
Abstract
Plaque psoriasis is an immune-mediated skin disease that affects roughly 3% of adults in the United States. Advances over the past 20 years in understanding the immune-mediated pathophysiology of psoriasis have led to the development of targeted biologic therapies for this condition. Currently, biologic medications approved for the treatment of plaque psoriasis include tumor necrosis factor α inhibitors, interleukin (IL)-17 or IL-17 receptor inhibitors, IL-12/23 inhibitors, and IL-23 inhibitors. Tildrakizumab-asmn is a monoclonal antibody that targets the p19 subunit of IL-23 and is approved for use in adult patients with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy. This article reviews the current pharmacologic, efficacy, and safety data on tildrakizumab-asmn.
Introduction
Plaque psoriasis is a chronic, immune-mediated skin disease that affects roughly 3% of adults in the United States.Citation1 Psoriasis may have significant disease comorbidities and a substantial negative impact on quality of life.Citation2 Mild or localized psoriasis may be managed with topical treatments, whereas systemic or biologic therapies are warranted for more severe or widespread disease.Citation3 The development of targeted biologic therapies as a systemic treatment option for psoriasis is a result of advances in the understanding of the immune-mediated pathophysiology of psoriasis over the past 20 years.Citation4 Currently, biologic medications approved for the treatment of plaque psoriasis include tumor necrosis factor alpha (TNF-α) inhibitors, interleukin (IL)-17 and IL-17 receptor (IL-17R) inhibitors, IL-12/23 inhibitors, and IL-23p19 inhibitors. The newest psoriasis treatment options approved by the US Food and Drug Administration (US FDA) are the anti-IL-17 biologics secukinumab (Cosentyx®, Novartis International AG, Basel, Switzerland) and ixekizumab (Taltz®, Eli Lilly and Co., Indianapolis, IN, USA), the anti-IL-17R biologic brodalumab (Siliq®, Valeant Pharmaceuticals, Bridgewater Township, NJ, USA), and the anti-IL-23p19 biologics guselkumab (Tremfya®, Janssen, Beerse, Belgium) and tildrakizumab-asmn (Ilumya®, Sun Pharmaceuticals, Mumbai, India). This review will focus on the most current evidence for tildrakizumab-asmn.
Rationale for the targeting of IL-23p19 to treat psoriasis
Although the pathophysiology of psoriasis is complex and incompletely understood, recent evidence that the IL-23/IL-17 axis plays a key role in psoriasis has led to the development of biologic treatments that target this pathway specifically.Citation5,Citation6 IL-23 is a heterodimeric cytokine consisting of a p19 and a p40 subunits, predominantly expressed by macrophages and dendritic cells.Citation7,Citation8 The p40 subunit of IL-23 is shared with IL-12, and is the target of another biologic for psoriasis, ustekinumab (Stelara®, Janssen). Recent studies suggest that targeting IL-23 alone may result in a more favorable risk–benefit profile in comparison to targeting both IL-23 and IL-12.Citation9 While IL-23 appears critical for IL-17 differentiation,Citation6 IL-12 is implicated in a host of other processes in the body, including host defense against infection and malignancy.Citation10 For this reason, the p19 subunit of IL-23, which is not shared with IL-12, became a target of drug development.Citation11
IL-23 is implicated as a “master cytokine” in inflammatory skin disease because it induces differentiation of type 17 helper T lymphocytes (Th17 cells), which in turn secrete pro-inflammatory cytokines, including IL-17A, IL-17F, and IL-22.Citation8 This results in increased keratinocyte proliferation and differentiation, which clinically presents as psoriatic epidermal hyperplasia.Citation12,Citation13 Further evidence for the role of IL-23 in psoriasis comes from messenger RNA data, which show the overexpression of IL-23p19 and IL-12/23p40 in psoriatic skin.Citation14 Favorable results from clinical trials testing new molecules targeting the IL-23p19 subunit further validate the important role of IL-23 in moderate-to-severe plaque psoriasis.Citation10
The first FDA approved biologic of the IL-23p19 class was guselkumab (Tremfya®, Janssen), which was approved in July 2017. Tildrakizumab was approved in 2018. Risankizumab (BI 655066, AbbVie),Citation15 a third IL-23p19 inhibitor, is currently in advanced stages of clinical evaluation.
Tildrakizumab
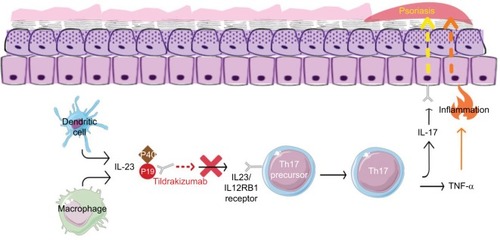
Tildrakizumab-asmn (tildrakizumab) is a humanized (from mouse) IgG1κ monoclonal antibody that selectively inhibits the p19 subunit of IL-23 and neutralizes its function (). The suffix “-asmn” is used to differentiate this originator biologic from future biosimilar versions of tildrakizumab.Citation16 In March 2018, tildrakizumab was approved by the US FDA for use in adult patients with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy.Citation17 It is recommended to evaluate patients for tuberculosis infection prior to initiating treatment. Tildrakizumab treatment may increase the risk of infection. The use of live vaccines is contraindicated for patients on tildrakizumab. Tildrakizumab is contraindicated in patients with a hypersensitivity reaction to tildrakizumab or to any of its excipients.
Figure 1 The mechanism of action of tildrakizumab.
Abbreviation: IL, interleukin; Th17, type 17 helper T lymphocytes; TNF-α, tumor necrosis factor alpha.
FDA approval for tildrakizumab was based on pooled data from three placebo-controlled clinical trials in which a total of 705 subjects received tildrakizumab at the FDA-approved dosing schedule.Citation17 Tildrakizumab is given by subcutaneous (SC) injection at a dose of 100 mg every 12 weeks, after the completion of initiation dosing, which consists of 100 mg at week 0 and week 4. Tildrakizumab is available in prefilled syringes, each containing 1 mL of 100 mg/mL of the antibody.
With regard to immunogenicity, ~6.5% of study participants treated with 100 mg tildrakizumab developed antibodies to tildrakizumab.Citation17 Of these, ~40% developed neutralizing antibodies, which were associated with lower serum concentrations of tildrakizumab and reduced efficacy.
Pharmacologic parameters of tildrakizumab
Pharmacokinetic studies of tildrakizumab in plaque psoriasis show that both the maximum concentration (Cmax) and the area under the curve (AUC) increase proportionally from doses of 50 to 200 mg or 0.1 to 10 mg/kg (0.5–2 times the FDA-approved recommended SC dosage).Citation17 The bioavailability of tildrakizumab is ~73%–80% following SC injection. The peak plasma concentration is 8.1 µg/mL, with a peak plasma time of ~6 days. Steady-state concentrations were achieved in week 16 following subcutaneously administered tildrakizumab at a dose of 100 mg, which is the approved recommended dosage. The mean steady-state trough concentrations ranged from 1.22 to 1.47 (SD), ±0.94 and ±1.12 µg/mL). The geometric mean steady-state Cmax was 8.1 µg/mL or 34%. Tildrakizumab concentrations were lower in subjects with higher body weight, though there is no indication for weight-based dose adjustments. The half-life (t1/2) iŝ 20.2–28.2 days. The volume of distribution is 10.8 L. Clearance occurs at a rate of 0.32 L/day (38%).
Per US prescribing information, no formal pharmacodynamic studies have been conducted for tildrakizumab.Citation17 Metabolism of tildrakizumab is also not characterized. However, the medication is expected to degrade into small peptides and amino acids through catabolic pathways, similar to endogenous IgG. The effects of renal and hepatic impairment on tildrakizumab have not been studied. Tildrakizumab has not been studied in special populations such as geriatric, pediatric, breastfeeding, or pregnant populations.
Clinical trials
The remainder of this article will review the clinical trial data for tildrakizumab in further detail ().
Table 1 Clinical trials of tildrakizumab for plaque psoriasis
Phase I
An exploratory, proof-of-concept, three-part, multiple-dose, randomized, placebo-controlled patient- and evaluator-blind, multi-center Phase I study of tildrakizumab was performed to determine the safety, tolerability, and pharmacokinetics of intravenous (IV) tildrakizumab at rising doses in adults with moderate-to-severe psoriasis.Citation13 A secondary objective of the trial was to evaluate the efficacy of tildrakizumab on psoriatic skin disease.
A total of 77 subjects were enrolled in this Phase I study. The study subjects included males (61/77, 79%) and females (16/77, 21%) who were predominantly white (69/77, 90%), ranging from 22 to 65 years of age (mean age of 47.3, SD 11.4), and mean weight of 97.67 kg (SD 20.23).
Part 1 of this trial examined safety, tolerability, and pharmacokinetics of four ascending doses of IV injections of tildrakizumab in 24 subjects divided into four ascending dose cohorts. The subjects were randomized to receive 0.1 mg/kg (n=3), 0.5 mg/kg (n=3), 3 mg/kg (n=6), or 10 mg/kg (n=6) tildrakizumab, or placebo dose (n=6) at weeks 0, 8, and 12. Subjects were followed up for 196 days after the first dose.
Part 2 examined the efficacy of higher doses of IV tildrakizumab in a larger number of subjects. Forty subjects received three doses of either 3 mg/kg (n=15) or 10 mg/kg (n=14) of IV tildrakizumab or placebo (n=11) given at weeks 0, 4, and 8. Subjects were followed up for 364 days after the first dose.
Part 3 evaluated the efficacy of lower doses of IV tildrakizumab. Twelve subjects were randomized in a 2:1:1 ratio to receive 0.05 mg/kg IV tildrakizumab (n=6), 0.1 kg/mg IV tildrakizumab (n=3), or placebo (n=3) at weeks 0, 8, and 12. Three treatments were administered in a parallel fashion at weeks 0, 8, and 12.
The primary efficacy end point was the percentage change in Psoriasis Area and Severity Index (PASI) score from baseline at 4 or 8 weeks after the last treatment day (week 16 for parts 1, 2, and 3). Throughout the study, study subjects were monitored by safety assessments that included vital signs, ECGs, clinical laboratory measurements, and adverse event (AE) screening. PASI scores were determined at screening, baseline, and throughout the study at specified intervals until day 112 in part 1, and day 364 in parts 2 and 3. Serum pharmacokinetic samples were collected at specified intervals in all three parts of the study. Pre-dose (within 3–7 days of initial dose) and post-dose biopsies (4 weeks after the last dose) were obtained from a subset of subjects treated with placebo (n=5), 3 mg/kg of tildrakizumab (n=10), 10 mg/kg of tildrakizumab (n=7), and healthy volunteers (n=5) for exploratory histological, immunohistochemical, and gene expression analyses. Skin biopsies were also assessed using a Histopathological Psoriasis Severity Score (HPSS),Citation18 in order to quantify the overall global effect of tildrakizumab on the epithelium, vasculature, and infiltrates of psoriatic skin.
Results
Pharmacodynamics
Tildrakizumab showed dose-related efficacy in the treatment of plaque psoriasis. Subjects treated with IV tildrakizumab 0.05–10 mg/kg had a mean (placebo-corrected) PASI score reduction of 50%–80% by week 14 (day 112), which sustained through week 28 (day 196). A majority of subjects on 3 or 10 mg/kg doses achieved PASI 75, with a large proportion achieving PASI 90, by day 112. Subjects treated with 3 or 10 mg/kg of IV tildrakizumab maintained at least PASI 50 by week 44 (day 308, 36 weeks after the last dose), whereas those in part 3 treated with 0.05 or 0.1 mg/kg did not. However, this Phase I study was not sufficiently powered to show a clear dose–response.
Safety and tolerability
Overall, IV tildrakizumab was well tolerated in all doses evaluated. The maximum dose evaluated was 10 mg/kg IV once monthly. The most common AEs included headache (tildrakizumab-treated: 11/57 [19%], placebo: 3/20 [15%], upper respiratory infection: 11/57 [19%] and 3/20 [15%], nasopharyngitis: 10/57 [18%] and 2/20 [10%], and cough: 9/57 [16%] and 3/20 [15%]). Of the eleven SAEs reported in eight subjects, ten were thought to be unrelated to treatment and one SAE was deemed possibly related to treatment (convulsions in a subject who received 10 mg/kg tildrakizumab 17 days prior).
Pharmacokinetics
Based on the cumulative results of parts 1, 2, and 3, the mean half-life (t1/2) of tildrakizumab ranged from 20.2 to 26.9 days. For doses of 0.05–10 mg/kg, the mean clearance ranged from 1.57 to 2.50 mL/day. No dose-related trends were observed for either half-life or clearance. Using a one-way analysis of variance dose proportionality test in part 2 only, dose proportionality for the AUC and Cmax was shown for 3 and 10 mg/kg doses of tildrakizumab. Tildrakizumab also demonstrated linear exposure over the administered doses.
Immunogenicity
A majority of the tildrakizumab-treated subjects tested negative for antidrug antibodies prior to treatment (n=51). After tildrakizumab treatment, 18% (9/51) of the pretreatment ADA-negative subjects had at least one posttreatment ADA-positive sample. Fifty-six percent (5/9) of these subjects showed lower tildrakizumab exposure than ADA-negative subjects. There was no difference in PASI response or AEs reported in subjects with ADA-positive samples.
Exploratory histological, immunohistochemical, and gene expression analyses
After treatment with tildrakizumab, epithelial hyperplasia in the psoriatic lesions improved to the levels found in non-lesional skin (as measured by a decrease in epithelial thickness), but not fully to that of normal, healthy skin. There was a significant reduction in the total HPSS (mean reduction 67%, 95% CI: 53%–81%). The HPSS was significantly reduced in both the 3 and 10 mg/kg treated groups (pre- and post-dose HPSS 16.5±1.3 [mean ± SEM] to 6.2±61.9 [P<0.01] and 20.0±2.5–4.4±1.2 [P<0.05], respectively). Each of the epithelial, vascular, and inflammatory cell parameters was significantly reduced after tildrakizumab treatment (P<0.01 for the epithelial score of 3 mg/kg, P<0.05 for all others), whereas reductions for these parameters in the placebo group were not significant.
Post-dose biopsies showed significant reduction mitosis within the suprabasal epidermal layer, as measured by cell proliferation (Ki67+ cells per mm) and keratinocyte differentiation (keratin 16 score) after treatment with tildrakizumab, but not with placebo.
The levels of inflammatory infiltrate of psoriatic skin lesions significantly decreased after treatment with tildrakizumab. Specifically, epidermal CD4+ and CD8+ T cells, as well as dermal myeloid dendritic cells, plasmacytoid dendritic cells, and CD15+ neutrophils were significantly reduced after three doses of 10 mg/kg tildrakizumab (P<0.05). Epidermal CD4+ T cells, dermal plasmacytoid dendritic cells, and neutrophils, but not epidermal CD8 T cells or dermal myeloid DCs, decreased after three doses of 3 mg/kg tildrakizumab (P<0.01). Langerhans cells were not significantly increased in psoriatic skin lesions at baseline, nor were they significantly reduced after tildrakizumab treatment.
IL-23p19 expression decreased to nearly undetectable levels after treatment with tildrakizumab, though the reduction was not significant as compared to placebo.
IL-23-associated psoriatic skin gene expression (IL-19, IL-20, S100A7, LCN2, CCL20, and CXCL8) was significantly reduced in skin biopsies of subjects treated with at least one dose of 3 or 10 mg/kg tildrakizumab (P<0.05), but not in the subjects treated with placebo. The decreases in gene expression correlated with decreased epithelial thickness (mm), decreased keratinocyte proliferation (measured by Ki67 cells per mm), normalized keratinocyte differentiation (measured by keratin 16 score), and reduced CD4 T cell and neutrophil infiltrates after treatment with tildrakizumab.
Phase II
A three-part, Phase IIb, randomized, double-blinded, placebo-controlled, parallel-design study of subcutaneously administered tildrakizumab (NCT01225731) in adult subjects with moderate-to-severe chronic plaque psoriasis clinical trial was performed at 64 sites in the United States, Canada, Europe, and Japan.Citation10
This Phase II trial enrolled 355 subjects, and 266 subjects completed the study. Study participants were men and women with baseline moderate-to-severe plaque psoriasis (PASI ≥12; BSA ≥10; PGA of moderate, marked, or severe) for at least 6 months.
In part 1 (weeks 0–16) of this trial, subjects were randomized in a 1:2:2:2:1 ratio to receive SC injections of tildrakizumab at doses of 5 mg (n=42), 25 mg (n=92), 100 mg (n=89), or 200 mg (n=86) or placebo (n=46). Doses were administered at weeks 0 and 4. At the end of part 1, subjects were assessed for their responder status (based on week 16 PASI response) in order to determine their treatment for part 2. Responders had at least a 75% reduction in PASI score from baseline.
During part 2 (weeks 16–52), all subjects received active treatment with tildrakizumab. Subjects on placebo in part 1 were started on 100 mg of tildrakizumab in part 2. Non-responders in the 5 and 25 mg groups were increased to 100 mg, while those in 100 mg groups were increased to 200 mg, and those in the 200 mg group remained at that dose. Responders on 5 or 25 mg tildrakizumab continued at their same doses through week 52. Responders on 200 or 100 mg of tildrakizumab were re-randomized to either continue their current dose or switch to a reduced dose of 100 or 25 mg, respectively.
Part 3 (weeks 52–72) consisted of a follow-up period after treatment was discontinued at week 52.
The primary efficacy end point was a reduction in PASI score of at least 75% from baseline to week 16. The impact on quality of life was monitored by use of the Dermatology Life Quality Index (DLQI). Periodic blood sampling was performed to evaluate the pharmacokinetics and development of ADA to tildrakizumab. AEs, ECG, clinical safety, and vital signs were monitored throughout the study.
Results
Clinical efficacy
At week 16, PASI 75 responses were achieved in 33%, 64%, 66%, and 74% of patients in the 5 mg (n=42), 25 mg (n=90), 100 mg (n=89), and 200 mg (n=86) tildrakizumab dose groups, respectively, compared to 4.4% for placebo (P≤0.001 for each active treatment group compared to placebo). In addition, PASI 90 at week 16 was achieved in 12%, 25%, 39%, and 52% of the tildrakizumab groups, respectively. The median time to PASI 75 was 85 days in the 25 mg group, 84 days in the 100 mg group, and 57 days in the 200 mg group (P≤0.001 compared to placebo). PASI 75 response rates at week 12 were 61% (n=54/89) in the 100 mg group and 72% (n=62/82) in the 200 mg group, compared to 4.4% for placebo (n=2/45) (P≤0.001 for each treatment group vs placebo).
Of the responders at week 16, results from part 2 of the study showed that PASI 75 response was maintained at week 52 in 97% of subjects who continued on doses of 100 mg (n=30/31) or 200 mg (n=29/32) throughout parts 1 and 2, compared to 70% of those who received a dose reduction from 100 to 25 mg (n=21/30). In part 2, there was a significant loss in efficacy in those who received a reduction from 100 to 25 mg compared to those who remained on 100 mg (P<0.005). There was no significant difference in those who remained on 200 mg in parts 1 and 2 compared to those who were decreased from 200 to 100 mg in part 2; PASI 75 was maintained at week 52 in 97% (29/32) and 85% (27/32), respectively.
In part 3, PASI 75 was maintained 20 weeks after study drug discontinuation in 96% of subjects on 100 mg and 93% on 200 mg tildrakizumab, in contrast to 68% of those whose dose was reduced from 200 to 100 mg at week 16. Eight subjects with a week 52 PASI 75 response experienced relapse in part 3 (25 mg, n=4; 100 mg, n=3; 200 mg, n=1).
Pharmacokinetics
The mean concentration of tildrakizumab was usually higher in responders than in nonresponders, and increased proportionally with dose. The exposure in the groups receiving 200 mg of tildrakizumab (the highest dose studied) was similar for both responders and nonresponders.
Immunogenicity
Of the 355 subjects enrolled, 46 had positive baseline ADA assays performed prior to receiving tildrakizumab. Of the subjects with baseline negative ADA screening, 25 developed at least one positive ADA after receiving a dose of tildrakizumab, and 10 of these 25 tested positive for neutralizing antibodies. Sixteen of the 46 (35%) subjects with positive ADA screens had qualitatively reduced tildrakizumab levels. The development of neutralizing antibodies did not appear related to the development of AEs.
Safety profile
Out of all randomized participants who received one or more doses of treatment, AEs were reported at a rate of 65%, 66%, and 40% in parts 1, 2, and 3, respectively. In all treatment arms, the overall incidence of AEs was generally similar and did not differ from placebo. No significant, clinically meaningful differences in clinical safety, ECGs, or vital signs were observed in patients receiving tildrakizumab compared with placebo. The most frequently reported AEs in parts 1–3 were nasopharyngitis and headache. There was a low rate of AEs of special interest, including serious infections, malignancy, major cardiovascular events, drug-related hypersensitivity, and drug-related injection site reactions. A total of 23 subjects reported SAEs (part 1, n=4; part 2, n=14, part 3, n=5). SAEs thought to be related to tildrakizumab treatment included bacterial arthritis, lymphedema, melanoma, stroke, epiglottitis, and knee infection. There was one death in the study, thought to be unrelated to study treatment. The study concluded that tildrakizumab was well tolerated and generally safe for 52 weeks of treatment.
Quality of life
The mean change in DLQI score from baseline to week 16 was −4.9, −9.2, −8.5, and −8.8 in the 5, 25, 100, and 200 mg tildrakizumab groups, respectively, compared with 1.0 in the placebo group. A DLQI score of 0 or 1 at week 16 was achieved by 32%, 57%, 52%, and 57% of patients in the tildrakizumab groups, respectively, compared to 0% in the placebo group. A 5-point reduction from baseline DLQI score at week 16 occurred in 52%, 70%, 65%, and 73% of the 5–200 mg tildrakizumab groups, respectively, compared with 19% of the placebo group. Symptoms/Feelings and Daily Activities were the DLQI subcategories that were most improved after treatment. Overall, the improvement in quality of life correlated to the clinical improvement in each treatment group.
Phase III (reSURFACE 1 and reSURFACE 2)
Tildrakizumab demonstrated efficacy in patients with moderate-to-severe chronic plaque psoriasis in two pivotal Phase III, three-part, parallel group, randomized, double-blind clinical trials, reSURFACE 1 (NCT01722331) and reSURFACE 2 (NCT01729754).Citation19 Based on data from the Phase IIb trial,Citation10 dosing consisted of 100 or 200 mg of tildrakizumab given subcutaneously at weeks 0 and 4 (initiation therapy) and every 12 weeks thereafter (maintenance therapy). Tildrakizumab was compared with placebo in reSURFACE one and placebo or etanercept in reSURFACE 2.Citation19
Participants in reSURFACE 1 (N=772) and reSURFACE 2 (N=1090) were 18 years of age or older with moderate-to-severe psoriasis, as defined by BSA ≥10% or PASI ≥12, and were considered candidates for phototherapy or systemic therapy. The baseline demographics of the study participants were similar in all groups; the majority were white males, with an average age of 44–48 years, who had not previously been on biologic therapy.Citation19 The most common previous medical condition in these patients was hypertension. The average baseline PASI score was around 20, with roughly 30% BSA involvement.Citation19
Part 1 of both studies comprised the first 12-week treatment period. In reSURFACE 1, study subjects were randomized in a 2:2:1 ratio to tildrakizumab 200 mg, tildrakizumab 100 mg, or placebo at weeks 0 and 4. In reSURFACE 2, study subjects were randomized in a 2:2:1:2 ratio to tildrakizumab 200 mg, tildrakizumab 100 mg, placebo, or etanercept 50 mg twice weekly.
Part 2 was a 16-week period (weeks 12–28) in which the placebo group in each study was re-randomized in a 1:1 ratio to tildrakizumab 200 or 100 mg starting with initiation dosing at week 12, while tildrakizumab groups continued maintenance dosing, with their next dose at week 16. In reSURFACE 2, the etanercept group received one dose weekly. Subjects from all groups who had a PASI response of <50% (non-responders) at week 28 were discontinued so as to maintain masking.Citation19
In part 3, subjects in both studies received either tildrakizumab or placebo until week 64. At week 28 in reSURFACE 1, tildrakizumab 200 and 100 mg responders (PASI ≥75) and partial responders (PASI ≥50 to PASI <75) were re-randomized to continue the same treatment, switch to a different dose of tildrakizumab, or switch to placebo. Subjects who experienced relapse on placebo or a different dose were re-treated with tildrakizumab upon relapse. In reSURFACE 2, part 3 also evaluated etanercept non-responders (PASI ≤50) and partial responders (PASI ≤50–75) at week 28 who were switched to tildrakizumab 200 mg until week 52. The duration of part 3 was 36 weeks (week 28–64) in reSURFACE 1 and 24 weeks (weeks 28–52) in reSURFACE 2. An optional long-term safety extension study of 4 years duration was then performed, followed by a follow-up/washout period of 20 weeks.
Study assessments included clinical response evaluations (PASI, PGA), safety evaluations, and quality of life measures (DLQI) throughout the study. Efficacy and safety measurements were done at baseline and weeks 4, 8, 12, 16, 22, and 28 in parts 1 and 2. In part 3, efficacy was assessed at weeks 32, 36, 40, 46, and 52 in both studies, and efficacy and safety were assessed at week 64 in reSURFACE 1.
The primary objective of the reSURFACE studies was to evaluate the efficacy and safety of tildrakizumab after 12 weeks. The primary efficacy endpoint was the proportion of patients achieving PASI 75 at week 12, for both the 100 and 200 mg doses, as compared to placebo. The co-primary endpoints were the proportion of patients achieving PASI 75 and PGA scores of “clear” or “minimal” with at least a two-grade reduction from baseline at week 12. In reSURFACE 2, PASI 75 and PGA responses at week 28 were also key secondary endpoints. The PASI 75 response in patients receiving continuous treatment with tildrakizumab from baseline to the end of week 64 in reSURFACE 1 or week 52 in reSURFACE 2 was also assessed as a secondary outcome. An additional secondary endpoint in both studies was the proportion of subjects with a DLQI score of either 0 or 1 by weeks 12 and 28.
Results
Clinical efficacy
The efficacy results from both the doses of tildrakizumab evaluated in the reSURFACE trials are summarized in . The efficacy data of the FDA-approved tildrakizumab (100 mg) as compared to placebo or placebo and etanercept are summarized in . In patients treated with 100 mg of tildrakizumab, 64% of subjects in reSURFACE 1 (n=197) and 61% (n=188) of patients in reSURFACE 2 achieved PASI 75 at week 12, compared to 6% (n=9) or 6% (n=9) and 48% (n=313), compared to placebo in reSURFACE 1 or compared to placebo and etanercept in reSURFACE 2, respectively. The results were significant (P<0.001) for comparisons of each tildrakizumab group to placebo. The peak efficacy of tildrakizumab occurred between weeks 22 and 28 in both trials.
Table 2 Phase III efficacy data of tildrakizumab from reSURFACE 1 and reSURFACE 2
Table 3 Phase III clinical trial efficacy data of tildrakizumab 100 mg at week 12
Safety profile
In both reSURFACE trials, the proportion of subjects reporting at least one AE in weeks 0–12 was similar in all treatment groups (tildrakizumab 200 mg: 42%/49%; tildrakizumab 100 mg: 47%/44%; placebo: 48%/55%; etanercept: 54%). The most common AEs in both reSURFACE trials were nasopharyngitis and upper respiratory tract infections. In reSURFACE 2, injection site erythema was more common in etanercept than tildrakizumab (tildrakizumab 200 mg: 1%; tildrakizumab 100 mg: 1%; placebo: 1%; etanercept: 9%). The rate of severe infections, malignancies, skin cancers, major cardiovascular events, and drug-related hypersensitivity reactions was low in both studies, and there was no significant difference between the active treatment groups. Discontinuation due to AEs was not frequently reported. The overall rate of SAEs in the first 12 weeks was low and comparable among the treatment groups (tildrakizumab 200 mg: 3%/2%; tildrakizumab 100 mg: 2%/1%; placebo: 1%/3%; etanercept: 2%). One death occurred in reSURFACE 2 in a subject with alcoholic cardiomyopathy and steatohepatitis; this patient completed part 1 only, with the last date of study medication on day 30, and the death occurring on day 96, though the cause of death was not determined.
Quality of life
In concordance with clinical improvements, there were significant improvements in mean changes in DLQI for all tildrakizumab-treated groups in the reSURFACE trials. Prior to treatment, the average baseline DLQI score for participants was 13–15, indicating severe impact. There was significantly greater number of subjects treated with either dose of tildrakizumab reporting DLQI scores of 0 or 1 (meaning no impact of skin disease on quality of life) when compared with placebo at 12 weeks (reSURFACE 1: 42% and 44% vs 5%; P<0.001; reSURFACE 2: 40% and 47% vs 8%, P<0.001). The proportion of tildrakizumab-treated subjects achieving these scores increased at week 28 (100 mg: 52%; 200 mg: 57%; placebo to 100 mg: 52%; placebo to 200 mg: 56%). In the reSURFACE 2 trial, tildrakizumab 200 mg was associated with significantly higher rates of patients achieving DLQI scores of 0/1 at week 12 compared to etanercept 50 mg (47% vs 36%; P=0.0029). DLQI scores of either 0 or 1 at week 28 were achieved by a significantly greater proportion of patients in both the tildrakizumab groups compared to patients treated with etanercept (100 mg: 54% (P=0.0003); 200 mg: 65% (P<0.0001) vs 39%.
Full results from extension studies of the original reSURFACE studies have not yet been reported.
Tildrakizumab compared to other biologics for psoriasis
With regard to efficacy, the class of newer biologic agents targeting IL-17 or IL-23 was all highly effective for psoriasis and are superior to older biologic agents targeting TNF-α. In clinical trials, tildrakizumab demonstrated superior efficacy over an anti-TNF-α agent, etanercept ().Citation19 However, when compared with newer biologic agents, the efficacy data from tildrakizumab is less favorable. Within the class of IL-23p19 inhibitors, and also in comparison to IL-17 inhibitors, tildrakizumab appears to be the least efficacious as measured by PASI scores, though no head-to-head studies comparing these biologics are yet available ().
Table 4 Efficacy of biologics targeting IL-23p19 and IL-17/17R for plaque psoriasis
From a safety standpoint, there is evidence for a favorable safety profile of tildrakizumab. Tildrakizumab’s safety profile is primarily characterized by non-serious AEs; nasopharyngitis and upper respiratory tract infections were the most common AEs in clinical trials. The dosing profile of tildrakizumab is also highly favorable, with maintenance dosing only once every 12 weeks. This is tied for the lowest frequency of any biologic currently approved for psoriasis, with the other being ustekinumab.
Use in other diseases
In addition to psoriasis, the IL-23, IL-17, and TNF pathways are implicated as important to the pathogenesis of psoriatic arthritis and axial spondyloarthropathies.Citation8 Tildrakizumab is under investigation in Phase II clinical trials for ankylosing spondylitis (NCT02980705) and psoriatic arthritis (NCT02980692).
Summary
Tildrakizumab is an IL-23p19 subunit inhibitor approved for use in adults with moderate-to-severe plaque psoriasis. FDA approval of tildrakizumab was based on evidence from several key clinical trials. A Phase I randomized placebo-controlled proof-of-concept study showed that IV tildrakizumab at doses of 0.05–10 mg/kg produced clinical improvement in patients with psoriasis.Citation13 Tildrakizumab also demonstrated significant improvement in the histological measures of psoriasis.Citation13 A dose range finding Phase IIb study led to the testing of tildrakizumab 100 and 200 mg.Citation10 Both Phase III studies met the primary efficacy endpoints, demonstrating significant clinical improvement with tildrakizumab (100 and 200 mg) compared to placebo as measured by PASI 75 response and PGA of 0/1 after the initial two doses.Citation19 Long-term safety and efficacy data, not only from tildrakizumab but also from the many other new biologics targeting IL-17 and IL-23, are needed to establish the role of tildrakizumab in the therapeutic armamentarium for psoriasis.
Disclosure
The authors report no conflicts of interest in this work.
References
- RachakondaTDSchuppCWArmstrongAWPsoriasis prevalence among adults in the United StatesJ Am Acad Dermatol201470351251624388724
- KimballABJacobsonCWeissSVreelandMGWuYThe psychosocial burden of psoriasisAm J Clin Dermatol20056638339216343026
- MenterAGottliebAFeldmanSRGuidelines of care for the management of psoriasis and psoriatic arthritis: Section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologicsJ Am Acad Dermatol200858582685018423260
- NickoloffBJNestleFORecent insights into the immunopathogenesis of psoriasis provide new therapeutic opportunitiesJ Clin Invest2004113121664167515199399
- PuigLThe role of IL 23 in the treatment of psoriasisExpert Rev Clin Immunol201713652553428165883
- TonelGConradCLaggnerUCutting edge: a critical functional role for IL-23 in psoriasisJ Immunol2010185105688569120956338
- LeeETrepicchioWLOestreicherJLIncreased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgarisJ Exp Med2004199112513014707118
- NestleFOKaplanDHBarkerJPsoriasisN Engl J Med2009361549650919641206
- FitchEHarperESkorchevaIKurtzSEBlauveltAPathophysiology of psoriasis: recent advances on IL-23 and Th17 cytokinesCurr Rheumatol Rep20079646146718177599
- PappKThaçiDReichKTildrakizumab (MK-3222), an anti-interleukin-23p19 monoclonal antibody, improves psoriasis in a phase IIb randomized placebo-controlled trialBr J Dermatol2015173493093926042589
- LevinAAGottliebABSpecific targeting of interleukin-23p19 as effective treatment for psoriasisJ Am Acad Dermatol201470355556124373779
- LowesMARussellCBMartinDATowneJEKruegerJGThe IL-23/T17 pathogenic axis in psoriasis is amplified by keratinocyte responsesTrends Immunol201334417418123291100
- KoppTRiedlEBangertCClinical improvement in psoriasis with specific targeting of interleukin-23Nature2015521755122222625754330
- ChanJRBlumenscheinWMurphyEIL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesisJ Exp Med2006203122577258717074928
- PappKABlauveltABukhaloMRisankizumab versus ustekinumab for moderate-to-severe plaque psoriasisN Engl J Med2017376161551156028423301
- YangEJBeckKMLiaoWTildrakizumab-asmn: What’s in a Name?Am J Clin Dermatol201819329129229687361
- Tildrakizumab US prescribing information2018 Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761067s000lbl.pdfAccessed April 21, 2018
- AckermanABBoerABenninBHistologic Diagnosis of Inflammatory Skin Diseases: An Algorithmic Method Based on Pattern Analysis3rd edNew York, NYArdor Scribendi2005
- ReichKPappKABlauveltATildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trialsLancet20173901009127628828596043
- BlauveltAPappKAGriffithsCEEfficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trialJ Am Acad Dermatol201776340541728057360
- ReichKArmstrongAWFoleyPEfficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: Results from the phase III, double-blind, placebo- and active comparator-controlled VOYAGE 2 trialJ Am Acad Dermatol201776341843128057361
- PappKAReichKPaulCA prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasisBr J Dermatol2016175227328626914406
- BlauveltAPappKALebwohlMGRapid onset of action in patients with moderate-to-severe psoriasis treated with brodalumab: a pooled analysis of data from two phase 3 randomized clinical trials (AMAG-INE-2 and AMAGINE-3J Am Acad Dermatol201777237237428711089
- LangleyRGElewskiBELebwohlMSecukinumab in plaque psoriasis – results of two phase 3 trialsN Engl J Med2014371432633825007392
- GordonKBBlauveltAPappKAPhase 3 trials of ixekizumab in moderate-to-severe plaque psoriasisN Engl J Med2016375434535627299809