Abstract
Background
Obstructive sleep apnea (OSA) occurs in 4% of middle-aged men and 2% of middle-aged women in the general population, and the prevalence is even higher in specific patient groups. OSA is an independent risk factor for a variety of cardiovascular diseases. Endothelial injury could be the pivotal determinant in the development of cardiovascular pathology in OSA. Endothelial damage ultimately represents a dynamic balance between the magnitude of injury and the capacity for repair. Bone marrow–derived endothelial progenitor cells (EPCs) within adult peripheral blood present a possible means of vascular maintenance that could home to sites of injury and restore endothelial integrity and normal function.
Methods
We summarized pathogenetic mechanisms of OSA and searched for available studies on numbers and functions of EPCs in patients with OSA to explore the potential links between the numbers and functions of EPCs and OSA. In particular, we tried to elucidate the molecular mechanisms of the effects of OSA on EPCs.
Conclusion
Intermittent hypoxia cycles and sleep fragmentation are major pathophysiologic characters of OSA. Intermittent hypoxia acts as a trigger of oxidative stress, systemic inflammation, and sympathetic activation. Sleep fragmentation is associated with a burst of sympathetic activation and systemic inflammation. In most studies, a reduction in circulating EPCs has emerged. The possible mechanisms underlying the decrease in the number or function of EPCs include prolonged inflammation response, oxidative stress, increased sympathetic activation, physiological adaptive responses of tissue to hypoxia, reduced EPC mobilization, EPC apoptosis, and functional impairment in untreated OSA. Continuous positive airway pressure (CPAP) therapy for OSA affects the mobilization, apoptosis, and function of EPCs through preventing intermittent hypoxia episodes, improving sleep quality, and reducing systemic inflammation, oxidative stress levels, and sympathetic overactivation. To improve CPAP adherence, the medical staff should pay attention to making the titration trial a comfortable first CPAP experience for the patients; for example, using the most appropriate ventilators or proper humidification. It is also important to give the patients education and support about CPAP use in the follow-up, especially in the early stage of the treatment.
Introduction
Obstructive sleep apnea (OSA) is a common condition characterized by repeated episodes of upper airway obstruction that result in interruptions of breathing during sleep, recurring episodes of hypoxemia, sleep fragmentation, and daytime sleepiness. OSA affects 3%–7% of adult men, 2%–5% of adult women,Citation1–Citation3 and up to 4% of children.Citation3,Citation4 At all ages, even in children, it is associated with complications in different organ systems, such as cardiovascular morbidity, hypertension, obesity, dyslipidemia, and insulin resistance.Citation5–Citation8 Moreover, both in children and adults, OSA causes behavioral and neuropsychological deficits in the central nervous system, including daytime sleepiness, depression,Citation9 impaired memory,Citation10 mood disorders, cognition deficiencies,Citation11 and even nocturnal enuresis.Citation12 Cognition deficiencies in patients with OSA have typically been found in attention and vigilance, memory and learning, executive functions, and simulated driving, in which endothelial dysfunctions could be the most intriguing explanation.Citation4,Citation13 There is evidence showing that sleep parameters can rapidly be normalized with continuous positive airway pressure (CPAP) treatment, but those deficits in cognitive performance often persist.Citation4,Citation13 OSA is also an independent risk factor for a variety of cardiovascular diseases such as atherosclerosis, hypertension, and coronary heart disease.Citation14,Citation15 The maintenance of an intact vascular endothelium is critical for preservation of the integrity of the vascular system. Endothelial injury could be the pivotal determinant in the development of cardiovascular pathology in OSA.Citation16–Citation22 One of the major pathophysiologic mechanisms of vascular injury is the endothelial damage from intermittent hypoxia (IH) with OSA pattern. Endothelial damage ultimately represents a dynamic balance between the magnitude of injury and the capacity for repair. The balance between the damage and repair ultimately determines the progression of cardiovascular diseases.
Vascular endothelium has a finite lifespan. Endothelial cells are shed into the circulation in both healthy and disease states, and a mechanism must exist by which these cells can be replaced.Citation23 It conventionally has been thought that this was exclusively accomplished by the proliferation and migration of resident mature endothelial cells adjacent to regions of injury.Citation24 However, the discovery of bone marrow–derived endothelial progenitor cells (EPCs) within adult peripheral blood presented another possible means of vascular maintenance; namely, a reservoir of circulating cells that could home to sites of injury and restore endothelial integrity and normal function. In 1997, Asahara et al described for the first time a population of putative EPCs in human peripheral blood.Citation25 In this study, selected circulating CD34-positive cells in human peripheral blood moved into the foci of vascular injury and differentiated into vascular endothelial cells. Further studies from this group showed that a specific population of bone marrow cells, now identified as EPCs, is recruited to the foci of vascular injury and neovascular formation, and these cells differentiate into vascular endothelial cells in both physiologic and pathologic neovascular formations.Citation26 Since then, accumulating evidence has indicated that EPCs support the integrity of the vascular endothelium and take part in repair processes throughout the cardiovascular system.Citation25,Citation27,Citation28 EPCs contribute to endothelial repair and neovascularization not only by physically integrating into the endothelial layer but also by excretion of paracrine factors that can stimulate the proliferation of resident endothelial cells,Citation29 which is of paramount importance in neovascularization.Citation30–Citation33
Logically, then, if the circulating progenitor pool represents an important source of endothelial cells for “repair,” a reduction in the number of progenitors might be expected to have a negative effect on endothelial function. There are several studies that have been carried out on EPCs in OSA, but the results currently available on the role of EPCs in OSA are controversial. EPCs have been reported as increased, decreased, or unchanged in OSA. In most of these studies, however, a reduction in circulating EPCs has emerged. Up until now, the mechanisms underlying the decrease in number or function of EPCs in patients with OSA have not been fully elucidated. In this review, we describe our current understanding of the effects of OSA on the number and function of EPCs and focus on the molecular mechanisms. Clarification and reinforcement of the repair mechanisms of EPCs may ameliorate endothelial damages and reduce OSA-related morbidities. In addition, pathophysiological insight will be provided for improvement of the repairing process after endothelial damage in patients with OSA.
EPCs
EPCs are premature circulating cells that are mainly derived from bone marrow and are endowed with the capacity both to be mobilized from bone marrow into the bloodstream in response to growth factors and cytokine release and to differentiate into mature endothelial cells and be involved in postnatal vasculogenesis and reendothelialization after endothelial damage.
In 1997, Asahara and colleagues demonstrated for the first time that some special purified CD34-positive hematopoietic progenitor cells from peripheral blood could differentiate, ex vivo, into an endothelial phenotype; the cells were then named EPCs.Citation25 Since then, different markers have been used to describe in vivo circulating EPCs. EPCs are positive for CD34 or the more immature marker protein CD133. Recent studies have shown that expression of the CD34 surface antigen is shared by EPCs, hematopoietic progenitor cells, and mature endothelial cells.Citation34 As they mature, EPCs lose the CD133 marker and acquire vascular endothelial growth factor (VEGF) receptor 2 (VEGFR2), also known as kinase insert domain-containing receptor (KDR).Citation35 The combination of CD34+ VEGFR2+, CD34+ CD133+, CD133+ VEGFR2+, and CD34+ CD133+ VEGFR2+ has been used by different investigators to describe in vivo circulating EPCs.Citation36 Numerous investigators have found that double-positive CD34+ VEGFR2+ cells can behave as EPCs. However, because some mature endothelial cells also coexpress CD34 and VEGFR2, better markers may be needed. The stem cell marker CD133 may be a more precise marker for defining subpopulations of cells that represent EPCs. Unlike the progenitor marker CD34, CD133 is not expressed on mature endothelial cells. A study group has shown that double-positive CD34+ CD133+ EPCs have high proliferative capacity and give rise to endothelial colonies in culture.Citation37 CD133+ VEGFR2+ dual-positive cells have been found colonizing the luminal surfaces of left ventricular assist devices explanted from humans, suggesting these cells may play a role in endothelial repair. An intriguing hypothesis is that triple CD133+ CD34+ VEGFR2+ cells represent more primitive EPCs with high proliferative potential, which then turn into CD133− CD34+ VEGFR2+ EPCs with a more limited proliferative capacity.
Recent data have suggested that at least two EPC sub-populations can be grown from peripheral blood mononuclear cells; namely, early EPCs, which display paracrine actions, and late-outgrowth EPCs, which are characterized by high proliferative potential, promoting angiogenesis in different ways.Citation33 Early EPCs contribute to angiogenesis in a paracrine fashion but fail to form vascular networks and to incorporate in endothelial-like structures in a newly developed angiogenesis assay. Late-outgrowth EPCs contribute to angiogenesis by directly incorporating into newly formed vascular networks but fail to stimulate angiogenesis in a paracrine fashion.Citation33
Bone marrow–derived EPC studies in patients with OSA
The studies on EPCs in OSA have been carried out in both adult and child patients. The number and functional activity of circulating EPCs are affected not only by different cardiovascular risk factors such as hypertension, obesity, hypercholesterolemia, diabetes, and smoking, but also by different physiological conditions, such as age, sex, cigarette use, and physical inactivity.Citation38–Citation40 Almost all currently available studies on EPCs in OSA recruited participants who were free of any other known cardiovascular risk factors. In these studies, patients with OSA and healthy control patients are matched for age, sex, and body mass index (BMI), and in addition, patients and control subject participants were similar in blood pressure, fasting blood glucose, and total cholesterol levels.Citation28–Citation41
The cumulative results currently available on the role of EPCs in OSA are controversial. Data from five recent studies reported a decrease in EPCs in patients with OSA. The study by de la Peña et alCitation41 reported that the percentage of EPCs (CD34+ VEGFR2+ cells) was significantly lower in patients with OSA who were free of any other known cardiovascular risk factor than in healthy control patients matched for age and sex. Endothelial function was not different between patients with OSA and control patients. No significant correlation between circulating EPCs and apnea-hypopnea index (AHI) was found in patients with OSA. Similarly, in 2008, Jelic et alCitation42 reported that baseline EPC (CD34+ CD133+ cells) levels were lower in patients with OSA than in control patients. CPAP therapy increased EPC levels to those of control participants when patients adhered to CPAP for more than 4 hours daily. EPC levels remained unchanged when patients used CPAP for less than 4 hours daily or declined CPAP. Another study by Jelic et alCitation43 in 2009 shared similar results. The authors reported that before treatment, EPC (CD34+ CD133+ VEGFR2+ cells) levels, a marker of endothelial repair capacity, were lower and endothelial microparticle levels (EMPs), a marker of endothelial apoptosis, were greater in patients with OSA than in control patients. Levels of EPCs and EMPs were inversely related. After effective treatment (CPAP >4 hours daily), EPC levels were similar in patients with OSA and control patients. Levels of EMP and EPC were unchanged in patients who declined CPAP and in a single patient who used CPAP for less than 4 hours daily. The authors concluded that OSA alone impairs endothelial repair capacity and promotes endothelial apoptosis.
Murri et alCitation44 found that EPCs (CD34+ CD133+ VEGFR2+ cells) were lower in the patients with OSA than in the control patients. There was a significant negative correlation between EPC levels and the severity of OSA, and the EPC levels correlated negatively with the levels of oxidative stress markers, but positively with markers of protection against oxidation. After 1 month of CPAP treatment, EPC levels increased and oxidative stress variables decreased. In a study in children, performed by Kheirandish-Gozal et al,Citation45 80 children with OSA and 20 control patients matched for BMI, age, sex, and ethnicity were recruited. Despite similar OSA severity, EPC (CD34+ CD133+ VEGFR2+ cells) counts were significantly lower among the 20 children with OSA with endothelial dysfunction when compared with either the 20 children without endothelial dysfunction or the control patients. Furthermore, EPC levels were significantly and inversely correlated with the magnitude of endothelial dysfunction, but neither EPCs nor the magnitude of endothelial dysfunction were associated with AHI. In contrast with these findings, Kizawa et alCitation46 reported that individuals with OSA had a threefold increase in EPCs (CD34+ CD133+ cells) in their blood circulation compared with the control group. After CPAP treatment, this increase was suppressed. Martin et alCitation18 found there were no significant differences in circulating EPCs (CD34+ CD133+ cells) between patients with OSA free of any other known cardiovascular risk factor and healthy control patients matched for age and BMI, respectively. Similarly, Yun et alCitation47 also found that EPC levels did not differ between patients with OSA and non-OSA patients. CPAP compliance did not affect EPC levels. The levels of EMPs in patients with OSA were significantly higher than those in the non-OSA group.
There are several possible reasons for this discrepancy. First, investigators in different studies assessed circulating EPCs, using different methods. Some studiesCitation18,Citation41–Citation48 assessed circulating EPCs using flow cytometry alone. One studyCitation47 assessed circulating EPCs by the assay of endothelial colony-forming units. It is possible that the assay of endothelial colony-forming unitsCitation48 might be more specific, and therefore more likely to detect differences between patients with OSA and control patients. Second, the EPCs studied by one group are not necessarily the same cell type as those of another. One of the major limitations in studying EPCs is the lack of unifying phenotypic markers that are employed by different investigators. Thus, different investigators employ different marker combinations for the assessment of EPCs: CD34+ VEGFR2+,Citation41 CD34+ 133+,Citation42,Citation44,Citation46 or CD34+ CD133+ VEGFR2+.Citation43–Citation45 Third, the recruited participants are different. For example, Kizawa et alCitation46 recruited only male subjects and excluded the possibility of the cyclical mobilization of EPCs, whereas other studies recruited both female and male participants; several reports have demonstrated that the influence of the menstrual cycle affects the number and function of EPCs.Citation49,Citation50 Fourth, these studies were performed on only a relatively small number of participants. Furthermore, the numbers of circulating EPCs that can be identified from peripheral blood samples are small.Citation51 Thus, because of the relatively rare event analysis, the sample sizes in these studies may have been too small to detect differences. Fifth, because hypoxia is critical to the changes of circulating EPCs, it is possible that participants with more profound nocturnal desaturation, such as those with lower resting lung volumesCitation52 or longer apnea duration,Citation53 might demonstrate altered numbers of circulating EPCs. Even though some studiesCitation41 reported no significant correlation between circulating EPCs, and AHI was found in patients with OSA, this might be explained by the narrow range of disease severity of the patients studied here (all of whom had severe OSA). Finally, these studies were done at the time of diagnosis of OSA. No one knows how long OSA had existed in those participants. It is possible there is a threshold duration of OSA that is necessary for the changes in circulating EPCs to be observed.
Pathogenetic mechanisms of OSA and the effects of OSA on EPCs ()
OSA is a common condition characterized by repeated episodes of upper airway obstruction that result in interruptions of breathing during sleep, recurring episodes of hypoxemia, sleep fragmentation, and excessive daytime sleepiness. These episodes induce cyclical alterations of arterial oxygen saturation and desaturation, which is referred to as hypoxia/reoxygenation or IH. The IH cycle is the major pathophysiologic character of OSA. IH acts as a trigger of oxidative stress, systemic inflammation, and sympathetic activation. Sleep fragmentation is associated with a burst of sympathetic activationCitation54 and increased levels of inflammatory markers such as C-reactive protein (CRP),Citation55 interleukin 6 (IL-6), and tumor necrosis factor-alpha (TNF-α).Citation56 Inflammatory responses induced by IH could activate oxidative stress in OSA. In turn, increased oxidative stress will lead to activation of nuclear factor (NF)-κB, and hence increased expression of a number of downstream NF-κB target genes; for example, proinflammatory cytokines, such as TNF-α, IL-6, and IL-8, as well as adhesion molecules such as intercellular adhesion molecule 1.Citation57
Figure 1 Potential molecular mechanisms through which obstructive sleep apnea has effects on endothelial progenitor cells.
Abbreviations: NF-κB, nuclear factor κB; ASK-1, apoptosis signal regulating kinase 1; JNK, Jun N-terminal kinase; Bax/Bcl-2, the ratio of Bax protein to Bcl-2 protein; EPC, endothelial progenitor cell; ROS, reactive oxygen species; eNOS, endothelial nitric oxide synthase; NO, nitric oxide; Ang II, angiotensin II; (COX)-2, cyclooxygenase-2; PGE2, prostaglandin E2; MMP9, matrix metalloproteinase -9; sKitL, soluble Kit ligand; HIF-1, hypoxia inducible factor-1; VEGF, Vascular endothelial growth factor; EPO, erythropoietin; SDF-1, stroma-derived factor-1.
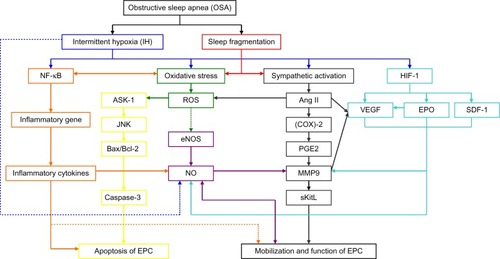
Effects of systemic inflammation on EPCs in OSA
Systemic inflammation could be caused directly by IH or caused indirectly by both oxidative stress and sympathetic activation in OSA. Increased reactive oxygen species (ROS) productionCitation57 may cause increased expression of inflammatory cytokines through activation of NF-κB, and hence increased expression of a number of downstream NF-κB target genes. In addition, increased sympathetic activity can cause an increase in inflammatory cytokines by increasing free fatty acid levels in the absence of ROS.Citation58,Citation59 Most studies show that patients with OSA have higher levels of circulating inflammatory markers, such as CRP and/or TNF-α and/or IL-6 and/or IL-8,Citation60–Citation66 with a significant fall after effective CPAP therapy.Citation60,Citation67,Citation68 Patients with OSA have lower levels of circulating anti-inflammatory cytokines such as IL−10. IL-10 correlates negatively with the severity of OSA and can inhibit the production of many pro-inflammatory cytokines such as IL-6.Citation66
Previous reports have suggested that prolonged inflammation response has been implicated with reduced EPCs mobilization, cell apoptosis, and functional impairment.Citation69,Citation70 Inflammatory response may affect EPCs mobilization. Increasing evidence indicates that EPC mobilization is closely correlated with variations in the levels of some inflammatory factors.Citation71 High levels of TNF-α contribute to a reduction in EPC number.Citation72 A positive association between CRP levels and circulating EPCs has been documented in patients with stable coronary artery disease, suggesting that a systemic inflammatory state stimulates EPC mobilization in these patients.Citation71 In a study, circulating EPCs were diminished in IL-10 genetically deficient mice as compared with wild-type mice, which suggests that IL-10 plays a crucial role in the mobilization of EPCs.Citation73 Inflammatory response may be implicated with EPC apoptosis, and a clinical study demonstrated that CRP is associated with apoptosis of EPCs in vitro.Citation74 In addition, EPCs that are mobilized in response to inflammatory stimulation may be functionally impaired.Citation69 CRP exerts direct inhibitory effects on EPC differentiation and survival, whereas EPCs exposed to CRP exhibit decreased angiogenic activity.Citation75
Increasing evidence indicates that a transient, restricted, or low-grade inflammation induces EPC mobilization, whereas prolonged or excessive or high-grade inflammatory stimuli, such as observed in OSA, has the opposite effect.Citation70,Citation76 Although the mechanisms regulating this effect are still unclear, the possible mechanisms may be that prolonged exposure of bone marrow to increased proinflammatory stimulation may lead to exhaustion of the EPC pool. The association between inflammation and EPCs is largely circumstantial and observational.Citation76 Further clinical studies are required to elucidate the exact mechanisms by which inflammation affects EPC mobilization and functional activity.
Effects of oxidative stress on EPCs in OSA
Oxidative stress is a known feature of OSACitation77 and is thought to be mainly caused by cyclical hypoxia/reoxygenation. Oxidative stress in OSA can also occur via activated inflammatory responses induced by hypoxia and by an increased sympathetic activity. IH and sleep fragmentationCitation54 lead to increased sympathetic activity, which stimulates the renin-angiotensin-aldosterone system (RAAS), resulting in elevated angiotensin II (Ang II), which is known to increase oxidative stress.
Oxidative stress may play a crucial role in EPC mobilization and functional bioactivity.Citation26 Increased superoxide generation reduces EPC levels and impairs EPC function.Citation78 In a rat model of myocardial infarction, increased production of ROS, which is the major oxidative stress marker, is associated with reduced EPC levels.Citation79 Some clinical studies have shown that conditions associated with increased oxidative stress have been associated with decreased EPC numbers in the peripheral circulation.Citation80 Murri et alCitation44 found that the levels of oxidative stress markers correlated negatively with levels of EPCs, whereas markers of protection against oxidation correlated positively with the levels of EPCs. After 1 month of CPAP treatment, oxidative stress variables decreased and EPC levels increased. In another study, incubation of EPCs with high levels of hydrogen peroxide (H2O2) induces apoptosis, profoundly reducing the numbers of EPCs.Citation82 There is increasing evidence that oxidative stress reduces and impairs EPC functioning.Citation76 Thum et alCitation78 found that increased production of ROS was associated with reduced EPC levels and impaired EPCs function. Conditions associated with increased oxidative stress lead to the mobilization of functionally defective EPCs, which have a lesser capability to mobilize, migrate, and incorporate into existing vasculature.Citation83 Therefore, it is clear that conditions associated with increased oxidative stress not only decrease the absolute numbers of circulating EPCs but also impair EPC function, with deleterious effects on vascular homeostasis.Citation76 It is still unclear whether a direct association exists between ROS and functional bioactivity of EPCs.
Effects of IH/oxidative stress on EPCs by NO unavailability
IH in OSA reduces endothelial NO production directly. L-arginine is the substrate for NO production by endothelial nitric oxide synthase (eNOS). Biosynthesis of NO from L-arginine is an oxygen-dependent process, and hypoxia might influence NO formation in vascular beds directly.Citation84 Hypoxia also can increase arginase II activity in endothelial cells, which degrades L-arginine. The plasma levels of L-arginine increase after a single night of CPAP therapy in patients with OSA.Citation85 Meanwhile, increasing oxidative stress caused by repetitive episodes of hypoxia/reoxygenation reduces endothelial NO production at the transcriptional and posttranscriptional levels indirectly.Citation86 Increased oxidative stress reduces and destabilizes eNOS messenger RNA (mRNA), in part via the Rho kinase pathway, in human venous and pulmonary artery endothelial cellsCitation82 and reduces endothelial NO production at the transcriptional level.Citation87 Prolonged oxidative stress such as that observed in untreated OSA reduces eNOS enzymatic activity by suppressing eNOS phosphorylation.Citation88 Tetrahydro-biopterin is a cofactor critical for NO production by eNOS.Citation89 Increased oxidative stress limits the availability of cofactors required for NO production. When this cofactor is depleted in conditions of increased oxidative stress, eNOS, a main source of basal endothelial NO production, preferentially promotes superoxide production, which hastens NO degradation and thereby reduces NO availability.Citation90
Mobilization of EPC from the bone marrow entails adequate NO production. Impaired recruitment of EPCs from the bone marrow is likely to be related to depressed NO production and activity in patients with OSA.Citation91 eNOS is essential for mobilization of EPCs.Citation91 Mice deficient in eNOS (Nos3−/−) show reduced VEGF-induced EPC mobilization. Interestingly, mice deficient in eNOS (Nos3−/−) also have reduced basal expression and activity of matrix metalloproteinase 9 (MMP-9). MMP-9 is a major target for NO, which activates MMP-9 by S-nitrosylation.Citation92 MMP-9 is required for stem cell mobilization.Citation93 VEGF-induced mobilization of EPCs in mice is not observed in mice deficient in MMP-9. In addition, mobilization of EPCs in response to VEGF administration is significantly inhibited by coadministration of a synthetic MMP inhibitor.Citation94 Thus, MMP-9 activation is a decisive checkpoint for recruitment of EPCs.Citation94 MMP-9 appears to be essential for the regulation of EPCs in response to various stimuli, but it must be activated by NO. MMP-9 degrades the extracellular matrix and transforms membrane-bounded Kit ligand (KitL, also known as stem cell factor) to soluble Kit ligand, triggering subsequent movement of cKit-positive stem cells, including EPCs, to the circulation.Citation94–Citation96
In addition to VEGF, oxidative stress also upregulates transcription of other angiogenic factors, such as stroma-derived factor-1 (SDF-1) or erythropoietin (EPO), in EPCs.Citation97
Effects of increased sympathetic activation on EPCs in OSA
IHCitation98 and sleep fragmentationCitation54 lead to increased sympathetic activity, which stimulates RAAS axis, resulting in elevated Ang II and aldosterone in OSA. Untreated OSA is associated with an upregulation of the RAAS. Ang II derived from leukocytes, especially peripheral blood mononuclear cells, is significantly increased in patients with OSA compared with control patients.Citation99 There is ample evidence that Ang II is involved in endothelial damage and atherogenesis via multiple mechanisms.Citation100,Citation101 Some of the harmful consequences of Ang II can be mediated by impairment of EPCs. A thorough analysis of Ang II-induced effects in EPCs regulation and function, and especially involved molecular mechanisms, has not been undertaken.
Effects of Ang II on EPCs by excessive generation of ROS
Accumulated evidence has shown that Ang II is implicated in a wide variety of pathologies of cardiovascular diseases.Citation102,Citation103 Prominent evidence among those featuring pathologies mediated by Ang II is the excessive generation of ROS.Citation26,Citation104 Ang II was shown to be a potent stimulus for ROS generation.
On one hand, as mentioned earlier, because ROS is thought to play an important role in the decrease in NO bioavailability, accumulation of ROS, especially resulting from the RAAS, leads to inhibition of the mobilization of EPCs from bone marrow. On the other hand, accumulation of ROS, especially resulting from the RAAS, also affects senescence and/or apoptosis of EPCs. Ang II was shown to induce the senescence of EPCs.Citation105 Endtmann et alCitation106 demonstrated that Ang II, through the angiotensin 1 receptor, induces oxidative stress (or ROS) and then activates the redox-sensitive apoptosis signal regulating kinase 1 (ASK-1)–dependent proapoptotic signaling pathways in early-outgrowth EPCs. Ang II enhances phosphorylation of ASK-1, activates c-Jun N-terminal kinase and p38-mitogen-activated protein kinase, and then decreases expression of antiapoptotic Bcl-2 and increases expression of proapoptotic Bax, leading to activation of caspase 3 and apoptosis of EPCs. ASK-1 is a member of the mitogen-activated protein kinase family, which activates both c-Jun N-terminal kinase and p38-mitogen-activated protein kinase pathways.Citation107 p38-mitogen-activated protein kinase inhibition in vitro and in vivo improves the number and functional capacities of bone marrow–derived EPCs, which is associated with reduced atherosclerosis in atherosclerotic mice.Citation108 ASK-1 constitutes a pivotal signaling pathway in stress-induced apoptosis, especially in the context of oxidative stress.Citation109 Ang II-induced activation of ASK-1 and caspase 3, resulting in apoptosis, is mediated through the induction of oxidative stress, because both effects are inhibited by coincubation with an antioxidant.Citation106
Effects of Ang II on EPCs by VEGF
A prominent physiological adaptive response of tissue to hypoxia, such as the IH condition under OSA, is angiogenesis, the formation of new blood vessels and increasing the blood supply.Citation110 VEGF promotes hypoxia-induced angiogenesis in vitro and in vivo, which has been shown to be upregulated by Ang II.Citation111 On one hand, Ang II induces expression of VEGF. Numerous reports have shown that VEGF expression is significantly increased in the plasma of patients with OSACitation99,Citation112 and is induced by Ang II in peripheral blood EPCs.Citation99 On the other hand, Ang II stimulates VEGFR2 mRNA and protein expression in human EPCs, resulting in enhanced VEGF-induced proliferation of EPCs and vascular network formation in a Matrigel assay.Citation113
Effects of Ang II on EPCs by MMP-9
Ang II may induce the expression of the inflammatory cyclooxygenase 2 gene and influence the extracellular matrix turnover by regulating the activity of prostaglan-din E2-dependent metalloproteinase in vascular cells.Citation114 Recently, Tazaki et al demonstrated that serum MMP-9 is increased in patients with OSA when compared with normal participants.Citation115 They speculated that elevated serum MMP-9 might induce vascular events in patients with OSA. MMP-9 is essential for homing and differentiation of EPCs on endothelial sites where they are required.Citation116 VEGF-induced mobilization of EPCs in mice is significantly inhibited by coadministration of a synthetic metalloproteinase inhibitor. Mobilization of EPCs in response to VEGF administration was not observed in mice deficient in MMP-9. NO appears to be essential for regulation of EPCs in response to various stimuli, but this process depends on the activation of MMP-9. Thus, MMP-9 activation is a decisive checkpoint for the recruitment of EPCs.Citation94
In addition, EPCs have been shown to express the Ang II type 1 receptor, suggesting that direct effects of Ang II on EPCs are possible.Citation117
Effects of HIF-1 signaling axis on EPCs
In patients with OSA, oxygen saturation may repeatedly decrease during the apneic events. A prominent physiological adaptive response of tissue to hypoxia is neovascularization and increasing the blood supply.Citation110 Under hypoxic conditions, transcription factors such as hypoxia inducible factor 1 (HIF-1) are activated, leading to increased transcription of proangiogenic proteins including VEGF, SDF-1, and EPO,Citation118 which mobilize EPCs, and finally, contributes to neovascularization.Citation119
At the mRNA level, the HIF-1 gene is constitutively expressed and not significantly upregulated by hypoxia. At the transcriptional level, however, hypoxia markedly increases the levels of HIF-1 protein.Citation120 Genes encoding vascular VEGF, SDF-1, and EPO are all under the control of HIF-1.Citation121 There is an HIF-1 binding site in the SDF-1 gene, in the promoter of the VEGF gene, and in the enhancer of the EPO gene. All of these genes are induced by IH, both in vivo and in vitro.Citation122
Effects of VEGF on EPCs
Numerous reports have shown that plasma levels of VEGF are elevated in patients with OSA.Citation41,Citation99,Citation111,Citation123 VEGF expression is markedly increased in patients with OSA, largely because of the effects of HIF-1 on VEGF transcription.Citation124 Apart from HIF-1 stimulation, VEGF expression is stimulated by Ang II in peripheral blood mononuclear cells.Citation99,Citation111 Furthermore, a study indicated that the vascular endogenous EPO/EPO receptor system also plays an important role for upregulation of the VEGF/VEGF receptor system.
The role of VEGF in EPC mobilization has been widely studied in both mice and humans, showing that, after acute ischemic injury, plasma levels of VEGF increase rapidly, leading to a 50-fold increase in EPC percentage in peripheral blood.Citation28 In animal models, exogenous administration of VEGF promotes mobilization of EPCs into the peripheral circulation.Citation125 Treatment with VEGF was reported to double the number of circulating EPCs in humans.Citation126 Gene transfer of VEGF into ischemic tissue increases circulating EPCs to levels more than two times higher than the baseline level.
However, a study reported an increase in plasma VEGF levels and a reduction in circulating EPCs in patients with OSA without any known cardiovascular risk factors compared with healthy participants of a similar age and BMI.Citation41 Plasma levels of VEGF are elevated in patients with OSA, and VEGF could promote mobilization of EPCs into the peripheral circulation, but levels of circulating EPCs in patients with OSA are reduced. Possible explanations include that VEGF activation may constitute an adaptive mechanism to the repetition of nocturnal hypoxic events, which may potentially contribute to counterbalancing the occurrence of OSA-related cardiovascular disease. Increased VEGF concentrations in OSA may reflect a physiological effort to mobilize EPCs in these patients and can represent an early event in the natural history of the disease.Citation41
Both experimental and clinical studies have demonstrated that VEGF significantly affects the kinetics of EPCs.Citation125,Citation127 VEGF stimulates VEGFR1 and VEGFR2 present on EPCs and activates MMP-9, which is essential for the homing and differentiation of EPCs.Citation128 VEGF has been shown to strongly induce Akt phosphorylation in endothelial cells. Akt is a serine threonine protein kinase that is activated by a number of growth factors and cytokines in a phosphatidylinositol 3 kinase (PI3 K)-dependent manner. Importantly, the PI3 K/Akt pathway plays a significant role in mediating VEGF biological activity. Dimmeler et al have shown that VEGF induces EPC differentiation via the PI3 K/Akt pathway.Citation129
Effects of SDF-1 on EPCs
Several factors have been shown to influence EPC mobilization and homing to hypoxic tissue, including chemok-ines,Citation35 angiogenic cytokines, and pharmacologic agents. SDF-1 is one such chemokine that is considered to play an important role in EPC homing and recruitment for hypoxic neovascularization.Citation130 SDF-1 is a chemokine of the cysteine-X-cysteine (CXC) family that binds to the chemokine receptor, cysteine-X-cysteine chemokine receptor (CXCR) 4, on target cells, which is produced within the bone marrow. The SDF-1/CXCR4 interaction is another important pathway in the mobilization of EPCs from the bone marrow. Studies have indicated that SDF-1 and its CXCR4 play a critical role in progenitor cell homing, mobilization, and differentiation.Citation131,Citation132 SDF-1 is capable of enhancing the recruitment and mobilization of EPCs to damaged endothelium during postnatal vasculogenesis.Citation133 Overexpression of SDF-1 in ischemic tissues has been found to enhance EPC recruitment from peripheral blood and to induce neoangiogenesis in ischemic tissues.Citation134 The number of circulating EPCs can be increased by SDF-1 gene transfer, using the adenovirus infection technique.Citation94,Citation124 Recent evidence also suggests that SDF-1 is a driving force for EPCs differentiation.Citation132
One study reported that plasma levels of SDF-1 are positively associated with EPC number and function in response to acute ischemic events, suggesting a role of SDF-1 in EPC mobilization and differentiation in humans.Citation135 However, data from another study showed that SDF-1 levels are inversely, rather than positively, associated with circulating EPC numbers.Citation136 One of the possible explanations for these controversial results is that the relationship of SDF-1 with EPC homing, mobilization, and differentiation in the acute phase is different from that in a normal situation. Studies of mouse ischemia models showed that the number of EPCs in peripheral blood was lower but the level of SDF-1 was much higher at 14 days after ischemia compared with control mice,Citation132 suggesting that EPCs are mobilized into peripheral blood from bone marrow after the onset of ischemia, but at a later stage, the numbers of mobilized EPCs in peripheral blood decrease because of their homing to the ischemic site.
Effects of EPO on EPCs
Plasma EPO increases exponentially with the degree of hypoxia in humans. Imagawa demonstrated that serum levels in patients with OSA were approximately 2-fold higher than those in normal patients.Citation137
Of note, EPO serum levels correlate with the number and function of EPCs isolated from both bone marrow and peripheral blood, suggesting that EPO may regulate EPCs in vivo. Administration of exogenous EPO induces mobilization and proliferation of EPCs. In contrast, both VEGF concentrations and recruitment of EPCs ischemic muscle are significantly enhanced in wild-type mice but are significantly impaired in mice that lack the EPO receptor system. These results further suggest that EPO may be important for VEGF secretion, EPC mobilization, and angiogenesis in vivo.Citation138 The authors indicate that the vascular endogenous EPO/EPO receptor system also plays an important role in angiogenesis in response to hind-limb ischemia through upregulation of VEGF/VEGF receptor system by recruiting EPCs.Citation138
Effects of continuous CPAP therapy on EPCs
CPAP continues to be the standard, primary, and first-line therapy for patients with OSA.Citation139 CPAP consists of an air pressure source that keeps a constant positive pressure in the airway through the respiratory cycle. The airflow is delivered through a nasal or oronasal interface and maintains patency of the upper airway. CPAP has been found to be highly efficacious, reducing OSA symptoms, including daytime sleepiness, sympathetic neural activation, and blood pressure, improves cognitive function and quality of life,Citation140–Citation142 and promoting mobilization of EPCs.Citation42,Citation43 However, there is a distinction between efficacy and effectiveness. Efficacy is the effect in the laboratory or under ideal circumstances, regardless of treatment adherence. CPAP demonstrates very good efficacy. Effectiveness is the effect in daily life, which depends on patient compliance with CPAP therapy.Citation143
CPAP adherence is defined as more than 4 hours per night of “mask-on” time, based on data that suggest that more than 4–5 hours of CPAP usage per night results in improvement in Epworth Sleepiness Scale scores.Citation144,Citation145 Adequate adherence to CPAP is essential for achievement of the benefits of CPAP treatment. Observational studies have reported that patients with OSA who refused or did not adhere to CPAP therapy experienced higher rates of myocardial infarction, stroke, and death compared with CPAP adherents.Citation146–Citation149 In another study, CPAP produced a modest reduction in blood pressure in patients with hypertension and OSA, but continued use of CPAP for 5.3 hours per day or longer could cause significant reductions in blood pressure for patients with incompletely controlled hypertension. Stepnowsky and Dimsdale demonstrated that higher rates of compliance (ie, >4 hours of usage per night) resulted in an improvement in the respiratory disturbance index, oxygen desaturation index, and arousal index.Citation150 Studies by Jelic et alCitation42,Citation43 reported that baseline EPC levels were lower in patients with OSA than in control patients. CPAP therapy increased EPC levels to those of control patients when patients adhered to CPAP for more than 4 hours daily. EPC levels remained unchanged when patients used CPAP for less than 4 hours daily or declined CPAP altogether. Although the mechanisms of how CPAP affects mobilization of EPCs are still unclear, the possible mechanisms may be through preventing hypoxia/reoxygenation or IH episodes, improving sleep quality, reducing oxidative stress levels, systemic inflammation,Citation151–Citation154 and excessive sympathetic activation,Citation155 which have been implicated for impaired EPCs with reduced mobilization, increased cell apoptosis, and damaged EPC function.
In fact, although CPAP provides effective treatment for OSA, patient adherence remains challenging. CPAP adherence was low. In one study, when individuals without follow-up were assumed to be nonadherent, the overall adherence rate was only 30.4%.Citation156 Another two retrospective surveys showed that the percentages of patients with good CPAP adherence were 56.8%Citation157 and 54.3%,Citation158 respectively. As reported in studies, male sex,Citation159 higher levels of education,Citation160 smoking,Citation161 nocturia,Citation161 benign prostatic hypertrophy,Citation161 and depressive symptomatologyCitation159 were predictors of poor CPAP adherence, and increasing age,Citation162–Citation164 higher incomes, higher AHI values,Citation159 and initial educational program were predictors of good CPAP adherence. Somers et alCitation159 found that increased length of time from the initial visit to receiving the CPAP machine was associated with poorer compliance. Therefore, efforts should be made to try to minimize the length of time between the initial visit and receiving CPAP treatment to improve compliance. Wang et alCitation165 found that only half of the patients having initial CPAP titration trial remained adherent to the CPAP treatment, and the other half of the patients either never initiated the CPAP treatment or had abandoned CPAP treatment. Wolkove et alCitation166 similarly found that 31% of patients do not commence treatment after polysomnography (PSG) diagnosis and CPAP trial. To improve CPAP adherence, the medical staff should pay attention to making the titration trial a comfortable first CPAP experience for the patients, such as using the most appropriate ventilators or proper humidification. It is also important to give the patients education and support about CPAP use in the follow-up, especially in the early stages of the treatment.
Conclusion
We conclude that IH cycle and sleep fragmentation are major pathophysiologic characters of OSA. IH acts as a trigger of oxidative stress, systemic inflammation, and sympathetic activation. Sleep fragmentation is associated with a burst of sympathetic activation and systemic inflammation. EPCs have been reported as decreased, increased, or unchanged. However, in most studies, a reduction in circulating EPCs has emerged and EPC functions are damaged. The possible mechanisms underlying the decrease in number or function of EPCs are that prolonged inflammation response, oxidative stress, increased sympathetic activation, and physiological adaptive response of tissue to hypoxia are implicated with reduced EPC mobilization, increased cell apoptosis, and functional impairment in untreated OSA. CPAP therapy for OSA affects EPCs through preventing IH episodes, improving sleep quality, reducing systemic inflammation, oxidative stress levels, and excess sympathetic activation.
Acknowledgments
This study was supported by the grants from the National Natural Science Foundation of China (No 81270144, 30800507, 81170071).
Disclosure
The authors report no conflicts of interest in this work
References
- FengJChenBYPrevalence and incidence of hypertension in obstructive sleep apnea patients and the relationship between obstructive sleep apnea and its confoundersChin Med J (Engl)2009122121464146819567173
- YoungTPeppardPEGottliebDJEpidemiology of obstructive sleep apnea: a population health perspectiveAm J Respir Crit Care Med200216591217123911991871
- LumengJCChervinRDEpidemiology of pediatric obstructive sleep apneaProc Am Thorac Soc 15200852242252
- FengJWuQZhangDChenBYHippocampal impairments are associated with intermittent hypoxia of obstructive sleep apneaChin Med J (Engl)2012125469670122490498
- LavieLOxidative stress – a unifying paradigm in obstructive sleep apnea and comorbiditiesProg Cardiovasc Dis200951430331219110132
- LévyPBonsignoreMREckelJSleep, sleep-disordered breathing and metabolic consequencesEur Respir J200934124326019567607
- SateiaMJNeuropsychological impairment and quality of life in obstructive sleep apneaClin Chest Med200324224925912800782
- CarotenutoMSantoroNGrandoneAThe insulin gene variable number of tandemrepeats (INS VNTR) genotype and sleep disordered breathing in childhood obesityJ Endocrinol Invest200932975275519574727
- CarotenutoMBruniOSantoroNDel GiudiceEMPerroneLPascottoAWaist circumference predicts the occurrence of sleep-disordered breathing in obese children and adolescents: a questionnaire-based studySleep Med20067435736116713341
- CarotenutoMEspositoMParisiLDepressive symptoms and childhood sleep apnea syndromeNeuropsychiatr Dis Treat201236937322977304
- DaulatzaiMADeath by a thousand cuts in Alzheimer’s disease: hypoxia – the prodromeNeurotox Res201324221624323400634
- CarotenutoMEspositoMPascottoAFacial patterns and primary nocturnal enuresis in childrenSleep Breath201115222122720607423
- StaatsRStollPZinglerDVirchowJCLommatzschMRegulation of brain-derived neurotrophic factor (BDNF) during sleep apnoea treatmentThorax200560868869216061712
- NietoFJYoungTBLindBKAssociation of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health StudyJAMA2000283141829183610770144
- MarinJMCarrizoSJVicenteEAgustiAGLong-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational studyLancet200536594641046105315781100
- AmraBKarbasiEHashemiMHoffmann-CastendiekBGolshanMEndothelial dysfunction in patients with obstructive sleep apnoea independent of metabolic syndromeAnn Acad Med Singapore200938546146419521652
- BayramNACiftciBKelesTEndothelial function in normotensive men with obstructive sleep apnea before and 6 months after CPAP treatmentSleep200932101257126319848355
- MartinKStanchinaMKouttabNHarringtonEORoundsSCirculating endothelial cells and endothelial progenitor cells in obstructive sleep apneaLung2008186314515018401642
- TrzepizurWGagnadouxFAbrahamPMicrovascular endothelial function in obstructive sleep apnea: Impact of continuous positive airway pressure and mandibular advancementSleep Med200910774675219147401
- DragerLFBortolottoLAFigueiredoACSilvaBCKriegerEMLorenzi-FilhoGObstructive sleep apnea, hypertension, and their interaction on arterial stiffness and heart remodelingChest200713151379138617494787
- Yim-YehSRahangdaleSNguyenATVascular dysfunction in obstructive sleep apnea and type 2 diabetes mellitusObesity (Silver Spring)2011191172220523303
- DragerLFBortolottoLAFigueiredoACKriegerEMLorenziGFEffects of continuous positive airway pressure on early signs of atherosclerosis in obstructive sleep apneaAm J Respir Crit Care Med2007176770671217556718
- DimmelerSZeiherAMVascular repair by circulating endothelial progenitor cells: the missing link in atherosclerosis?J Mol Med (Berl)2004821067167715322703
- CarmelietPMoonsLStassenJMVascular wound healing and neointima formation induced by perivascular electric injury in miceAm J Pathol199715027617769033288
- AsaharaTMuroharaTSullivanAIsolation of putative progenitor endothelial cells for angiogenesisScience199727553029649679020076
- YaoEHYuYFukudaNOxidative stress on progenitor and stem cells in cardiovascular diseasesCurr Pharm Biotechnol20067210110816724944
- OrlicDKajsturaJChimentiSBodineDMLeriAAnversaPTransplanted adult bone marrow cells repair myocardial infarcts in miceAnn N Y Acad Sci2001938221229 discussion 229–23011458511
- GillMDiasSHattoriKVascular trauma induces rapid but transient mobilization of VEGFR2(+)AC133(+) endothelial precursor cellsCirc Res200188216717411157668
- HungHSShyuWCTsaiCHHsuSHLinSZTransplantation of endothelial progenitor cells as therapeutics for cardiovascular diseasesCell Transplant20018910031012
- SahooSKlychkoEThorneTExosomes from human CD34(+) stem cells mediate their proangiogenic paracrine activityCirc Res2011109772472821835908
- ScheubelRJHoltzJFriedrichIParacrine effects of CD34 progenitor cells on angiogenic endothelial sproutingInt J Cardiol2010139213414119008002
- MorishitaTUzuiHNakanoANumber of endothelial progenitor cells in peripheral artery disease as a marker of severity and association with pentraxin-3, malondialdehyde-modified low-density lipoprotein and membrane type-1 matrix metalloproteinaseJ Atheroscler Thromb201219214915822123215
- SievekingDPBuckleACelermajerDSNgMKStrikingly different angiogenic properties of endothelial progenitor cell subpopulations: insights from a novel human angiogenesis assayJ Am Coll Cardiol200851666066818261686
- ZammarettiPZischAHAdult ‘endothelial progenitor cells’. Renewing vasculatureInt J Biochem Cell Biol200537349350315618004
- UrbichCDimmelerSEndothelial progenitor cells: characterization and role in vascular biologyCirc Res200495434335315321944
- FadiniGPBaessoIAlbieroMSartoreSAgostiniCAvogaroATechnical notes on endothelial progenitor cells: ways to escape from the knowledge plateauAtherosclerosis2008197249650318249408
- QuiriciNSoligoDCanevaLServidaFBossolascoPDeliliersGLDifferentiation and expansion of endothelial cells from human bone marrow CD133(+) cellsBr J Haematol2001115118619411722432
- MasudaHKalkaCTakahashiTEstrogen-mediated endothelial progenitor cell biology and kinetics for physiological postnatal vasculogenesisCirc Res2007101659860617656679
- FadiniGPde KreutzenbergSVCoracinaACirculating CD34+ cells, metabolic syndrome, and cardiovascular riskEur Heart J200627182247225516912055
- KondoTHayashiMTakeshitaKSmoking cessation rapidly increases circulating progenitor cells in peripheral blood in chronic smokersArterioscler Thromb Vasc Biol20042481442144715191940
- de la PeñaMBarcelóABarbeFEndothelial function and circulating endothelial progenitor cells in patients with sleep apnea syndromeRespiration2008761283217921670
- JelicSPadelettiMKawutSMInflammation, oxidative stress, and repair capacity of the vascular endothelium in obstructive sleep apneaCirculation2008117172270227818413499
- JelicSLedererDJAdamsTEndothelial repair capacity and apoptosis are inversely related in obstructive sleep apneaVasc Health Risk Manag2009590992019997572
- MurriMGarcía-DelgadoRAlcázar-RamírezJEffect of CPAP on oxidative stress and circulating progenitor cell levels in sleep patients with apnea-hypopnea syndromeRespir Care201156111830183621605479
- Kheirandish-GozalLBhattacharjeeRKimJClairHBGozalDEndothelial progenitor cells and vascular dysfunction in children with obstructive sleep apneaAm J Respir Crit Care Med20101821929720203242
- KizawaTNakamuraYTakahashiSSakuraiSYamauchiKInoueHPathogenic role of angiotensin II and oxidised LDL in obstructive sleep apnoeaEur Respir J20093461390139819574336
- YunCHJungKHChuKIncreased circulating endothelial microparticles and carotid atherosclerosis in obstructive sleep apneaJ Clin Neurol201062899820607048
- HillJMZalosGHalcoxJPCirculating endothelial progenitor cells, vascular function, and cardiovascular riskN Engl J Med2003348759360012584367
- FadiniGPde KreutzenbergSAlbieroMGender differences in endothelial progenitor cells and cardiovascular risk profile: the role of female estrogensArterioscler Thromb Vasc Biol2008285997100418276910
- RobbAOMillsNLSmithIBInfluence of menstrual cycle on circulating endothelial progenitor cellsHum Reprod200924361962519088108
- RafiiSCirculating endothelial precursors: mystery, reality, and promiseJ Clin Invest20001051171910619857
- StanchinaMLMalhotraAFogelRBThe influence of lung volume on pharyngeal mechanics, collapsibility, and genioglossus muscle activation during sleepSleep200326785185614655919
- WhiteDPPathogenesis of obstructive and central sleep apneaAm J Respir Crit Care Med2005172111363137016100008
- BlasiAJoJAValladaresEJuarezRBaydurAKhooMCAutonomic cardiovascular control following transient arousal from sleep: a time-varying closed-loop modelIEEE Trans Biomed Eng2006531748216402605
- Meier-EwertHKRidkerPMRifaiNEffect of sleep loss on C-reactive protein, an inflammatory marker of cardiovascular riskJ Am Coll Cardiol200443467868314975482
- IrwinMRWangMCampomayorCOCollado-HidalgoAColeSSleep deprivation and activation of morning levels of cellular and genomic markers of inflammationArch Intern Med2006166161756176216983055
- LavieLObstructive sleep apnoea syndrome – an oxidative stress disorderSleep Med Rev200371355112586529
- HückingKHamilton-WesslerMEllmererMBergmanRNBurst-like control of lipolysis by the sympathetic nervous system in vivoJ Clin Invest2003111225726412531882
- NguyenMTSatohHFavelyukisSJNK and tumor necrosis factor-alpha mediate free fatty acid-induced insulin resistance in 3T3-L1 adipocytesJ Biol Chem200528042353613537116085647
- RyanSTaylorCTMcNicholasWTPredictors of elevated nuclear factor-kappaB-dependent genes in obstructive sleep apnea syndromeAm J Respir Crit Care Med2006174782483016840748
- KobayashiKNishimuraYShimadaTEffect of continuous positive airway pressure on soluble CD40 ligand in patients with obstructive sleep apnea syndromeChest2006129363263716537861
- TaumanRO’BrienLMGozalDHypoxemia and obesity modulate plasma C-reactive protein and interleukin-6 levels in sleep-disordered breathingSleep Breath2007112778417171553
- GozalDCrabtreeVMSans CapdevilaOWitcherLAKheirandish-GozalLC-reactive protein, obstructive sleep apnea, and cognitive dysfunction in school-aged childrenAm J Respir Crit Care Med2007176218819317400731
- KhalyfaACapdevilaOSBuazzaMOSerperoLDKheirandish-GozalLGozalDGenome-wide gene expression profiling in children with non-obese obstructive sleep apneaSleep Med2009101758618261956
- LiAMChanMHYinJC-reactive protein in children with obstructive sleep apnea and the effects of treatmentPediatr Pulmonol2008431344018041751
- SahlmanJMiettinenKPeuhkurinenKKuopio Sleep Apnoea GroupThe activation of the inflammatory cytokines in overweight patients with mild obstructive sleep apnoeaJ Sleep Res201019234134820040038
- YamauchiMTamakiSTomodaKEvidence for activation of nuclear factor kappaB in obstructive sleep apneaSleep Breath200610418919317013605
- MinoguchiKYokoeTTanakaAAssociation between lipid per-oxidation and inflammation in obstructive sleep apnoeaEur Respir J200628237838516880368
- WernerNNickenigGInfluence of cardiovascular risk factors on endothelial progenitor cells: limitations for therapy?Arterioscler Thromb Vasc Biol200626225726616322535
- AndreouITousoulisDTentolourisCAntoniadesCStefanadisCPotential role of endothelial progenitor cells in the pathophysiology of heart failure: clinical implications and perspectivesAtherosclerosis2006189224725416860805
- GeorgeJGoldsteinEAbashidzeSCirculating endothelial progenitor cells in patients with unstable angina: association with systemic inflammationEur Heart J200425121003100815191769
- ChenYHLinSJLinFYHigh glucose impairs early and late endothelial progenitor cells by modifying nitric oxide-related but not oxidative stress-mediated mechanismsDiabetes20075661559156817389326
- KrishnamurthyPThalMVermaSInterleukin-10 deficiency impairs bone marrow-derived endothelial progenitor cell survival and function in ischemic myocardiumCirc Res2011109111280128921959218
- SugawaraJMitsui-SaitoMHayashiCDecrease and senescence of endothelial progenitor cells in patients with preeclampsiaJ Clin Endocrinol Metab20059095329533215956081
- SuhWKimKLChoiJHC-reactive protein impairs angiogenic functions and decreases the secretion of arteriogenic chemo-cytokines in human endothelial progenitor cellsBiochem Biophys Res Commun20043211657115358216
- TousoulisDAndreouIAntoniadesCTentolourisCStefanadisCRole of inflammation and oxidative stress in endothelial progenitor cell function and mobilization: therapeutic implications for cardiovascular diseasesAtherosclerosis2008201223624718599065
- SchulzRMahmoudiSHattarKEnhanced release of superoxide from polymorphonuclear neutrophils in obstructive sleep apnea. Impact of continuous positive airway pressure therapyAm J Respir Crit Care Med20001622 Pt 156657010934088
- ThumTFraccarolloDSchultheissMEndothelial nitric oxide synthase uncoupling impairs endothelial progenitor cell mobilization and function in diabetesDiabetes200756366667417327434
- ThumTFraccarolloDGaluppoPBone marrow molecular alterations after myocardial infarction: Impact on endothelial progenitor cellsCardiovasc Res2006701506016480696
- WatsonTGoonPKLipGYEndothelial progenitor cells, endothelial dysfunction, inflammation, and oxidative stress in hypertensionAntioxid Redox Signal20081061079108818315493
- HungYCSavaVMBlagodarskyVAHongMYHuangGSProtection of tea melanin on hydrazine-induced liver injuryLife Sci20037291061107112495784
- UrbichCKnauAFichtlschererSFOXO-dependent expression of the proapoptotic protein Bim: pivotal role for apoptosis signaling in endothelial progenitor cellsFASEB J200519897497615824087
- TepperOMGalianoRDCaplaJMHuman endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structuresCirculation2002106222781278612451003
- LeemanMde BeylVZBiarentDMaggioriniMMélotCNaeijeRInhibition of cyclooxygenase and nitric oxide synthase in hypoxic vasoconstriction and oleic acid-induced lung injuryAm J Respir Crit Care Med19991595 Pt 11383139010228099
- LavieLHefetzALuboshitzkyRLaviePPlasma levels of nitric oxide and L-arginine in sleep apnea patients: effects of nCPAP treatmentJ Mol Neurosci2002115763
- LiaoJKZuluetaJJYuFSPengHBCoteCGHassounPMRegulation of bovine endothelial constitutive nitric oxide synthase by oxygenJ Clin Invest1995966266126668675632
- AntoniadesCShirodariaCWarrickN5-methyltetrahydrofolate rapidly improves endothelial function and decreases superoxide production in human vessels: effects on vascular tetrahydrobiopterin availability and endothelial nitric oxide synthase couplingCirculation2006114111193120116940192
- TanakaTNakamuraHYodoiJBloomETRedox regulation of the signaling pathways leading to eNOS phosphorylationFree Radic Biol Med20053891231124215808421
- ChannonKMTetrahydrobiopterin: regulator of endothelial nitric oxide synthase in vascular diseaseTrends Cardiovasc Med200414832332715596110
- LaursenJBSomersMKurzSEndothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterinCirculation200110391282128811238274
- AicherAHeeschenCMildner-RihmCEssential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cellsNat Med20039111370137614556003
- GuZKaulMYanBS-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell deathScience200229755841186119012183632
- DimmelerSZeiherAMReactive oxygen species and vascular cell apoptosis in response to angiotensin II and pro-atherosclerotic factorsRegul Pept2000901–3192510828488
- HeissigBHattoriKDiasSRecruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligandCell2002109562563712062105
- UmemuraTHigashiYEndothelial progenitor cells: therapeutic target for cardiovascular diseasesJ Pharmacol Sci200810811618776710
- LiuZJVelazquezOCHyperoxia, endothelial progenitor cell mobilization, and diabetic wound healingAntioxid Redox Signal200810111869188218627349
- JanicBArbabASThe role and therapeutic potential of endothelial progenitor cells in tumor neovascularizationScientificWorldJournal2010101088109920563532
- LesskeJFletcherECBaoGUngerTHypertension caused by chronic intermittent hypoxia – influence of chemoreceptors and sympathetic nervous systemJ Hypertens19971512 Pt 2159316039488210
- TakahashiSNakamuraYNishijimaTSakuraiSInoueHEssential roles of angiotensin II in vascular endothelial growth factor expression in sleep apnea syndromeRespir Med20059991125113116085213
- WassmannSNickenigGPathophysiological regulation of the AT1-receptor and implications for vascular diseaseJ Hypertens Suppl2006241S15S2116601568
- DaughertyARateriDLLuHInagamiTCassisLAHypercholesterolemia stimulates angiotensin peptide synthesis and contributes to atherosclerosis through the AT1A receptorCirculation2004110253849385715596561
- SteckelingsUMRompeFKaschinaEUngerTThe evolving story of the RAAS in hypertension, diabetes and CV disease: moving from macrovascular to microvascular targetsFundam Clin Pharmacol200923669370319817870
- VijanSGAngiotensin-converting enzyme inhibitors (ACEIs), not angiotensin receptor blockers (ARBs), are preferred and effective mode of therapy in high cardiovascular risk patientsJ Indian Med Assoc2009107317818219810392
- CubbonRMKahnMBWheatcroftSBEffects of insulin resistance on endothelial progenitor cells and vascular repairClin Sci (Lond)2009117517319019630751
- YinTMaXZhaoLChengKWangHAngiotensin II promotes NO production, inhibits apoptosis and enhances adhesion potential of bone marrow-derived endothelial progenitor cellsCell Res200818779279918560380
- EndtmannCEbrahimianTCzechT2011Angiotensin II impairs endothelial progenitor cell number and function in vitro and in vivo: implications for vascular regenerationHypertension58339440321825227
- TobiumeKMatsuzawaATakahashiTASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosisEMBO Rep20012322222811266364
- SeegerFHSeddingDLangheinrichACHaendelerJZeiherAMDimmelerSInhibition of the p38 MAP kinase in vivo improves number and functional activity of vasculogenic cells and reduces atherosclerotic disease progressionBasic Res Cardiol2010105338939719911112
- LiuHZhangHIlesKEThe ADP-stimulated NADPH oxidase activates the ASK-1/MKK4/JNK pathway in alveolar macrophagesFree Radic Res200640886587417015265
- AdairTHGayWJMontaniJPGrowth regulation of the vascular system: evidence for a metabolic hypothesisAm J Physiol19902593 Pt 2R393R4041697737
- TamaratRSilvestreJSKubisNEndothelial nitric oxide synthase lies downstream from angiotensin II-induced angiogenesis in ischemic hindlimbHypertension200239383083511897773
- KählerCMWechselbergerJMolnarCPriorCSerum levels of vascular endothelial growth factor are elevated in patients with obstructive sleep apnea and severe night time hypoxiaAm J Respir Crit Care Med200316719293 author reply 9312502484
- ImanishiTHanoTNishioIAngiotensin II potentiates vascular endothelial growth factor-induced proliferation and network formation of endothelial progenitor cellsHypertens Res200427210110815005273
- BrillaCGZhouGRuppHMaischBWeberKTRole of angiotensin II and prostaglandin E2 in regulating cardiac fibroblast collagen turnoverAm J Cardiol199576138D13D
- TazakiTMinoguchiKYokoeTIncreased levels and activity of matrix metalloproteinase-9 in obstructive sleep apnea syndromeAm J Respir Crit Care Med2004170121354135915361365
- AsaharaTTakahashiTMasudaHVEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cellsEMBO J199918143964397210406801
- HaznedarogluICOztürkMATowards the understanding of the local hematopoietic bone marrow renin-angiotensin systemInt J Biochem Cell Biol200335686788012676173
- DuRLuKVPetritschCHIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasionCancer Cell200813320622018328425
- AsaharaTMasudaHTakahashiTBone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularizationCirc Res199985322122810436164
- HuangLEAranyZLivingstonDMBunnHFActivation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunitJ Biol Chem19962715032253322598943284
- SemenzaGLWangGLA nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activationMol Cell Biol19921212544754541448077
- BunnHFPoytonROOxygen sensing and molecular adaptation to hypoxiaPhysiol Rev19967638398858757790
- CherniackEPVascular endothelial growth factor and sleep apnea: clutching at straws in the nightRespiration200741171817191001
- ShweikiDItinASofferDKeshetEVascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesisNature199235963988438451279431
- MooreMAHattoriKHeissigBMobilization of endothelial and hematopoietic stem and progenitor cells by adenovector-mediated elevation of serum levels of SDF-1, VEGF, and angiopoietin-1Ann N Y Acad Sci20019383645 discussion 45–4711458524
- KalkaCMasudaHTakahashiTVascular endothelial growth factor(165) gene transfer augments circulating endothelial progenitor cells in human subjectsCirc Res200086121198120210864908
- KhakooAYFinkelTEndothelial progenitor cellsAnnu Rev Med2005567910115660503
- DéryMAMichaudMDRichardDEHypoxia-inducible factor 1: regulation by hypoxic and non-hypoxic activatorsInt J Biochem Cell Biol200537353554015618010
- DimmelerSAicherAVasaMHMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathwayJ Clin Invest20018108339139711489932
- UrbichCAicherAHeeschenCSoluble factors released by endothelial progenitor cells promote migration of endothelial cells and cardiac resident progenitor cellsJ Mol Cell Cardiol200539573374216199052
- MöhleRBautzFRafiiSMooreMABruggerWKanzLThe chemokine receptor CXCR-4 is expressed on CD34+ hematopoietic progenitors and leukemic cells and mediates transendothelial migration induced by stromal cell-derived factor-1Blood19989112452342309616148
- De FalcoEPorcelliDTorellaARSDF-1 involvement in endothelial phenotype and ischemia-induced recruitment of bone marrow progenitor cellsBlood2004104123472348215284120
- GargRTellezAAlviarCGranadaJKleimanNSLevEIThe effect of percutaneous coronary intervention on inflammatory response and endothelial progenitor cell recruitmentCatheter Cardiovasc Interv200872220520918651648
- HiasaKIshibashiMOhtaniKGene transfer of stromal cell-derived factor-1alpha enhances ischemic vasculogenesis and angiogenesis via vascular endothelial growth factor/endothelial nitric oxide synthase-related pathway: next-generation chemokine therapy for therapeutic neovascularizationCirculation2004109202454246115148275
- XiaoQKiechlSPatelSEndothelial progenitor cells, cardiovascular risk factors, cytokine levels and atherosclerosis – results from a large population-based studyPLoS One2007210e97517925881
- XiaoQYeSOberhollenzerFSDF1 gene variation is associated with circulating SDF1alpha level and endothelial progenitor cell number: the Bruneck StudyPLoS One200312e4061
- ImagawaSYamaguchiYHiguchiMLevels of vascular endothelial growth factor are elevated in patients with obstructive sleep apnea--hypopnea syndromeBlood2001Aug 159841255125711493479
- NakanoMSatohKFukumotoYImportant role of erythropoietin receptor to promote VEGF expression and angiogenesis in peripheral ischemia in miceCirc Res2007100566266917293480
- BasnerRCContinuous positive airway pressure for obstructive sleep apneaN Engl J Med2007356171751175817460229
- BallesterEBadiaJRHernándezLEvidence of the effectiveness of continuous positive airway pressure in the treatment of sleep apnea/hypopnea syndromeAm J Respir Crit Care Med199915924955019927363
- Campos-RodriguezFPerez-RonchelJGrilo-ReinaALima-AlvarezJBenitezMAAlmeida-GonzalezCLong-term effect of continuous positive airway pressure on BP in patients with hypertension and sleep apneaChest200713261847185217925415
- SopkovaZDorkovaZTkacovaRPredictors of compliance with continuous positive airway pressure treatment in patients with obstructive sleep apnea and metabolic syndromeWien Klin Wochenschr200912111–1239840419626298
- FlayBREfficacy and effectiveness trials (and other phases of research) in the development of health promotion programsPrev Med19861554514743534875
- StradlingJRDaviesRJIs more NCPAP better?Sleep200023Suppl 4S150S15310893091
- WeaverTEMaislinGDingesDFRelationship between hours of CPAP use and achieving normal levels of sleepiness and daily functioningSleep200730671171917580592
- BuchnerNJSannerBMBorgelJRumpLCContinuous positive airway pressure treatment of mild to moderate obstructive sleep apnea reduces cardiovascular riskAm J Respir Crit Care Med2007176121274128017673692
- Campos-RodriguezFPeña-GriñanNReyes-NuñezNMortality in obstructive sleep apnea-hypopnea patients treated with positive airway pressureChest2005128262463316100147
- DohertyLSKielyJLSwanVMcNicholasWTLong-term effects of nasal continuous positive airway pressure therapy on cardiovascular outcomes in sleep apnea syndromeChest200512762076208415947323
- PekerYHednerJNorumJKraicziHCarlsonJIncreased incidence of cardiovascular disease in middle-aged men with obstructive sleep apnea: a 7-year follow-upAm J Respir Crit Care Med2002166215916512119227
- StepnowskyCJDimsdaleJEDose-response relationship between CPAP compliance and measures of sleep apnea severitySleep Med20023432933414592195
- MermigkisCBouloukakiIMermigkisDCRP evolution pattern in CPAP-treated obstructive sleep apnea patients. Does gender play a role?Sleep Breath201216381381921881894
- SchizaSEMermigkisCPanagiotisPC-reactive protein evolution in obstructive sleep apnoea patients under CPAP therapyEur J Clin Invest2010401196897520629709
- IshidaKKatoMKatoYAppropriate use of nasal continuous positive airway pressure decreases elevated C-reactive protein in patients with obstructive sleep apneaChest2009136112512919255295
- BuriokaNMiyataMFukuokaYEndoMShimizuEDay-night variations of serum interleukin-6 in patients with severe obstructive sleep apnea syndrome before and after continuous positive airway pressure (CPAP)Chronobiol Int200825582783418780208
- ZieglerMGMillsPJLoredoJSAncoli-IsraelSDimsdaleJEEffect of continuous positive airway pressure and placebo treatment on sympathetic nervous activity in patients with obstructive sleep apneaChest2001120388789311555525
- JooMJHerdegenJJSleep apnea in an urban public hospital: assessment of severity and treatment adherenceJ Clin Sleep Med20073328528817561598
- LindbergECarterNGislasonTJansonCRole of snoring and daytime sleepiness in occupational accidentsAm J Respir Crit Care Med2001164112031203511739131
- MasaJFRubioMFindleyLJHabitually sleepy drivers have a high frequency of automobile crashes associated with respiratory disorders during sleepAm J Respir Crit Care Med20001624 Pt 11407141211029353
- SomersMLPetersonESharmaSYaremchukKContinuous positive airway pressure adherence for obstructive sleep apneaISRN Otolaryngol2011201194358623724263
- Nino-MurciaGMcCannCCBliwiseDLGuilleminaultCDementWCCompliance and side effects in sleep apnea patients treated with nasal continuous positive airway pressureWest J Med198915021651692658326
- WeaverTEChasensERContinuous positive airway pressure treatment for sleep apnea in older adultsSleep Med Rev20071129911117275370
- BudhirajaRParthasarathySDrakeCLEarly CPAP use identifies subsequent adherence to CPAP therapySleep200730332032417425228
- Simon-TuvalTReuveniHGreenberg-DotanSOksenbergATalATarasiukALow socioeconomic status is a risk factor for CPAP acceptance among adult OSAS patients requiring treatmentSleep200932454555219413149
- SinDDMayersIManGCPawlukLLong-term compliance rates to continuous positive airway pressure in obstructive sleep apnea: a population-based studyChest2002121243043511834653
- WangYGaoWSunMChenBAdherence to CPAP in patients with obstructive sleep apnea in a Chinese populationRespir Care201257223824321762553
- WolkoveNBaltzanMKamelHDabrusinRPalayewMLong-term compliance with continuous positive airway pressure in patients with obstructive sleep apneaCan Respir J200815736535918949106