated for by the presence of the other wild-type subunits. This can be explained by using a simple statistical probability model. If the co-assembly of wild-type and mutant Kir6.2 subunits is independent and random and follows a binomial distribution, as a single channel is made of 4 Kir6.2 subunits, then only 1 out of 16 channels in the heterozygous population will contain all 4 mutant Kir6.2 subunits that display a significant decrease in ATP sensitivity ().Citation4 The other 15 channels (4/16 with 3 mutant subunits; 6/16 with 2 mutant subunits; 4/16 with 1 mutant subunit, and 1/16 with 0 mutant subunits) will have comparable ATP sensitivity to the channel containing all wild-type subunits, so the resulting ATP sensitivity of heterozygous population is very close, but not identical, to that of a pure wild-type channel population. However, this small shift of ATP sensitivity in the heterozygous channel population leads to NDM for the following reasons. Under physiological conditions, intracellular concentration of ATP is in the range of 1–5 mM, such that KATP channels exhibit very low activity. Additionally, the β-cell membrane possesses a high electrical resistance such that only a small reduction in ATP sensitivity to the channel results in a small increase in KATP channel activity that holds the β-cell membrane potential in a more polarized state and suppresses insulin secretion.Citation23 Therefore, even a very modest reduction in heterozygous KATP channel ATP sensitivity can lead to significantly impaired insulin secretion resulting in NDM.
In direct contrast, if the mutation in question increases intrinsic KATP channel Po (in the absence of ATP), there will be a significant reduction in ATP sensitivity in heterozygous population compared to wild-type population, as the presence of one single mutant subunit will increase the intrinsic Po of KATP channels. (For example, Q52R of Kir6.2 subunit. Channel Po were 0.53 ± 0.02, 0.70 ± 0.03, and 0.83 ± 0.01 for wild type, heterozygous, and homomeric Q52R channels, respectively. P < 0.001 and P < 0.001, vs wild-type, respectively.)Citation4 This can be explained by using the same statistical model described in detail earlier. Fifteen out of 16 channels will contain at least one mutant subunit in a heterozygous channel population () and exhibit a marked decrease in ATP sensitivity. Thus, the ATP sensitivity of heterozygous population is significantly reduced compared to that of wild-type population and is associated with a more severe DEND syndrome phenotype.Citation17 This provides a rational explanation as to why mutations that increase the intrinsic Po produce a more severe clinical phenotype, such as DEND, whereas mutations that decrease ATP-binding affinity lead to a milder clinical phenotype such as PNDM.
The specific location of mutations within either subunit also correlates well with the severity of the clinical phenotype. In general, mutations in Kir6.2 subunit are typically associated with PNDM, iDEND, and DEND, whereas mutations in SUR1 subunit are more frequently associated with TNDM. This may be accounted for by the overriding ability of ATP to inhibit channel activity within wild-type Kir6.2 subunits even when there is an enhanced stimulatory effect of Mg-ADP via the effects of a SUR1 subunit activation mutation causing TNDM.Citation74 Furthermore, although some activation KATP channel mutations lead to transient diabetes, these patients are at increased risk of developing T2D later in life. Interestingly, the common genetic variants E23K in KCNJ11 and S1369A in ABCC8 form a haplotype and are associated with an increased risk to T2D.Citation98–Citation100 The precise molecular mechanisms that underlie this increased risk likely result from even subtler alterations of ATP/ADP sensitivityCitation101 than those described for the monogenic mutations that cause overt forms of NDM.
While there is good evidence for a clear genotype–phenotype relationship with several activation mutations in KATP channels, the association between phenotype and genotype is not absolute, as there is often a different severity of clinical phenotype among patients carrying the same mutation. This strongly implies that there are other factors (such as underlying polygenic diabetes risk, diet, or environment) that influence the development of clinical phenotype besides the presence of a single KCNJ11 or ABCC8 mutation in NDM patients.Citation102,Citation103
Pharmacotherapy for NDM patients carrying KCNJ11 and ABCC8 mutations
Before the discovery that mutations in a number of genes underlie NDM, daily insulin therapy was the only effective treatment for patients. As mentioned earlier, NDM can be the result of mutations in multiple genes (eg, KCNJ11, ABCC8, GCK, INS, FOXP3, EIF2AK3, and ZAC/HYMAI).Citation104 Since 2004, many NDM patients with either KCNJ11 or ABCC8 mutations have been successfully treated with a pharmacological approach, removing the requirement for insulin injections.Citation105,Citation106 The SU drugs, a class of KATP channel inhibitor, have been widely used as a treatment of T2D for over 50 years. SUs bind directly to the KATP channel complex, leading to channel closure and subsequent stimulation of insulin secretion (). Recent studies now demonstrate that glycemic control in NDM patients with KATP channel activation mutations can be managed with SU therapy alone.Citation107,Citation108 Therefore, SUs should be considered as an attractive alternative therapy to treat NDM patients carrying mutations in KCNJ11 and ABCC8 genes. As mutations in a number of genes can cause NDM and the causal mutation in each patient may differ greatly, a pharmacogenomic approach to treatment may be possible to “tailor” SU therapy based on specific NDM genotype.
SUs can be classified according to their historical discovery with first generation SUs, including tolazamide, tolbutamide, and chlorpropamide, and second generation SUs, including glyburide, glipizide, and glimepiride (). Compared to the first generation SUs, the second generation SUs are now more commonly used in the treatment of NDM as they are more potent and tend to have a longer duration of action.
Figure 6 Structures and binding-site classification of clinically used sulfonylurea drugs.
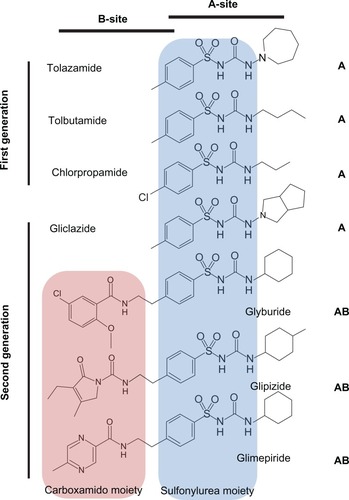
There are two SU-binding sites within the KATP channel complexCitation109 that have been identified to date. The “A-site” is located in the intracellular loops connecting TM segments 14–16 on SUR1 subunit. The “B-site” is composed of the intracellular loop between TM segments 5–6 in the SUR1 subunit and the N-terminus in Kir6.2 subunit (). Therefore, SUR1 subunit possesses a bipartite pocket with distinct A- and B-binding sites. Furthermore, SUs can be classified as A-site or AB-site drugs based on where they bind to the channel (). The A site within SUR1 subunit binds the SU moiety, whereas the B site binds the non-SU carboxamido moiety of the molecule.Citation109 Most of the first generation SUs (eg, tolbutamide and chlorpropamide) are A-site drugs, whereas the majority of the second generation SUs (eg, glyburide, glipizide, and glimepiride) are AB-site drugs, which also accounts for the higher potency of the second generation SUs. An exception to this is gliclazide, which is an A-site SU with potency comparable to the AB-site SUs.
A key issue in the optimization of SU therapy in NDM is whether mutant KATP channels can be inhibited by SUs in a similar concentration range to wild-type channels. For NDM patients with SUR1 subunit mutations, there are no reports of mutations in the ABCC8 altering SU inhibition. This may be because NDM patients with mutant SUR1 subunits often exhibit a milder clinical NDM phenotype. Therefore, SU therapy should be effective for most NDM patients with SUR1 subunit mutations.
In contrast, several studies suggest that NDM patients with Kir6.2 subunit mutations respond differently to SU therapy,Citation110–Citation112 which is likely related to the underlying molecular mechanisms of mutations that alter KATP channel function. Mutations that reduce binding affinity of inhibitory ATP causing TNDM or PNDM show adequate efficacy of SUs.Citation113 However, mutations that enhance intrinsic channel Po causing DEND or iDEND have a reduced inhibitory efficacy of SUs.Citation114 In general, the greater the ability of a specific mutation to increase the intrinsic channel Po, the higher the SU dosage required to achieve the same level of channel inhibition seen in with mutations causing PNDM and TNDM.Citation115,Citation116 As SUs are unable to sufficiently inhibit KATP channels with mutations that cause a greatly enhanced intrinsic channel Po, DEND patients with activation mutations in Kir6.2 subunit often require a combination of SU and insulin therapy rather than SU therapy alone. SU dosage for NDM patients can be quite high (up to 2.5 mg/kg/day of glyburide) compared with the dosage for patients with T2D (∼0.2 mg/kg/day).
SUs are extensively metabolized in the liver, primarily by the cytochrome P450 2C9 enzyme encoded by the CYP2C9 gene. To date, several pharmacogenomic studies have focused on the influence of common gene variants in the CYP2C9 gene on SU pharmacokinetics.Citation117–Citation119 As the activity of cytochrome P450 2C9 variants correlates well with serum levels of SUs, patients carrying CYP2C9 variants that reduce cytochrome P450 2C9 enzymatic activity possess elevated serum levels of SUs.Citation120 Therefore, screening for these common CYP2C9 variants may provide additional information as to whether a NDM patient carrying a KCNJ11 or ABCC8 gene mutation may respond better to SU therapy.
Although insulin therapy may control glucose homeostasis in NDM patients with mutant KATP channels, it does not restore the normal KATP channel activity in nonpancreatic tissues such as the brain and skeletal muscle. On the other hand, SUs can inhibit KATP channels in many tissues such as the central nervous system (CNS), ameliorating the neurological dysfunction observed in iDEND/DEND.Citation121–Citation123
One potential concern is that the dosage for SUs needed to adequately control glucose homeostasis may not be enough to resolve neurological symptoms, as SUs have to cross the blood–brain barrier to exert inhibitory effect on CNS KATP channels. However, several studies showed that improvements in mental and motor function were found in patients carrying mutant KATP channels with DEND syndrome treated with SUs.Citation121–Citation123 These observations suggest that SUs are able to cross the blood–brain barrier at concentrations sufficient to inhibit KATP channels in the CNS.
Several recent studies now show that early diagnosis and treatment of DEND patients carrying KCNJ11 or ABCC8 gene mutations with SU therapy could reduce or even prevent the neurological dysfunction in addition to dramatically improving glycemic control.Citation121–Citation123 Most DEND patients who have successfully transferred to SU therapy were children at the time of transfer. Therefore, if the causal mutation is on either KCNJ11 or ABCC8 genes, then an early switch to SU therapy may minimize the extent of neurological problems. This also emphasizes the importance of early screening for mutations in KCNJ11 and ABCC8 genes in those NDM patients with neurological features.
Traditionally, PNDM patients with mutations in other genes such as GCK gene encoding glucokinase are treated with insulin therapy. A recent study reported that a PNDM patient with the T168A mutation in glucokinase exhibited modest responsiveness to SU therapy.Citation124 Furthermore, MODY patients with mutations in HNF1α (hepatocyte nuclear factor) are very sensitive to SU therapy and many of them have been successfully transferred to SU therapy from insulin therapy.Citation125,Citation126 Therefore, the use of SU therapy should also be considered in NDM patients with mutations in genes other than KCNJ11 and ABCC8. These findings also highlight the central role that KATP channels play in regulating insulin secretion.
SUs exhibit differential potencies on KATP channels with different subunit compositions that are often expressed in a variety of tissues.Citation127–Citation130 The majority of first generation SUs and gliclazide (A site) are more selective for KATP channels containing the SUR1 subunit as found in the pancreas and CNS (IC50 was 50 nmol/L for gliclazide on KATP channels containing SUR1 subunits).Citation131 Thus, KATP channels containing either SUR2A or SUR2B subunits (heart/skeletal/smooth muscle) would not be inhibited by these SUs at the same concentration (IC50 was 0.8 mmol/L for gliclazide on KATP channels containing SUR2A subunits).Citation131 As second generation SUs (AB site) are nonselective, they will inhibit all KATP channels with similar potency (IC50 s were 3, 5.4, and 7.3 nmol/L for glimepiride on KATP channels containing SUR1, SUR2A, and SUR2B subunits, respectively).Citation132
Recent studies implicate a role for skeletal muscle (Kir6.2 and SUR2A) KATP channels in peripheral insulin sensitivity.Citation133,Citation134 In NDM patients with Kir6.2 activation mutations, overactive KATP channels in skeletal muscle (Kir6.2 and SUR2A) may reduce insulin sensitivity in addition to decreasing insulin secretion, further contributing to the development of NDM.Citation135 Inhibition of skeletal muscle KATP channels with Kir6.2 activation mutations with SUs may increase peripheral insulin sensitivity.Citation136 This notion is supported by studies showing that better glycemic control is achieved with AB-site SUs, compared with A-site SUs.Citation116,Citation123,Citation128,Citation137 This is because skeletal muscle and β-cell are inhibited by AB-site SUs as both insulin secretion and insulin sensitivity are achieved. Therefore, AB-site SUs may be the better choice for NDM patients with Kir6.2 activation mutations.
In clinical practice, the two major treatments for NDM patients are insulin therapy and oral SUs and treatment for individual patient varies depending on the genetic cause of NDM.Citation138 For 50% of PNDM patients and 10% of TNDM patients carrying mutant KATP channels, SU therapy is an attractive alternative to insulin therapy. However, for other PNDM patients carrying mutations in PTF1A, EIF2AK3, FOXP3 and 80% of TNDM patients carrying mutations in chromosome 6q24 (eg, ZAC/HYMAI), SU responsiveness is minimal and the patients’ only option is insulin therapy.Citation3 Therefore, it is important to diagnose the underlying genetic cause of NDM to fully optimize treatment.Citation139 Genetic testing is not only important for the correct diagnosis but may now also be used in the optimization of treatment in a large number of PNDM and TNDM patients with KATP channel mutations.Citation140
Conclusion
KATP channels play a central physiological role in pancreatic β-cells, where they act as key regulators of insulin secretion in response to changes in plasma glucose. Inactivation or activation mutations in KATP channels lead to altered KATP channel activity, producing a phenotype of either HI or NDM. With respect to KATP channel mutations in NDM, the severity of the clinical phenotype correlates well with the magnitude of KATP channel activation.
To date, it is estimated that ∼90% of NDM patients carrying KATP channel activation mutations can discontinue daily insulin injections and show improved glycemic control when they are switched to a high-dose SU therapy. Besides improving the quality of life for NDM patients, switching from insulin injection to SU therapy can also reduce neurological symptoms associated with patients with more severe forms of NDM (iDEND/DEND).
Furthermore, genetic information, coupled with clinical factors, may help to improve the treatment of NDM by aiding in the appropriate selection of therapeutic strategies (insulin injection, or SU therapy, or a combination of both) and a more accurate adjustment of SU dosage. Future research will likely lead to improved glycemic control by the development of a rational pharmacogenomic approach to “tailor” SU therapy based on an NDM patient’s individual genotype.
Disclosure
The authors report no conflicts of interest in this work.
References
- WiedemannBSchoberEWaldhoerTIncidence of neonatal diabetes in Austria-calculation based on the Austrian Diabetes RegisterPediatr Diabetes2010111182319496964
- SlingerlandASShieldsBMFlanaganSEReferral rates for diagnostic testing support an incidence of permanent neonatal diabetes in three European countries of at least 1 in 260,000 live birthsDiabetologia20095281683168519499210
- HattersleyATPearsonERMinireview: pharmacogenetics and beyond: the interaction of therapeutic response, beta-cell physiology, and genetics in diabetesEndocrinology200614762657266316556760
- ProksPAntcliffJFLippiatJGloynALHattersleyATAshcroftFMMolecular basis of Kir6.2 mutations associated with neonatal diabetes or neonatal diabetes plus neurological featuresProc Natl Acad Sci U S A2004101175391754415583126
- StøyJEdghillELFlanaganSEInsulin gene mutations as a cause of permanent neonatal diabetesProc Natl Acad Sci U S A2007104150401504417855560
- ColomboCPorzioOLiuMSeven mutations in the human insulin gene linked to permanent neonatal/infancy-onset diabetes mellitusJ Clin Invest20081182148215618451997
- NjølstadPRSovikOCuesta-MunozANeonatal diabetes mellitus due to complete glucokinase deficiencyN Eng J Med200134415881592
- NjølstadPRSagenJVBjorkhaugLPermanent neonatal diabetes caused by glucokinase deficiency: inborn error of the glucose-insulin signaling pathwayDiabetes2003522854286014578306
- EdghillELGloynALGorielyAOrigin of de novo KCNJ11 mutations and risk of neonatal diabetes for subsequent siblingsJ Clin Endocrinol Metab2007921773177717327377
- GloynALCummingsEAEdghillELPermanent neonatal diabetes due to paternal germline mosaicism for an activating mutation of the KCNJ11 gene encoding the Kir6.2 subunit of the beta-cell potassium adenosine triphosphate channelJ Clin Endocrinol Metab2004893932393515292329
- VaxillaireMPopulaireCBusiahKKir6.2 mutations are a common cause of permanent neonatal diabetes in a large cohort of French patientsDiabetes2004532719272215448107
- GloynALSiddiquiJEllardSMutations in the genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) in diabetes mellitus and hyperinsulinismHum Mutat20062722023116416420
- VaxillaireMDechaumeABusiahKNew ABCC8 mutations in relapsing neonatal diabetes and clinical featuresDiabetes2007561737174117389331
- BabenkoAPPolakMCaveHActivating mutations in the ABCC8 gene in neonatal diabetes mellitusN Engl J Med200635545646616885549
- ShieldJPFlanaganSEMackayDJMosaic paternal uniparental isodisomy and an ABCC8 gene mutation in a patient with permanent neonatal diabetes and hemihypertrophyDiabetes20085725525817942821
- AshcroftFMProksPSmithPAStimulus-secretion coupling in pancreatic β-cellsJ Cell Biochem199455Suppl54657929618
- ProksPGirardCHaiderSA gating mutation at the internal mouth of the Kir6.2 pore is associated with DEND syndromeEMBO Rep2005647047515864298
- AshcroftFMRorsmanPElectrophysiology of the pancreatic beta-cellProg Biophys Mol Biol198954871432484976
- CookDLHalesCNIntracellular ATP directly blocks K+ channels in pancreatic B-cellsNature19843112712736090930
- AshcroftFMHarrisonDEAshcroftSJGlucose induces closure of single potassium channels in isolated rat pancreatic beta-cellsNature19843124464486095103
- AshcroftFMATP-sensitive K channels and disease: from molecule to maladyAm J Physiol Endocrinol Metab2007293880889
- AshcroftFMATP-sensitive potassium channelopathies: focus on insulin secretionJ Clin Invest20051152047205816075046
- SeinoSMikiTPhysiological and pathophysiological roles of ATP-sensitive K+ channelsProg Biophys Mol Biol20038113317612565699
- MikiTLissBMinamiKATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasisNat Neurosci2001450751211319559
- LiDPChenSRPanHLAdenosine inhibits paraventricular pre-sympathetic neurons through ATP-dependent potassium channelsJ Neurochem2010113253054220096091
- Bahi-BuissonNEisermannMNivotSInfantile spasms as an epileptic feature of DEND syndrome associated with an activating mutation in the potassium adenosine triphosphate (ATP) channel, Kir6.2J Child Neurol20072291147115017890419
- AmorosoSSchmid-AntomarchiHFossetMGlucose, sulfonylureas, and neurotransmitter release: role of ATP-sensitive K+ channelsScience19902478528542305257
- ShyngSNicholsCGOctameric stoichiometry of the KATP channel complexJ Gen Physiol19971106556649382894
- ClementJPKunjilwarKGonzalezGAssociation and stoichiometry of KATP channel subunitsNeuron1997188278389182806
- InagakiNGonoiTClementJPReconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptorScience1995270116611707502040
- SakuraHAmmalaCSmithPACloning and functional expression of the cDNA encoding a novel ATP-sensitive potassium channel subunit expressed in pancreatic beta-cells, brain, heart and skeletal muscleFEBS Lett19953773383448549751
- InagakiNTsuuraYNambaNCloning and functional characterization of a novel ATP-sensitive potassium channel ubiquitously expressed in rat tissues, including pancreatic islets, pituitary, skeletal muscle, and heartJ Biol Chem1995270569156947890693
- ShiNQYeBMakielskiJCFunction and distribution of the SUR isoforms and splice variantsJ Mol Cell Cardiol200539516015978902
- Aguilar-BryanLNicholsCGWechslerSWCloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretionScience19952684234267716547
- ChutkowWASimonMCLe BeauMMCloning, tissue expression, and chromosomal localization of SUR2, the putative drug-binding subunit of cardiac, skeletal muscle, and vascular KATP channelsDiabetes199645143914458826984
- IsomotoSKondoCYamadaMA novel sulfonylurea receptor forms with BIR (Kir6.2) a smooth muscle type ATP-sensitive K+ channelJ Biol Chem199627124321243248798681
- SimJHYangDKKimYCATP-sensitive K+ channels composed of Kir6.1 and SUR2B subunits in guinea pig gastric myocytesAm J Physiol Gastrointest Liver Physiol2002282G137G14411751167
- AntcliffJFHaiderSProksPFunctional analysis of a structural model of the ATP-binding site of the KATP channel Kir6.2 subunitEMBO J20052422923915650751
- ProksPAshcroftFMModeling KATP channel gating and its regulationProg Biophys Mol Biol20099971918983870
- MasiaRKosterJCTuminiSAn ATP-binding mutation (G334D) in KCNJ11 is associated with a sulfonylurea-insensitive form of developmental delay, epilepsy, and neonatal diabetesDiabetes20075632833617259376
- AittoniemiJFotinouCCraigTJSUR1: a unique ATP-binding cassette protein that functions as an ion channel regulatorPhilos Trans R Soc Lond B Biol Sci2009364151425726718990670
- MikiTSeinoSRoles of KATP channels as metabolic sensors in acute metabolic changesJ Mol Cell Cardiol20053891792515910876
- NicholsCGShyngSLNestorowiczAAdenosine diphosphate as an intracellular regulator of insulin secretionScience1996272178517878650576
- GribbleFMTuckerSJAshcroftFMThe essential role of the Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxideEMBO J199716114511529135131
- ZingmanLVAlekseevAEBienengraeberMSignaling in channel/enzyme multimers: ATPase transitions in SUR module gate ATP-sensitive K+ conductanceNeuron20013123324511502255
- MikhailovMVCampbellJDde WetH3-D structural and functional characterization of the purified KATP channel complex Kir6.2-SUR1EMBO J2005244166417516308567
- ZerangueNSchwappachBJanYNA new ER trafficking signal regulates the subunit stoichiometry of plasma membrane KATP channelsNeuron19992253754810197533
- BienengraeberMAlekseevAEAbrahamMRATPase activity of the sulfonylurea receptor: a catalytic function for the KATP channel complexFASEB J2000141943195211023978
- ThomasPMYeYLightnerEMutation of the pancreatic islet inward rectifier Kir6.2 also leads to familial persistent hyperinsulinemic hypoglycemia of infancyHum Mol Genet19965180918128923010
- ThomasPMCoteGJWohllkNMutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancyScience19952684264297716548
- GloynALPearsonERAntcliffJActivating mutations in the gene encoding the ATP sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetesN Eng J Med200435018381849
- ColomboCDelvecchioMZecchinoCTransient neonatal diabetes mellitus is associated with a recurrent (R201H) KCNJ11 (KIR6.2) mutationDiabetologia200548112439244116205880
- YorifujiTNagashimaKKurokawaKThe C42R mutation in the Kir6.2 (KCNJ11) gene as a cause of transient neonatal diabetes, childhood diabetes, or later-onset, apparently type 2 diabetes mellitusJ Clin Endocrinol Metab20059063174317815784703
- RiedelMJSteckleyDCLightPECurrent status of the E23K Kir6.2 polymorphism: implications for type-2 diabetesHum Genet2005116313314515565284
- TarasovAINicolsonTJRivelineJPA rare mutation in ABCC8/SUR1 leading to altered ATP-sensitive K+ channel activity and beta-cell glucose sensing is associated with type 2 diabetes in adultsDiabetes20085761595160418346985
- DunneMJCosgroveKEShepherdRMHyperinsulinism in infancy: from basic science to clinical diseasePhysiol Rev20048423927514715916
- DunneMJKaneCShepherdRMFamilial persistent hyperinsulinemic hypoglycemia of infancy and mutations in the sulfonylurea receptorN Engl J Med1997336107037069041101
- ShimomuraKFlanaganSEZadekBAdjacent mutations in the gating loop of Kir6.2 produce neonatal diabetes and hyperinsulinismEMBO Mol Med2009116617720049716
- GlaserBThorntonPOtonkoskiTGenetics of neonatal hyperinsulinismArch Dis Child200082F79F86
- FlanaganSEClauinSBellanne-ChantelotCUpdate of mutations in the genes encoding the pancreatic beta-cell K(ATP) channel subunits Kir6.2 (KCNJ11) and sulfonylurea receptor 1 (ABCC8) in diabetes mellitus and hyperinsulinismHum Mutat20093017018018767144
- TaschenbergerGMougeyAShenSIdentification of a familial hyperinsulinism causing mutation in the sulfonylurea receptor 1 that prevents normal trafficking and function of KATP channelsJ Biol Chem2002277171391714611867634
- PartridgeCJBeechDJSivaprasadaraoAIdentification and pharmacological correction of a membrane trafficking defect associated with a mutation in the sulfonylurea receptor causing familial hyperinsulinismJ Biol Chem2001276359473595211457841
- YanFLinCWWeisigerESulfonylureas correct trafficking defects of ATP-sensitive potassium channels caused by mutations in the sulfonylurea receptorJ Biol Chem2004279110961110514707124
- HuopioHReimannFAshfieldRDominantly inherited hyperinsulinism caused by a mutation in the sulfonylurea receptor type 1J Clin Invest200010689790611018078
- MatsuoMTrappSTanizawaYFunctional analysis of a mutant sulfonylurea receptor, SUR1–1420C, that is responsible for persistent hyperinsulinemic hypoglycemia of infancyJ Biol Chem2000275411844119110993895
- ShyngSLFerrigniTShepardJBFunctional analyses of novel mutations in the sulfonylurea receptor 1 associated with persistent hyperinsulinemic hypoglycemia of infancyDiabetes199847114511519648840
- PinneySEMacMullenCBeckerSClinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutationsJ Clin Invest20081182877288618596924
- AmatoEDTammaroPCraigTJVariable phenotypic spectrum of diabetes mellitus in a family carrying a novel KCNJ11 gene mutationDiabet Med20082565165618544102
- ShimomuraKGirardCAProksPMutations at the same residue (R50) of Kir6.2 (KCNJ11) that cause neonatal diabetes produce different functional effectsDiabetes2006551705171216731833
- GirardCShimomuraKProksPFunctional analysis of six Kir6.2 (KCNJ11) mutations causing neonatal diabetesPflugers Arch200645332333217021801
- TarasovAIGirardCALarkinBFunctional analysis of two K ir6. 2 (KCNJ11) mutations, K170T and E322K, causing neonatal diabetesDiabetes Obes Metab20079Suppl 2465517919178
- KlupaTEdghillELNazimJThe identification of a R201H mutation in KCNJ11, which encodes Kir6.2, and successful transfer to sustained-release sulphonylurea therapy in a subject with neonatal diabetes: evidence for heterogeneity of beta cell function among carriers of the R201H mutationDiabetologia2005481029103115838686
- FlanaganSEEdghillELGloynALMutations in KCNJ11, which encodes Kir6.2, are a common cause of diabetes diagnosed in the first 6 months of life, with the phenotype determined by genotypeDiabetologia2006491190119716609879
- FlanaganSEPatchAMMackayDJMutations in ATP-sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthoodDiabetes2007561930193717446535
- SlingerlandASHurkxWNoordamKSulphonylurea therapy improves cognition in a patient with the V59M KCNJ11 mutationDiabet Med20082527728118307455
- DoyleDAMorais CabralJPfuetznerRAThe structure of the potassium channel: molecular basis of K+ conduction and selectivityScience199828069779525859
- HattersleyATAshcroftFMActivating mutations in Kir6.2 and neonatal diabetes: new clinical syndromes, new scientific insights, and new therapyDiabetes2005542503251316123337
- ProksPGirardCBaevreHFunctional effects of mutations at F35 in the NH2-terminus of Kir6.2 (KCNJ11), causing neonatal diabetes, and response to sulfonylurea therapyDiabetes20065561731173716731836
- ReimannFTuckerSJProksPInvolvement of the N-terminus of Kir6.2 in coupling to the sulphonylurea receptorJ Physiol199951832533610381582
- BabenkoAPBryanJSUR-dependent modulation of KATP channels by an N-terminal Kir6.2 peptide: defining intersubunit gating interactionsJ Biol Chem2002277439974400412213829
- KosterJCKurataHTEnkvetchakulDA DEND mutation in Kir6.2 (KCNJ11) reveals a flexible N-terminal region critical for ATP-sensing of the KATP channelBiophys J2008954689469718708460
- ProksPGirardCAshcroftFMFunctional effects of KCNJ11 mutations causing neonatal diabetes: enhanced activation by Mg-ATPHum Mol Genet2005142717272616087682
- TarasovAIWeltersHJSenkelSA Kir6.2 mutation causing neonatal diabetes impairs electrical activity and insulin secretion from INS-1 beta-cellsDiabetes2006553075308217065345
- MännikköRJefferiesCFlanaganSEInteraction between mutations in the slide helix of Kir6.2 associated with neonatal diabetes and neurological symptomsHum Mol Genet201019696397220022885
- GloynALReimannFGirardCRelapsing diabetes can result from moderately activating mutations in KCNJ11Hum Mol Genet20051492593415718250
- JohnSAWeissJNXieLHMolecular mechanism for ATP-dependent closure of the K+ channel Kir6.2J Physiol2003552233412860923
- ProksPArnoldALBruiningJA heterozygous activating mutation in the sulphonylurea receptor SUR1 (ABCC8) causes neonatal diabetesHum Mol Genet2006151793180016613899
- ProksPShimomuraKCraigTJMechanism of action of a sulphonylurea receptor SUR1 mutation (F132L) that causes DEND syndromeHum Mol Genet2007162011201917584766
- FangKCsanadyLChaKWThe N-terminal transmembrane domain (TMD0) and a cytosolic linker (L0) of sulphonylurea receptor define the unique intrinsic gating of KATP channelsJ Physiol200657637938916887879
- MasiaRde LeonDDMacMullenCA mutation in the TMD0–L0 region of sulfonylurea receptor-1 (L225P) causes permanent neonatal diabetes mellitus (PNDM)Diabetes2007561357136217317760
- PatchAMFlanaganSEBoustredCMutations in the ABCC8 gene encoding the SUR1 subunit of the KATP channel cause transient neonatal diabetes, permanent neonatal diabetes or permanent diabetes diagnosed outside the neonatal periodDiabetes Obes Metab20079Suppl 2283917919176
- BabenkoAPBryanJSUR domains that associate with and gate KATP pores define a novel gatekeeperJ Biol Chem2003278415774158012941953
- TammaroPGirardCMolnesJKir6.2 mutations causing neonatal diabetes provide new insights into Kir6.2-SUR1 interactionsEMBO J2005242318233015962003
- de WetHReesMGShimomuraKIncreased ATPase activity produced by mutations at arginine-1380 in nucleotide-binding domain 2 of ABCC8 causes neonatal diabetesProc Natl Acad Sci U S A2007104189881899218025464
- de WetHProksPLafondMA mutation (R826 W) in nucleotide-binding domain 1 of ABCC8 reduces ATPase activity and causes transient neonatal diabetesEMBO Rep2008964865418497752
- EllardSFlanaganSEGirardCAPermanent neonatal diabetes caused by dominant, recessive, or compound heterozygous SUR1 mutations with opposite functional effectsAm J Hum Genet20078137538217668386
- MarkworthESchwanstecherCSchwanstecherMATP4–mediates closure of pancreatic beta-cell ATP-sensitive potassium channels by interaction with 1 of 4 identical sitesDiabetes2000491413141810969823
- HaniEHBoutinPDurandEMissense mutations in the pancreatic islet beta cell inwardly rectifying K+ channel gene (KIR6.2/BIR): a meta-analysis suggests a role in the polygenic basis of type II diabetes mellitus in CaucasiansDiabetologia199841151115159867219
- GloynALWeedonMNOwenKRLarge-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetesDiabetes20035256857212540637
- BarrosoILuanJMiddelbergRPCandidate gene association study in type 2 diabetes indicates a role for genes involved in beta-cell function as well as insulin actionPLoS Biol20031E2014551916
- HammingKSSolimanDMatemiszLCCoexpression of the type 2 diabetes susceptibility gene variants KCNJ11 E23K and ABCC8 S1369A alter the ATP and sulfonylurea sensitivities of the ATP-sensitive K channelDiabetes2009582419242419587354
- SperlingMANeonatal diabetes mellitus: from understudy to center stageCurr Opin Pediatr200517451251816012265
- PolakMShieldJNeonatal and very-early-onset diabetes mellitusSemin Neonatol20049596515013476
- GreeleySATuckerSEWorrellHIUpdate in neonatal diabetesCurr Opin Endocrinol Diabetes Obes2010171131919952737
- SagenJVRaederHHathoutEPermanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: patient characteristics and initial response to sulfonylurea therapyDiabetes2004532713271815448106
- StanikJGasperikovaDPaskovaMPrevalence of permanent neonatal diabetes in Slovakia and successful replacement of insulin with sulfonylurea therapy in KCNJ11 and ABCC8 mutation carriersJ Clin Endocrinol Metab2007921276128217213273
- UK Prospective Diabetes Study GroupIntensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33)Lancet19983528378539742976
- The Diabetes Control and Complications Trial Research GroupThe effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitusN Eng J Med1993329977986
- WinklerMStephanDBiegerSTesting the bipartite model of the sulfonylurea receptor binding site: binding of A-, B-, and A B-site ligandsJ Pharmacol Exp Ther200732270170817495126
- AshcroftFMNew uses for old drugs: neonatal diabetes and sulphonylureasCell Metab20101117918120197050
- StoyJGreeleySAPazVPDiagnosis and treatment of neonatal diabetes: an United States experiencePediatr Diabetes2008945045918662362
- ProksPReimannFGreenNSulfonylurea stimulation of insulin secretionDiabetes200251Suppl 3S368S37612475777
- ZungAGlaserBNimriRGlibenclamide treatment in permanent neonatal diabetes mellitus due to an activating mutation in Kir6.2J Clin Endocrinol Metab2004895504550715531505
- MannaTDBattistimCRadonskyVGlibenclamide unresponsiveness in a brazilian child with permanent neonatal diabetes mellitus and DEND syndrome due to a C166Y mutation in KCNJ11 (Kir6.2) geneArq Bras Endocrinol Metabol20085281350135519169493
- KosterJCRemediMSDaoCATP and sulfonylurea sensitivity of mutant ATP-sensitive K channels in neonatal diabetes: implications for pharmacogenomic therapyDiabetes2005542645265416123353
- PearsonERFlechtnerINjolstadPRSwitching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutationsN Eng J Med2006355467477
- ChenKWangRWenSYRelationship of P450 2C9 genetic polymorphisms in Chinese and the pharmacokinetics of tolbutamideJ Clin Pharm Ther200530324124915896241
- KirchheinerJRootsIGoldammerMEffect of genetic polymorphisms in cytochrome p450 (CYP) 2C9 and CYP2C8 on the pharmacokinetics of oral antidiabetic drugs: clinical relevanceClin Pharmacokinet200544121209122516372821
- SuzukiKYanagawaTShibasakiTEffect of CYP2C9 genetic polymorphisms on the efficacy and pharmacokinetics of glimepiride in subjects with type 2 diabetesDiabetes Res Clin Pract200672214815416325295
- BeckerMLVisserLETrienekensPHCytochrome P450 2C9 *2 and *3 polymorphisms and the dose and effect of sulfonylurea in type II diabetes mellitusClin Pharmacol Ther200883228829217597710
- SlingerlandASNuboerRHadders-AlgraMImproved motor development and good long-term glycemic control with sulfonylurea treatment in a patient with the syndrome of intermediate developmental delay, early-onset generalised epilepsy and neonatal diabetes associated with the V59M mutation in the KCNJ11 geneDiabetologia2006492559256317047922
- MlynarskiWTarasovAGachASulfonylurea improves CNS function in a case of intermediate DEND syndrome caused by a mutation in KCNJ11Nat Clin Pract Neurol200731164064517982434
- KosterJCCadarioFPeruzziCThe G53D mutation in Kir6.2 (KCNJ11) is associated with neonatal diabetes and motor dysfunction in adulthood that is improved with sulfonylurea therapyJ Clin Endocrinol Metab2008931054106118073297
- TurkkahramanDBircanITribbleNDPermanent neonatal diabetes mellitus caused by a novel homozygous (T168A) glucokinase (GCK) mutation: initial response to oral sulphonylurea therapyJ Pediatr200815312212618571549
- SøvikONjølstadPFøllingIHyperexcitability to sulphonylurea in MODY3Diabetologia19984156076089628283
- PearsonERLiddellWGShepherdMSensitivity to sulphonylureas in patients with hepatocyte nuclear factor-1 alpha gene mutations: evidence for pharmacogenetics in diabetesDiabet Med200017754354510972586
- GribbleFMReimannFDifferential selectivity of insulin secret-agogues mechanisms, clinical implications, and drug interactionsJ Diabetes Complications200317Suppl 2111512623163
- GribbleFMReimannFSulphonylurea action revisited: the post-cloning eraDiabetologia20034687589112819907
- InagakiNGonoiTClementJPA family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channelsNeuron199616101110178630239
- GribbleFMTuckerSJSeinoSTissue specificity of sulfonylureas: studies on cloned cardiac and beta-cell K(ATP) channelsDiabetes1998479141214189726229
- GribbleFMAshcroftFMDifferential sensitivity of beta-cell and extrapancreatic K(ATP) channels to gliclazideDiabetologia199942784584810440127
- SongDKAshcroftFMGlimepiride block of cloned beta-cell, cardiac and smooth muscle K(ATP) channelsBr J Pharmacol2001133119319911325810
- MikiTMinamiKZhangLATP-sensitive potassium channels participate in glucose uptake in skeletal muscle and adipose tissueAm J Physiol Endocrinol Metab2002283E1178E118412388128
- ChutkowWASamuelVHansenPADisruption of SUR2-containing K(ATP) channels enhances insulin-stimulated glucose uptake in skeletal muscleProc Natl Acad Sci U S A200198117601176411562480
- SkupienJMaleckiMTMlynarskiWAssessment of insulin sensitivity in adults with permanent neonatal diabetes mellitus due to mutations in the KCNJ11 gene encoding Kir6.2Rev Diabet Stud200631172017491708
- MaleckiMTSkupienJKlupaTTransfer to sulphonylurea therapy in adult subjects with permanent neonatal diabetes due to KCNJ11-activating mutations: evidence for improvement in insulin sensitivityDiabetes Care200730114714917192350
- TessierDDawsonKTétraultJPGlibenclamide vs gliclazide in type 2 diabetes of the elderlyDiabet Med199411109749807895463
- HattersleyABruiningJShieldJISPAD Clinical Practice Consensus Guidelines 2006–2007. The diagnosis and management of monogenic diabetes in childrenPediatr Diabetes20067635236017212604
- PunPClarkRWanKWNeonatal diabetes mellitus: the impact of molecular diagnosisNeoReviews2010116e306e310
- DiStefanoJKWatanabeRMPharmacogenetics of anti-diabetes drugsPharmaceuticals2010382610264620936101