
Abstract
Primary distal renal tubular acidosis (dRTA) is a rare genetic disorder caused by impaired distal acidification due to a failure of type A intercalated cells (A-ICs) in the collecting tubule. dRTA is characterized by persistent hyperchloremia, a normal plasma anion gap, and the inability to maximally lower urinary pH in the presence of systemic metabolic acidosis. Common clinical features of dRTA include vomiting, failure to thrive, polyuria, hypercalciuria, hypocitraturia, nephrocalcinosis, nephrolithiasis, growth delay, and rickets. Mutations in genes encoding three distinct transport proteins in A-ICs have been identified as causes of dRTA, including the B1/ATP6V1B1 and a4/ATP6V0A4 subunits of the vacuolar-type H+-ATPase (H+-ATPase) and the chloride–bicarbonate exchanger AE1/SLC4A1. Homozygous or compound heterozygous mutations in ATP6V1B1 and ATP6V0A4 lead to autosomal recessive (AR) dRTA. dRTA caused by SLC4A1 mutations can occur with either autosomal dominant or AR transmission. Red blood cell abnormalities have been associated with AR dRTA due to SLC4A1 mutations, including hereditary spherocytosis, Southeast Asia ovalocytosis, and others. Some patients with dRTA exhibit atypical clinical features, including transient and reversible proximal tubular dysfunction and hyperammonemia. Incomplete dRTA presents with inadequate urinary acidification, but without spontaneous metabolic acidosis and recurrent urinary stones. Heterozygous mutations in the AE1 or H+-ATPase genes have recently been reported in patients with incomplete dRTA. Early and sufficient doses of alkali treatment are needed for patients with dRTA. Normalized serum bicarbonate, urinary calcium excretion, urinary low-molecular-weight protein levels, and growth rate are good markers of adherence to and/or efficacy of treatment. The prognosis of dRTA is generally good in patients with appropriate treatment. However, recent studies showed an increased frequency of chronic kidney disease (CKD) in patients with dRTA during long-term follow-up. The precise pathogenic mechanisms of CKD in patients with dRTA are unknown.
Introduction
Primary distal (type 1) renal tubular acidosis (dRTA) is a rare genetic disorder caused by impaired distal acidification due to a failure of type A intercalated cells (A-ICs) of the collecting tubule.Citation1 dRTA is characterized by persistent hyperchloremia, a normal plasma anion gap, and the inability to maximally lower urinary pH in the presence of systemic metabolic acidosis.Citation2 Common clinical features of dRTA include vomiting, failure to thrive, polyuria, hypercalciuria, hypocitraturia, nephrocalcinosis, nephrolithiasis, growth delay, and rickets.Citation1,Citation2 The clinical variant of dRTA that presents with inadequate urinary acidification without spontaneous metabolic acidosis is termed incomplete dRTA (idRTA).Citation2,Citation3
Mutations in genes encoding three distinct transport proteins have been identified as causes of dRTA: the B1/ATP6V1B1 and a4/ATP6V0A4 subunits of the vacuolar-type H+-ATPase (H+-ATPase) and the chloride–bicarbonate exchanger AE1/SLC4A1.Citation3 However, because a genetic cause is determined in only 70%–80% of patients with dRTA,Citation4,Citation5 mutations in additional genes are likely to cause dRTA.Citation3,Citation6
The clinical manifestations of patients with dRTA depend on the underlying gene mutations.Citation3 For example, the majority of patients with dRTA caused by mutations in genes encoding for H+-ATPase develop sensorineural hearing loss (SNHL).Citation6 Autosomal dominant (AD) dRTA with heterozygous AE1 gene mutations causes less severe clinical manifestations.Citation7 However, a recent study showed that clinical features are not specific indicators of the underlying causal gene.Citation5
Herein, we review the recent advances in the pathogenesis, underlying gene mutations, atypical clinical features, incomplete type, kidney stone formation, treatment, and long-term outcome of dRTA.
Mechanisms of renal acid excretion
The western diet plus endogenous metabolism generates 1 mmol/kg body weight per day of nonvolatile acids in adults.Citation8 An additional 1–2 mmol/kg body weight per day of nonvolatile acids is produced from the formation of hydroxyapatite from growth bone in children.Citation7 The kidneys maintain acid– base homeostasis by reabsorbing filtered HCO3−, excreting acid in the form of NH4+ and titratable acids, and producing new HCO3− to replenish that lost to metabolism.Citation3 The distal nephron is responsible for the secretion of H+ that is then buffered by NH3 and titratable acids, leading to urinary acidification.Citation6,Citation7
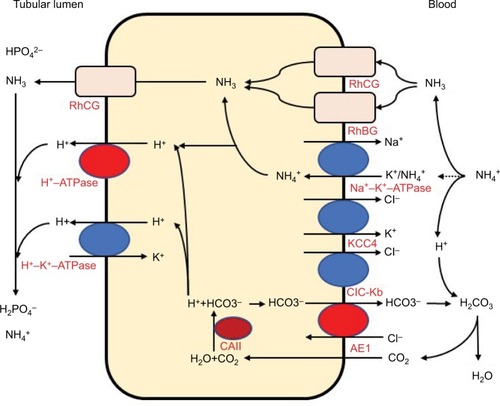
The distal convoluted tubules and cortical collecting ducts consist of principal cells, which reabsorb sodium via the epithelial sodium channels, acid-secreting A-ICs, and base-secreting type B intercalated cells (B-ICs). A-ICs mainly contribute to urinary acidification by generating new HCO3− and excreting NH into the urine ().Citation3,Citation9,Citation10 CO2 is hydrated by cytosolic carbonic anhydrase type II (CAII) that is present in all intercalated cells (ICs) of the collecting tubule and forms H+ and HCO3−. A-ICs secrete H+ by apical H+-ATPase with an additional contribution from H+–K+-ATPase. A-ICs release HCO3− into the blood in exchange for Cl− via the basolateral anion exchanger AE1. Cl− exits the cell by the potassium chloride co-transporter (KCC4) or the chloride channel (ClC-Kb).Citation7 B-ICs express an apical Cl—HCO3− exchanger, pendrin, and a basolateral H+-ATPase, responsible for net base secretion. Chronic acidosis converts B-ICs to A-ICs, which is mediated by an extracellular matrix protein hensin/DMBT1, and increases net acid secretion.Citation11,Citation12 Although the mechanism by which ICs sense a change in blood pH is unclear, Schwartz et al recently showed that principal cells respond to acid by producing SFD1 which regulates ICs subtype distribution.Citation13
Figure 1 Acid and ammonia secretion in type A intercalated cells in the distal nephron.
Because the capacity of the distal nephron to excrete acid as free H+ is limited, the distal nephron excretes the majority of acid coupled to urine buffers, ammonia (NH3), and titratable acids (mainly HPO42−). Therefore, the net acid secretion in urine is the sum of the ammonium (NH4+) charge plus the titratable acid (H2PO4−) charge minus the HCO3− charge.Citation8 Under normal conditions, the kidneys excrete approximately one-third to one-half of the net acids as titratable acids and one-half to two-thirds as NH4+. Under conditions of chronic acidosis or acid load, NH4+ excretion can increase several-fold to tenfold, while titratable acid excretion shows only a small increase.Citation8 Therefore, the excretion of NH4+ is the main mechanism for urine acidification under conditions of chronic acidosis.
In contrast to most urinary solutes, the majority of urinary NH3 is generated in the kidney.Citation14 NH +4 is generated from glutamine in the proximal tubular cells and is secreted into the urine in the proximal tubule lumen, which is coupled with HCO3− recovery resulting from glutamine metabolism. NH4+ is primarily reabsorbed by the Na–K–2Cl co-transporter NKCC2 in the thick ascending limb of the loop of Henle and is accumulated in the interstitium. Interstitial NH3/NH +4 is then secreted into the cortical and medullary collecting duct lumen by several mechanisms: diffusion of medullary NH3 across the basolateral and apical membranes into the lumen; basolateral Na+–K+-ATPase that transports NH + 4 into the cell by substituting NH4+ for K+ in the inner medullary collecting duct; NH3 uptake across the basolateral membrane via the rhesus blood group glycoproteins, RhBG and RhCG; and NH3 secretion across ICs via RhCG. Collecting duct NH3 secretion occurs by parallel NH3 and H+ transport, and apical H+ secretion involves both H+-ATPase and H+–K+-ATPase ().Citation8,Citation14,Citation15
Pathogenic mechanisms of dRTA
dRTA is caused by the failure of the kidney A-ICs to acidify the urine normally, which results from a dysfunction in any of the transporters involved in this process. Mutations in three transporter genes expressed in A-ICs have been identified as causes of dRTA, including the B1 (ATP6V1B1) and a4 (ATP6V0A4) subunits of H+-ATPase and the AE1/SLC4A1.Citation16 However, because mutations in these genes are identified in only 70%–80% of patients with dRTA,Citation4,Citation5 dRTA is also likely to be caused by mutations in other genes.Citation3,Citation17 In addition to its genetic component, dRTA can also be acquired.Citation7
Vacuolar H+-ATPase gene (ATP6V1B1 and ATP6V0A4) mutations
H+-ATPases are multisubunit enzymatic proton pumps, which consist of two domains, the V1 cytoplasmic domain (subunits A–H) and the V0 membrane domain composed of subunits a, d c″, c, and e.Citation18 The V0 domain mediates proton transfer and requires ATP hydrolysis by the V1 domain.Citation18,Citation19 The B1 subunit is expressed in the kidney, inner ear, epididymis, and ciliary body of the eye. The a4 subunit is only expressed in the kidney, inner ear, and epididymis. In the kidney, the B1 subunit is expressed only in ICs of the late distal tubule, connecting segment, and collecting duct, while the a4 subunit is expressed in the proximal tubule, loop of Henle, and ICs of the late distal tubule, connecting segment, and collecting duct.Citation20 Homozygous or compound heterozygous mutations in B1 and a4 lead to autosomal recessive (AR) dRTA.
Karet et al were the first to identify homozygous mutations in ATP6V1B1 in AR dRTA patients with early SNHL.Citation21 Subsequently, Smith et al detected homozygous mutations in ATP6V0A4 in AR dRTA patients without SNHL.Citation22 However, it became later obvious that a number of patients with AR dRTA with homozygous or compound heterozygous mutations in ATP6V0A4 developed SNHL in early childhoodCitation23 or in young adulthood.Citation24 Compound heterozygous mutations in ATP6V1B1 were also found in some patients with dRTA.Citation23,Citation25,Citation26
Premature termination codons, frameshift mutations, or splice-site mutations, which are predicted to disrupt the encoded protein, are found in most patients with ATP6V1B1 or ATP6V0A4 mutations, while missense mutations have been described in only a few patients.Citation21–Citation24 Experimental studies using rat inner medullary collecting duct cell culture or yeast models showed that dysfunction or impaired assembly of the B1 subunit with other protein complex subunits is the most common outcome of missense mutations in ATP6V1B1.Citation27,Citation28 Su et al recreated previously reported missense mutations of the a4 subunit G820RCitation22 and R807QCitation24 in yeast to examine the effects on protein expression. They found that the G820R mutation caused a loss of required phosphofructokinase-1 (PFK) binding to the a4 subunit, while the R807Q mutation resulted in a loss of pump protein.Citation29
In mouse models, mice deficient in the B1 subunit show more alkaline urine without systemic acidosis when fed a standard diet. These mice also develop more severe metabolic acidosis after an acid load compared to normal mice, indicating a failure of normal urinary acidification.Citation30 The B1-deficient mice do not develop hearing impairment or nephrocalcinosis. In contrast, mice lacking the a4 subunit demonstrate dRTA with severe metabolic acidosis, hypokalemia, early nephrocalcinosis, and severe hearing impairment with enlarged cochlear and endolymphatic ducts in the inner ear.Citation31,Citation32 In addition, these mice develop proximal renal tubular dysfunction with defective endocytic trafficking, proteinuria, and phosphaturia, which has not been reported in humans.Citation33 This may be due to compensatory changes in the a1, a2, and a3 subunits in the proximal tubules of dRTA patients with the ATP6V0A4 mutation.Citation17,Citation34
Chloride–bicarbonate exchanger AE1 gene (SLC4A1) mutations
The Cl—HCO3− exchanger AE1, encoded by SLC4A1, is expressed in the red blood cells (RBCs; eAE1, Band 3) and in the A-ICs of kidney (kAE1). SLC4A1 mutations can cause dRTA or RBC abnormalities including hereditary spherocytosis (HS), Southeast Asia ovalocytosis (SAO), hereditary stomatocytosis, and hereditary xerocytosis.Citation35 Most mutations cause either dRTA or RBC abnormalities, whereas only a few mutations lead to abnormalities in both.Citation3 dRTA caused by AE1 gene mutations can occur with either an AD or AR transmission ().Citation1,Citation6 AE1 mutations are rare and usually have an AD transmission in Caucasians, while AR dRTA is common in Asians.Citation17 The clinical symptoms are more severe and the age of onset is earlier in patients with AR dRTA compared to patients with AD dRTA.Citation7 Patients with AD dRTA can present with complete dRTA or idRTA, whereas patients with AR dRTA always present with complete dRTA.Citation16 Although RBC abnormalities have been associated with AR dRTA, hemolytic anemia is extremely rare in AD dRTA, and only one family with AD idRTA and HS due to a heterozygous splicing mutation (c.1432–1G>A, Band 3PRIBRAM) has been reported.Citation36
Table 1 SLC4A1 mutations in dRTA
AD AE1 gene (SLC4A1) mutations
Wrong et al reported a family with AD dRTA caused by a heterozygous mutation of R589H in AE1.Citation37 Subsequent studies described AD dRTA with AE1 gene mutations including R589H,Citation38–Citation40 R589C,Citation38 R589S,Citation39 S613F,Citation38 R901X,Citation39 A858D,Citation41 A888L+889X,Citation42 G609R,Citation43 D905Gfs15,Citation44 D905dup,Citation45 and M909T.Citation46
Experimental studies using transfected cell models showed that AE1 gene mutations caused normal or modestly reduced anion transport activity and impaired trafficking with retention in the endoplasmic reticulum (ER). Heterodimers with the wild-type AE1 polypeptide caused a dominant negative trafficking phenotype (R589H and S613F)Citation35,Citation47–Citation49 or mistargeting to the apical membranes or to both the apical and basolateral membranes (G609R, R901X, and M909T).Citation43,Citation46,Citation49–Citation51
In in vivo studies, mice lacking Ae1 exhibited complete dRTA with hemolytic anemia, while mice heterozygous for Ae1 showed no apparent defect.Citation52,Citation53 Mumtaz et al recently generated an Ae1 R607H knockin (KI) mouse, corresponding to the most common AD dRTA mutation in human AE1, R589H.Citation54 They found that both homozygous and heterozygous R607H KI mice exhibited idRTA without RBC abnormalities.Citation54 Mutant mice exhibited decreased levels of Ae1 in A-ICs, but preserved basolateral targeting of the mutant protein, and reduced expression of H+-ATPase due to impaired targeting to the apical membranes.Citation9,Citation54
AR AE1 gene (SLC4A1) mutations
Tanphaichitr et al first reported AR dRTA with a homozygous AE1 gene loss-of-function mutation, G701D, in two siblings in Thailand who had xerocytic hemolytic anemia with normal RBC AE1 activity.Citation55 AE1 G701D interacts with an RBC AE1 chaperon, glycophorin A (GPA), which rescues the mutant protein in RBCs. GPA is not expressed in renal ICs, which explains the normal erythroid AE1 expression in these patients.Citation35,Citation55 Subsequently, Yenchitsomanus et al reported a homozygous AE1 G701D mutation in five out of eight Thai families with AR dRTA with mild RBC morphological abnormalities without hemolytic anemia and suggested that AE1 G701D was a common molecular defect in Thailand.Citation56
SAO is a common hereditary condition in South East and Melanesia. SAO is caused by the AE1 (ΔAla400-Ala408) mutation, which causes ovalocytic erythrocytes to be resistant to invasion by the malarial parasite.Citation57 Vasuvattakul et al reported two patients from Northeast Thailand with AR dRTA and SAO resulting from compound heterozygous AE1 G701D/SAO mutations.Citation58 Bruce et al described AE1 gene mutations associated with AR dRTA and SAO in families from Malaysia and Papua New Guinea. The mutations included compound heterozygous AE1 G701D/SAO and ΔV850/SAO and homozygous AE1 ΔV850/ΔV850.Citation41 Patients with compound heterozygous AE1 A858D/SAO mutations in this study exhibited dRTA and SAO with AD transmission. Subsequently, other mutations were identified, including R602H/SAO from South Thailand,Citation59 Q759H/SAO from Malaysia,Citation60 and G701/SAO from the Philippines.Citation61 Although homozygous SAO was long thought to be lethal,Citation62 Picard et al recently reported a child with homozygous SAO mutations who had AR dRTA with severe dyserythropoietic and hemolytic anemia with SAO red cells.Citation63
Approximately 20% of HS cases are caused by heterozygous AE1 gene mutations.Citation64 Hence, HS can be associated with AR dRTA. Ribeiro et al reported a child with severe HS and AR dRTA caused by homozygous AE1 V488M (B and 3 Coimbra).Citation65 Toye et al reported that homozygous S667F (Band 3 Courcouronnes) causes HS and idRTA.Citation66 Subsequently, it was found that compound heterozygous E522K/G701DCitation67 and C479W (Band 3 Edmonton I)/G701DCitation68 and homozygous A858DCitation69 cause AR dRTA with HS.
Several other AE1 mutations have been reported to cause AR dRTA with RBC abnormalities other than SAO or HS. These include compound heterozygous ΔV850/A858D (red cells of bizarre shapes and anemia)Citation41 and G701D/A858D (ovalocytes and acanthocytes),Citation70 homozygous A858D (hemolytic anemia with elliptocytosis, stomatocytosis, and acanthocytosisCitation71 or acanthocytosis and echinocytosis),Citation72 and nonsense mutation of S447X, Band 3 nullVIENNA (severe hemolytic anemia).Citation73 In addition, AE1 mutations causing AR dRTA without RBC abnormalities have been reported, including homozygous ΔV850Citation41 and compound heterozygous G494S/G701DCitation44 and S773P/G701D.Citation59
Experimental studies using transfected cell models have revealed that these mutations cause Golgi retention due to impaired trafficking with loss of function, which is rescued by GPA (G701D),Citation48,Citation55,Citation74 misfolding and targeting for degradation (S773P),Citation48,Citation74 decreased AE1 function (ΔV850);Citation41 ER retention due to impaired trafficking with normal anion transport function, which is rescued by GPA (S667F),Citation66 mild trafficking impairment (E522K);Citation67 and ER retention due to impaired trafficking and misfolding (C479W).Citation68
Other candidate genes for dRTA
Because ~20% of patients with dRTA do not have mutations in genes for H+-ATPase or AE1, additional candidate genes have been identified in mouse models of dRTA.Citation3,Citation6 These include HKα2 (colonic H+–K+-ATPase),Citation75 Kcc4 (K+–Cl− co-transporter, Kcc4),Citation76 genes for the H+-ATPase C, G, and d subunits,Citation77 Foxi1 (the Forkhead transcription factor, Foxi1),Citation78 Rhcg (the ammonia transporter, Rhcg),Citation79,Citation80 Slc26a7 (Cl−–HCO3− exchanger, Slc26a7, co-localized with AE1),Citation81 DMBT1 (component of the pathway of acidosis-induced conversion of B-ICs into A-ICs, hensin),Citation11 GPR4 (proton sensing G protein-coupled receptor, GPR4),Citation82 NHE4 (Na+–H+ exchanger 4, NHE4),Citation83 SLC4A5 (Na+–HCO3− co-transporter, NBCe2),Citation84 Atp6ap2 (ATPase H+ transporting lysosomal accessory protein 2, atp6ap2),Citation85 and Ncoa7 (nuclear receptor coactivator 7, Ncoa7: an H+-ATPase interacting protein), as shown in .Citation86 Although most of these genes have not been previously identified in human disease, Enerbäck et al identified homozygous FOXI1mutations in two families with AR dRTA and early-onset SNHL.Citation87 Furthermore, a recent study found homozygous or compound heterozygous WDR72 (tryptophan–aspartate repeat domain 72, WDR72: a protein possibly associated with intracellular endocytic vesicle trafficking) mutations in two families with AR dRTA ().Citation88
Table 2 Candidate genes for distal renal tubular acidosis
Causes of acquired dRTA
The main causes of acquired dRTA include medications and, most commonly, autoimmune diseases, with Sjögren syndrome being the most frequent cause. Although the precise pathogenic mechanisms of dRTA development in Sjögren syndrome are unclear, several studies suggest that autoanti-bodies against carbonic anhydrase or A-ICs transporters are involved in the pathogenesis.Citation89 Systemic lupus erythematosus, thyroiditis, and renal transplantation have been reported as other autoimmune causes of dRTA.Citation2
Medications including amphotericin B, foscarnet, analgesic abuse, lithium, melphalan, and amiloride also cause dRTA.Citation2,Citation7 In the case of amphotericin B and foscarnet, the mechanisms mediating these effects involved increased membrane permeability in the collecting duct and increased mitochondrial dysfunction in renal tubular cells, respectively.Citation90 The mechanisms associated with other drugs are not clear.
Atypical clinical features of dRTA
Some patients with dRTA exhibit atypical clinical features, which include transient and reversible proximal tubular dysfunction and hyperammonemia.
Reversible proximal tubular dysfunction in patients with dRTA
Reversible proximal tubular dysfunction has been reported in patients with dRTA. This appears to involve defects in bicarbonate reabsorption,Citation91,Citation92 low-molecular-weight proteinuria,Citation93 hypouricemia with uricosuria, phosphaturia, and generalized aminoaciduria.Citation94,Citation95 Besouw et al recently reported that 16 of 24 patients with dRTA showed transient and partial Fanconi syndrome that resembled Dent disease or Low syndrome. Proximal tubular dysfunction was only seen in children with mutations in subunits of the H+-ATPase and in those with unknown mutations.Citation96
Although the exact mechanism underlying reversible proximal tubular dysfunction is unclear, it has been suggested to involve hypokalemic nephropathyCitation32,Citation94 and/or dysfunction of the receptor-mediated endosomal pathway in renal proximal tubule cells.Citation3,Citation97 The chloride transporter ClC-5 (2Cl−–H+ exchanger) is expressed in the apical endosomes of renal proximal tubules containing H+-ATPase. ClC-5 normally acts in the endosomal pathway by coupling with H+-ATPase. Mutations of the ClC-5 gene cause Dent disease.Citation98 Picollo et al demonstrated that low extracellular pH or acidosis inhibits ClC-5 function by reducing the driving force for 2Cl−–H+ exchange.Citation99 Therefore, inhibition of ClC-5 function due to systemic acidosis may lead to partial renal Fanconi syndrome in patients with dRTA.Citation97
Hyperammonemia in patients with dRTA
Hyperammonemia, first described by Miller and Schwartz in dRTA, was not initially recognized as an important clinical feature of dRTA.Citation100 Subsequently, Miura et al reported a high frequency of hyperammonemia in patients with dRTA (four of six patients with available data), suggesting that hyperammonemia is a common feature of dRTA.Citation25 Hyperammonemia was reported in 15 patients with dRTA in a recent systematic reviewCitation101 and in a case report.Citation102 In these patients, a negative correlation was observed between blood ammonia and bicarbonate levels, and alkali therapy resulted in a rapid normalization of ammonia levels.Citation101 Increased renal ammonia synthesis in response to acidosis, without appropriate ammonia excretion, may result in hyperammonemia in dRTA.Citation100,Citation101
Incomplete distal renal tubular acidosis
idRTA, first described in 1959, presents with inadequate urinary acidification without spontaneous metabolic acidosis.Citation103 Failure to acidify urinary pH <5.3 in the NH4Cl load was considered diagnostic for idRTA.Citation89 However, because urinary acidification capacity is a continuous trait, idRTA is not a distinct entity, and may be a variant of normal urinary acidification.Citation89,Citation104
Patients with idRTA commonly exhibit hypocitraturia, hypercalciuria, nephrocalcinosis, and nephrolithiasis.Citation105,Citation106 A wide range of idRTA prevalence has been reported in patients with recurrent urinary stones.Citation104 Bone abnormalities also have been frequently reported in patients with idRTA, including ricketsCitation107 or growth failureCitation108 in children and osteoporosis and osteopenia in adults.Citation109–Citation111 However, other studies found no association between idRTA and lower bone mass.Citation1,Citation65,Citation112
Although the molecular basis for idRTA is unknown in most patients, heterozygous mutations in SLC4A1 (Band 3PPRIBRAM and A858D),Citation36,Citation41 ATP6V1B1 (F468fsX487),Citation113 and ATP6V0A4 (S544L)Citation114 have been reported. Furthermore, Dhayat et al reported that recurrent kidney stone formers with H+-ATPase B1 subunit p.E161K single-nucleotide polymorphism have idRTA with an increased prevalence of calcium phosphate kidney stones.Citation115 A recent experimental study revealed that Ncoa7 (H+-ATPase interacting protein)-knockout mice have idRTA.Citation86 Another study showed that haploinsufficiency of Atp6v1b in mice causes idRTA.Citation116
Kidney stone formation in dRTA
The combination of hypercalciuria, hypocitraturia, and high urine pH contributes to the development of kidney stone formation and/or nephrocalcinosis in dRTA.Citation6
Because bicarbonate is depleted from the extracellular fluid in dRTA, buffering of the retained nonvolatile acids promotes the release of calcium phosphate from bone, which increases urinary excretion of calcium and phosphate in dRTA.Citation6,Citation7 Moreover, metabolic acidosis decreases the function and expression of the TRPV5 calcium channel in the distal tubule, independent of parathyroid hormone and vitamin D, which also contributes to hypercalciuria in dRTA.Citation117
Urinary citrate inhibits stone formation by complexing with calcium, inhibiting spontaneous nucleation, and preventing the growth of crystals.Citation118 Metabolic acidosis increases citrate reabsorption in the proximal tubule via increased activity of sodium-dependent dicarboxylate transporter 1.Citation17,Citation117,Citation118
A high urine pH increases the supersaturation of calcium phosphate in the tubular lumen, thereby increasing the risk of kidney stone formation.Citation117
Treatment
The primary objectives of dRTA treatment are correction of metabolic acidosis and avoidance of disease-related complications, which include failure to thrive, growth retardation, rickets, osteoporosis, nephrolithiasis, and nephrocalcinosis.Citation3,Citation16,Citation89 It is especially important to prevent nephrocalcinosis because progressive nephrocalcinosis may lead to chronic kidney disease (CKD) and end-stage renal disease in patients with dRTA.Citation89
Alkali in the form of sodium and/or potassium bicarbonate or citrate salts should be administered to maintain a normal serum bicarbonate concentration of >20 mEq/L in infants and >22 mEq/L in children and adults.Citation6,Citation89 However, excessive sodium bicarbonate will increase the extracellular volume and decrease the reabsorption of proximal tubular HCO3−, which increases the need for alkali.Citation6 In addition, because the increase in sodium intake after sodium citrate or sodium bicarbonate administration can result in increased urinary calcium excretion, potassium salts may be more effective than sodium salts for treatment of dRTA with kidney stones.Citation119,Citation120 As citrate salts can also correct hypocitraturia and prevent nephrolithiasis, potassium citrate is usually recommended.Citation16,Citation89
Young children with dRTA need higher doses of alkali because of the greater rate of acid production caused by the formation of hydroxyapatite associated with bone growth.Citation7 The amount of alkali needed usually decreases with age from as much as 5–8 mEq/kg/day in infants to 3–4 mEq/kg/day in children after the age of 6 years to 1–2 mEq/kg/day in adults.Citation6,Citation89,Citation92 A recent study showed that children with mutations in ATP6V1B1 or ATP6V0A4 generally needed higher doses of alkali compared to those with SLC4A1 mutations.Citation96 In patients with severe hypokalemia despite administration of potassium citrate, potassium supplementation may be required.Citation96
Dietary modifications to increase urinary citrate excretion may benefit dRTA patients with kidney stones and hypocitraturia. Dietary modifications include increased intake of fluid and citrus fruits, normal intake of calcium, and restricted intake of sodium, oxalate, animal proteins, and fructose.Citation118
Normalization of serum bicarbonate level, urinary calcium excretion, urinary low-molecular-weight protein levels (β2 microglobulin or α2 microglobulin), and growth rate are good markers for adherence to and/or adequacy of treatment.Citation6,Citation93,Citation96 In addition, abdominal ultrasonography should be regularly checked to detect nephrocalcinosis and nephrolithiasis.
Long-term outcome
The prognosis of dRTA is generally good in patients treated with early and sufficient doses of alkali, but alkali administration does not improve hearing impairment in patients with dRTA and SNHL.Citation16,Citation96 Moreover, recent studies showed an increased frequency of CKD in patients with dRTA during long-term follow-up. Besouw et al reported 9 of 24 (37.5%) children with dRTA developed CKD stage 2.Citation96 Palazzo et al also showed 16 of 51 (31.3%) patients with dRTA developed CKD during long-term follow-up and after puberty.Citation5 Although the precise pathogenic mechanisms of CKD in patients with dRTA are unknown, the combination of nephrocalcinosis and hypokalemia, which results in tubulointerstitial damage, or kidney damage after repeated episodes of dehydration and acute kidney injury have been suggested as potential causal factors.Citation5,Citation17
Disclosure
The author reports no conflicts of interest in this work.
References
- FryACKaretFEInherited renal acidosesPhysiology200722320221117557941
- Rodríguez SorianoJRenal tubular acidosis: the clinical entityJ Am Soc Nephrol20021382160217012138150
- MohebbiNWagnerCAPathophysiology, diagnosis and treatment of inherited distal renal tubular acidosisJ Nephrol201831451152228994037
- BesouwMTPBieniasMWalshPClinical and molecular aspects of distal renal tubular acidosis in childrenPediatr Nephrol201732698799628188436
- PalazzoVProvenzanoABecherucciFThe genetic and clinical spectrum of a large cohort of patients with distal renal tubular acidosisKidney Int20179151243125528233610
- Gil-PeñaHMejíaNSantosFRenal tubular acidosisJ Pediatr2014164469169824345454
- QuigleyRWolfMTRenal tubular acidosis in childrenAvnerEDHarmonWENiaudetPYoshikawaNEmmaFGoldsteinSLPediatric Nephrology7th edNew YorkSpringer-Verlag Berlin Heidelberg201612731306
- HammLLNakhoulNHering-SmithKSAcid–base homeostasisClin J Am Soc Nephrol201510122232224226597304
- TrepiccioneFProsperiFde La MotteLRNew findings on the pathogenesis of distal renal tubular acidosisKidney Dis20173398105
- CoreyHEEcksteinDRenal tubular acidosisRoncoCBellomoRKellumJARicciZCritical Care Nephrology3rd edPhiladelphiaElsevier2019405408
- GaoXEladariDLevielFDeletion of hensin/DMBT1 blocks conversion of beta- to alpha-intercalated cells and induces distal renal tubular acidosisProc Natl Acad Sci USA201010750218722187721098262
- Al-AwqatiQCell biology of the intercalated cell in the kidneyFEBS Lett2013587131911191423684635
- SchwartzGJGaoXTsuruokaSSDF1 induction by acidosis from principal cells regulates intercalated cell subtype distributionJ Clin Invest2015125124365437426517693
- WeinerIDVerlanderJWAmmonia transporters and their role in acid– base balancePhysiol Rev201797246549428151423
- WeinerIDMitchWESandsJMUrea and ammonia metabolism and the control of renal nitrogen excretionClin J Am Soc Nephrol20151081444145825078422
- BatlleDHaqueSKGenetic causes and mechanisms of distal renal tubular acidosisNephrol Dial Transplant201227103691370423114896
- KurtzIRenal tubular acidosis: H+/base and ammonia transport abnormalities and clinical syndromesAdv Chronic Kidney Dis201825433435030139460
- Blake-PalmerKGKaretFECellular physiology of the renal H+ATPaseCurr Opin Nephrol Hypertens200918543343819561496
- BretonSBrownDRegulation of luminal acidification by the V-ATPasePhysiology201328531832923997191
- WagnerCAFinbergKEBretonSMarshanskyVBrownDGeibelJPRenal vacuolar H+-ATPasePhysiol Rev20048441263131415383652
- KaretFEFinbergKENelsonRDMutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafnessNat Genet199921184909916796
- SmithANSkaugJChoateKAMutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearingNat Genet2000261717510973252
- Vargas-PoussouRHouillierPLe PottierNGenetic investigation of autosomal recessive distal renal tubular acidosis: evidence for early sensorineural hearing loss associated with mutations in the ATP6V0A4 geneJ Am Soc Nephrol20061751437144316611712
- StoverEHBorthwickKJBavaliaCNovel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing lossJ Med Genet2002391179680312414817
- MiuraKSekineTTakahashiKMutational analyses of the ATP6V1B1 and ATP6V0A4 genes in patients with primary distal renal tubular acidosisNephrol Dial Transplant20132882123213023729491
- MohebbiNVargas-PoussouRHegemannSCHomozygous and compound heterozygous mutations in the ATP6V1B1 gene in patients with renal tubular acidosis and sensorineural hearing lossClin Genet201383327427822509993
- YangQLiGSinghSKAlexanderEASchwartzJHVacuolar H+-ATPase B1 subunit mutations that cause inherited distal renal tubular acidosis affect proton pump assembly and trafficking in inner medullary collecting duct cellsJ Am Soc Nephrol20061771858186616769747
- FusterDGZhangJXieXSMoeOWThe vacuolar-ATPase B1 subunit in distal tubular acidosis: novel mutations and mechanisms for dysfunctionKidney Int200873101151115818368028
- SuYBlake-PalmerKGSorrellSHuman H+ATPase a4 subunit mutations causing renal tubular acidosis reveal a role for interaction with phosphofructokinase-1Am J Physiol Renal Physiol20082954F950F95818632794
- FinbergKEWagnerCABaileyMAThe B1-subunit of the H+ ATPase is required for maximal urinary acidificationProc Natl Acad Sci USA200510238136161362116174750
- NorgettEEGolderZJLorente-CánovasBInghamNSteelKPKaret FranklFEAtp6v0a4 knockout mouse is a model of distal renal tubular acidosis with hearing loss, with additional extrarenal phenotypeProc Natl Acad Sci USA201210934137751378022872862
- Lorente-CánovasBInghamNNorgettEEGolderZJKaret FranklFESteelKPMice deficient in H+-ATPase a4 subunit have severe hearing impairment associated with enlarged endolymphatic compartments within the inner earDis Model Mech20136243444223065636
- HenningsJCPicardNHuebnerAKA mouse model for distal renal tubular acidosis reveals a previously unrecognized role of the V-ATPase a4 subunit in the proximal tubuleEMBO Mol Med20124101057107122933323
- SchulzNDaveMHStehbergerPAChauTWagnerCADifferential localization of vacuolar H+-ATPases containing a1, a2, a3, or a4 (ATP6V0A1-4) subunit isoforms along the nephronCell Physiol Biochem2007201-410912017595521
- AlperSLMolecular physiology and genetics of Na+-independent SLC4 anion exchangersJ Exp Biol2009212Pt 111672168319448077
- RysaváRTesarVJirsaMBrabecVJarolímPIncomplete distal renal tubular acidosis coinherited with a mutation in the band 3 (AE1) geneNephrol Dial Transplant1997129186918739306337
- WrongOUnwinRFineLGCohenEThakkerRTannerMUnravelling of the molecular mechanisms of kidney stones. Report of a Meeting of Physicians and ScientistsLancet19963489041156115658950885
- BruceLJCopeDLJonesGKFamilial distal renal tubular acidosis is associated with mutations in the red cell anion exchanger (Band 3, AE1) geneJ Clin Invest19971007169317079312167
- KaretFEGainzaFJGyöryAZMutations in the chloride– bicarbonate exchanger gene AE1 cause autosomal dominant but not autosomal recessive distal renal tubular acidosisProc Natl Acad Sci USA19989511633763429600966
- JarolimPShayakulCPrabakaranDAutosomal dominant distal renal tubular acidosis is associated in three families with heterozygosity for the R589H mutation in the AE1 (band3) Cl−/HCO3−exchangerJ Biol Chem199827311638063889497368
- BruceLJWrongOToyeAMBand 3 mutations, renal tubular acidosis and South-East Asian ovalocytosis in Malaysia and Papua New Guinea: loss of up to 95% band 3 transport in red cellsBiochem J20003501415110926824
- CheiddeLVieiraTCLimaPRSaadSTHeilbergIPA novel mutation in the anion exchanger 1 gene is associated with familial distal renal tubular acidosis and nephrocalcinosisPediatrics20031126 Pt 11361136714654610
- RungrojNDevonaldMACuthbertAWA novel missense mutation in AE1 causing autosomal dominant distal renal tubular acidosis retains normal transport function but is mistargeted in polarized epithelial cellsJ Biol Chem200427914138331383814734552
- ZhangZLiuKXHeJWIdentification of two novel mutations in the SLC4A1 gene in two unrelated Chinese families with distal renal tubular acidosisArch Med Res201243429830422609520
- ShaoLXuYDongQLangYYueSMiaoZA novel SLC4A1 variant in an autosomal dominant distal renal tubular acidosis family with a severe phenotypeEndocrine201037347347820960171
- FryACSuYYiuVCuthbertAWTrachtmanHKaret FranklFEMutation conferring apical-targeting motif on AE1 exchanger causes autosomal dominant distal RTAJ Am Soc Nephrol20122371238124922518001
- QuiltyJALiJReithmeierRAImpaired trafficking of distal renal tubular acidosis mutants of the human kidney anion exchanger kAE1Am J Physiol Renal Physiol20022825F810F82011934690
- CordatEKittanakomSYenchitsomanusPTDominant and recessive distal renal tubular acidosis mutations of kidney anion exchanger 1 induce distinct trafficking defects in MDCK cellsTraffic20067211712816420521
- ToyeAMBantingGTannerMJRegions of human kidney anion exchanger 1 (kAE1) required for basolateral targeting of kAE1 in polarised kidney cells: mis-targeting explains dominant renal tubular acidosis (dRTA)J Cell Sci2004117Pt 81399141014996906
- QuiltyJACordatEReithmeierRAImpaired trafficking of human kidney anion exchanger (kAE1) caused by hetero-oligomer formation with a truncated mutant associated with distal renal tubular acidosisBiochem J2002368Pt 389590312227829
- DevonaldMASmithANPoonJPIhrkeGKaretFENon-polarized targeting of AE1 causes autosomal dominant distal renal tubular acidosisNat Genet200333212512712539048
- StehbergerPAShmuklerBEStuart-TilleyAKPetersLLAlperSLWagnerCADistal renal tubular acidosis in mice lacking the AE1 (band3) Cl–/HCO3– exchanger (slc4a1)J Am Soc Nephrol20071851408141817409310
- AkelAWagnerCAKovacikovaJEnhanced suicidal death of erythrocytes from gene-targeted mice lacking the Cl–/HCO3– exchanger AE1Am J Physiol Cell Physiol20072925C1759176717251326
- MumtazRTrepiccioneFHenningsJCIntercalated cell depletion and vacuolar H+-ATPase mistargeting in an Ae1 R607H knockin modelJ Am Soc Nephrol20172851507152027932475
- TanphaichitrVSSumboonnanondaAIdeguchiHNovel AE1 mutations in recessive distal renal tubular acidosis. Loss-of-function is rescued by glycophorin AJ Clin Invest199810212217321799854053
- YenchitsomanusPTVasuvattakulSKirdponSAutosomal recessive distal renal tubular acidosis caused by G701D mutation of anion exchanger 1 geneAm J Kidney Dis2002401212912087557
- JarolimPPalekJAmatoDDeletion in erythrocyte band 3 gene in malaria-resistant Southeast Asian ovalocytosisProc Natl Acad Sci USA1991882411022110261722314
- VasuvattakulSYenchitsomanusPTVachuanichsanongPAutosomal recessive distal renal tubular acidosis associated with Southeast Asian ovalocytosisKidney Int19995651674168210571775
- SritippayawanSSumboonnanondaAVasuvattakulSNovel compound heterozygous SLC4A1 mutations in Thai patients with autosomal recessive distal renal tubular acidosisAm J Kidney Dis2004441647015211439
- ChooKENicoliTKBruceLJTannerMJRuiz-LinaresAWrongOMRecessive distal renal tubular acidosis in Sarawak caused by AE1 mutationsPediatr Nephrol200621221221716252102
- AnacletoFEBruceLJClaytonPHegdeSResontocLPWrongODistal renal tubular acidosis in Filipino children, caused by mutations of the anion-exchanger SLC4A1 (AE1, Band 3) geneNephron Physiol201011421924
- LiuSCJarolimPRubinHLThe homozygous state for the band 3 protein mutation in Southeast Asian ovalocytosis may be lethalBlood19948410359035917949112
- PicardVProustAEveillardMHomozygous Southeast Asian ovalocytosis is a severe dyserythropoietic anemia associated with distal renal tubular acidosisBlood2014123121963196524652967
- JarolimPMurrayJLRubinHLCharacterization of 13 novel band 3 gene defects in hereditary spherocytosis with band 3 deficiencyBlood19968811436643748943874
- RibeiroMLAlloisioNAlmeidaHSevere hereditary spherocytosis and distal renal tubular acidosis associated with the total absence of band 3Blood20009641602160410942416
- ToyeAMWilliamsonRCKhanfarMBand 3 Courcouronnes (Ser667Phe): a trafficking mutant differentially rescued by wild-type band 3 and glycophorin ABlood2008111115380538918174378
- ChangYHShawCFJianSHHsiehKHChiouYHLuPJCompound mutations in human anion exchanger 1 are associated with complete distal renal tubular acidosis and hereditary spherocytosisKidney Int200976777478319625994
- ChuCWoodsNSawasdeeNBand 3 Edmonton I, a novel mutant of the anion exchanger 1 causing spherocytosis and distal renal tubular acidosisBiochem J2010426337938820028337
- SinhaRAgarwalIBawazirWMBruceLJDistal renal tubular acidosis with hereditary spherocytosisIndian Pediatr201350769369523942433
- KhositsethSSirikaneratAWongbenjaratKDistal renal tubular acidosis associated with anion exchanger 1 mutations in children in ThailandAm J Kidney Dis200749684185017533027
- ShmuklerBEKedarPSWarangPHemolytic anemia and distal renal tubular acidosis in two Indian patients homozygous for SLC4A1/AE1 mutation A858DAm J Hematol2010851082482820799361
- FawazNABeshlawiIOAl ZadjaliSdRTA and hemolytic anemia: first detailed description of SLC4A1 A858D mutation in homozygous stateEur J Haematol201288435035522126643
- KagerLBruceLJZeitlhoferPBand 3 nullVIENNA, a novel homozygous SLC4A1 p.Ser477X variant causing severe hemolytic anemia, dyserythropoiesis and complete distal renal tubular acidosisPediatr Blood Cancer2017643e26227
- KittanakomSCordatEAkkarapatumwongVYenchitsomanusPTReithmeierRATrafficking defects of a novel autosomal recessive distal renal tubular acidosis mutant (S773P) of the human kidney anion exchanger (kAE1)J Biol Chem200427939409604097115252044
- SimpsonAMSchwartzGJDistal renal tubular acidosis with severe hypokalaemia, probably caused by colonic H+-K+-ATPase deficiencyArch Dis Child200184650450711369570
- BoettgerTHübnerCAMaierHRustMBBeckFXJentschTJDeafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4Nature2002416688387487811976689
- SmithANBorthwickKJKaretFEMolecular cloning and characterization of novel tissue-specific isoforms of the human vacuolar H(+)-ATPase C, G and d subunits, and their evaluation in autosomal recessive distal renal tubular acidosisGene20022971-216917712384298
- BlomqvistSRVidarssonHFitzgeraldSDistal renal tubular acidosis in mice that lack the forkhead transcription factor Foxi1J Clin Invest2004113111560157015173882
- BiverSBelgeHBourgeoisSA role for Rhesus factor Rhcg in renal ammonium excretion and male fertilityNature2008456722033934319020613
- BourgeoisSBounoureLChristensenEIHaploinsufficiency of the ammonia transporter Rhcg predisposes to chronic acidosis: Rhcg is critical for apical and basolateral ammonia transport in the mouse collecting ductJ Biol Chem201328885518552923281477
- XuJSongPNakamuraSDeletion of the chloride transporter slc26a7 causes distal renal tubular acidosis and impairs gastric acid secretionJ Biol Chem200928443294702947919723628
- SunXYangLVTiegsBCDeletion of the pH sensor GPR4 decreases renal acid excretionJ Am Soc Nephrol201021101745175520798260
- BourgeoisSMeerLVWootlaBNHE4 is critical for the renal handling of ammonia in rodentsJ Clin Invest201012061895190420484819
- WenDYuanYCorneliusRJDeficient acid handling with distal RTA in the NBCe2 knockout mouseAm J Physiol Renal Physiol20153096F523F53026109087
- TrepiccioneFGerberSDGrahammerFRenal Atp6ap2/(Pro) renin receptor is required for normal vacuolar H+-ATPase function but not for the renin–angiotensin systemJ Am Soc Nephrol201627113320333027044666
- MerkulovaMPăunescuTGNairAVTargeted deletion of the Ncoa7 gene results in incomplete distal renal tubular acidosis in miceAm J Physiol Renal Physiol20183151F173F18529384414
- EnerbäckSNilssonDEdwardsNAcidosis and deafness in patients with recessive mutations in FOXI1J Am Soc Nephrol20182931041104829242249
- RungrojNNettuwakulCSawasdeeNDistal renal tubular acidosis caused by tryptophan–aspartate repeat domain 72 (WDR72) mutationsClin Genet201894540941830028003
- VallésPGBatlleDHypokalemic distal renal tubular acidosisAdv Chronic Kidney Dis201825430332030139458
- KittererDSchwabMAlscherMDBraunNLatusJDrug-induced acid–base disordersPediatr Nephrol20153091407142325370778
- SimónHOriveBZamoraIMendizabalSThe acidification defect in the syndrome of renal tubular acidosis with nerve deafnessActa Paediatr Scand1979682291295419999
- Rodriguez-SorianoJValloACastilloGOliverosRNatural history of primary distal renal tubular acidosis treated since infancyJ Pediatr198210156696767131138
- IgarashiTKawatoHKamoshitaSReversible low-molecular-weight proteinuria in patients with distal renal tubular acidosisPediatr Nephrol1990465935961708269
- WatanabeTProximal renal tubular dysfunction in primary distal renal tubular acidosisPediatr Nephrol2005201868815549407
- TasicVKornetiPGucevZHoppeBBlauNCheongHIAtypical presentation of distal renal tubular acidosis in two siblingsPediatr Nephrol20082371177118118386070
- BesouwMTPBieniasMWalshPClinical and molecular aspects of distal renal tubular acidosis in childrenPediatr Nephrol201732698799628188436
- WatanabeTRenal Fanconi syndrome in distal renal tubular acidosisPediatr Nephrol2017326109328293727
- SatohNSuzukiMNakamuraMFunctional coupling of V-ATPase and CLC-5World J Nephrol201761142028101447
- PicolloAMalvezziMAccardiAProton block of the CLC-5 Cl–/H+ exchangerJ Gen Physiol2010135665365920513761
- MillerSGSchwartzGJHyperammonaemia with distal renal tubular acidosisArch Dis Child19977754414449487970
- ClericettiCMMilaniGPLavaSAGBianchettiMGSimonettiGDGianniniOHyperammonemia associated with distal renal tubular acidosis or urinary tract infection: a systematic reviewPediatr Nephrol201833348549129134448
- GökceoğluAUTas¸arMAYalakiZGünes¸ABakırAAn infant with hypercalcemia and hyperammonia: inborn error of metabolism or not? QuestionsPediatr Nephrol2018
- WrongODaviesHEThe excretion of acid in renal diseaseQ J Med19592811025931313658353
- FusterDGMoeOWIncomplete distal renal tubular acidosis and kidney stonesAdv Chronic Kidney Dis201825436637430139463
- OstherPJBollerslevJHansenABEngelKKildebergPPathophysiology of incomplete renal tubular acidosis in recurrent renal stone formers: evidence of disturbed calcium, bone and citrate metabolismUrol Res19932131691738342250
- ArampatzisSRöpke-RiebenBLippunerKHessBPrevalence and densitometric characteristics of incomplete distal renal tubular acidosis in men with recurrent calcium nephrolithiasisUrol Res2012401535921713545
- SluysmansTVanoverscheldeJPMalvauxPGrowth failure associated with medullary sponge kidney, due to incomplete renal tubular acidosis type 1Eur J Pediatr1987146178803556185
- OduwoleAOGiwaOSArogundadeRARelationship between rickets and incomplete distal renal tubular acidosis in childrenItal J Pediatr2010365420699008
- WegerMDeutschmannHWegerWKotankoPSkrabalFIncomplete renal tubular acidosis in “primary” osteoporosisOsteoporos Int199910432532910692983
- WegerWKotankoPWegerMDeutschmannHSkrabalFPrevalence and characterization of renal tubular acidosis in patients with osteopenia and osteoporosis and in non-porotic controlsNephrol Dial Transplant200015797598010862634
- SromickiJJHessBAbnormal distal renal tubular acidification in patients with low bone mass: prevalence and impact of alkali treatmentUrolithiasis201745326326927412028
- PongchaiyakulCDomrongkitchaipornSStitchantrakulWChailurkitLORajatanavinRIncomplete renal tubular acidosis and bone mineral density: a population survey in an area of endemic renal tubular acidosisNephrol Dial Transplant200419123029303315479744
- ZhangJFusterDGCameronMAIncomplete distal renal tubular acidosis from a heterozygous mutation of the V-ATPase B1 subunitAm J Physiol Renal Physiol20143079F1063F107125164082
- ImaiEKanekoSMoriTOkadoTUchidaSTsukamotoYA novel heterozygous mutation in the ATP6V0A4 gene encoding the V-ATPase a4 subunit in an adult patient with incomplete distal renal tubular acidosisClin Kidney J20169342442827274828
- DhayatNASchallerAAlbanoGThe vacuolar H+-ATPase B1 subunit polymorphism p.E161K associates with impaired urinary acidification in recurrent stone formersJ Am Soc Nephrol20162751544155426453614
- BourgeoisSBettoniCBaronSWagnerCAHaploinsufficiency of the mouse Atp6v1b1 gene leads to a mild acid–base disturbance with implications for kidney stone diseaseCell Physiol Biochem20184731095110729843146
- AlexanderRTCordatEChambreyRDimkeHEladariDAcidosis and urinary calcium excretion: insights from genetic disordersJ Am Soc Nephrol201627123511352027468975
- ZuckermanJMAssimosDGHypocitraturia: pathophysiology and medical managementRev Urol200911313414419918339
- SakhaeeKNicarMHillKPakCYContrasting effects of potassium citrate and sodium citrate therapies on urinary chemistries and crystallization of stone-forming saltsKidney Int19832433483526645208
- LemannJGrayRWPleussJAPotassium bicarbonate, but not sodium bicarbonate, reduces urinary calcium excretion and improves calcium balance in healthy menKidney Int19893526886952540373