
Abstract:
Congenital hypothyroidism (CH), the most common pediatric endocrine disorder with an estimated prevalence of 1:2,000 to 1:4,000, is an under-recognized problem in countries without routine newborn screening (NBS) programs. Thyroid dysgenesis (TD) is the most common cause of primary CH accounting for approximately 85% of all cases; most of the remaining patients have dyshormonogenesis. Transient CH and CH with eutopic gland, are increasingly being identified after introduction of routine NBS. The clinical features of CH are often subtle resulting in delayed diagnosis and eventually poor intellectual outcome. In developed countries, detection by NBS and early initiation of treatment has largely eliminated the intellectual disability caused by this disorder. The lower screening thyroid stimulating hormone (TSH) cutoff and changes in birth demographics in some countries have been associated with an increase in the reported incidence of CH. However, the additional cases detected by the lower TSH cutoff tend to have either milder or transient hypothyroidism. Diagnosis of CH is made on the basis of serum concentrations of TSH and thyroxine (T4). Thyroid ultrasound, radionuclide scintigraphy, serum thyroglobulin (TG) levels and specific genetic tests help ascertaining the exact etiological diagnosis. Non-availability of later tests should not deter the pediatrician from initiation of treatment. Age at initiation of treatment and starting dose of levothyroxine are critical factors that determine the long-term outcome. Higher doses of levothyroxine at 10–15 μg/kg/day are required in infants, with titration based on T4 and TSH levels, which are repeated frequently. Coexistence of other congenital anomalies in children with CH adds to the morbidity. Approximately 70% of babies worldwide are not born in an area with an established NBS program and hence are not detected and treated early. Consequently, the economic burden of mental retardation due to CH remains a significant public health challenge in countries without NBS. The health burden owing to CH continues to be high even in countries with well-developed NBS.
Introduction
Congenital hypothyroidism (CH) is one of the leading causes of preventable mental and growth retardation, especially in underdeveloped and developing countries that do not routinely employ newborn screening (NBS). It has been broadly classified as primary (thyroid gland dysfunction) and secondary (pituitary gland dysfunction), permanent and transient types, and syndromic. Whatever the etiology, CH results in significant loss of productivity, generally considered to exceed the cost of a routine NBS program.Citation1 Approximately 28% of children with CH have intellectual disability (defined as IQ <70), with mean IQ of 85%, a leftward shift of one standard deviation.Citation2 Routine neonatal screening in developed countries has resulted in virtual disappearance of intellectual disability due to CH. The mean IQ has shown an increase by 10–20 points in patients with CH before and after introduction of screening programs in these countries.Citation2 Despite the documented impact of NBS programs since 1970s, lesser developed countries have failed to implement the same because of financial constraints, logistic issues, and poor commitment of policymakers. The incidence of CH varies widely from 1:2,000 to 1:4,000 in different countries and has shown a consistent increase over the past several decades. The disease burden remains very high in countries with huge populations and no NBS programs. Even in countries with well-developed NBS, the health burden due to CH continues to be high.Citation3
Epidemiology
Before the advent of NBS programs, the incidence of CH, usually detected after appearance of clinical features, was reported to range between 1:7,000 and 1:10,000.Citation4 With the NBS experience, the incidence is considered as 1:3,000 to 1:4,000 live births.Citation4 The differences in incidence are generally attributed to geographical and ethnic variations, but are more likely to be related to iodine deficiency or type of screening method used.Citation5 A very high incidence (1:914 to 1:1,985) has been reported from some countries with not so well-established NBS programs.Citation6,Citation7 In the USA, the incidence of CH has shown an increase (from approximately 1:4,100 to 1:2,350) over the past few decades.Citation8 The major reason for the increase is considered to be the inclusion of cases with transient hypothyroidism (TH).Citation8 A consistent increase (1:3,616 in 2005, 1:1,369 in 2006, and 1:1,030 in 2007) was noted in Brazil and was mainly attributed to reduction of neonatal thyroid stimulating hormone (TSH) value limits over time.Citation9 A high incidence (1:1,800 live births) was also found over a decade of usage of the NBS program in Greek Cypriot population.Citation10 A low incidence of 1:10,000 (after exclusion of TH) was recorded in France after about 2 decades of start of NBS.Citation11 Thus, the disease burden continues to be high worldwide.
Thyroid gland development in fetus
The thyroid gland is the first endocrine structure appearing during development. The median thyroid anlage, which forms the follicular cells of thyroid, appears on the floor of pharynx on day 20–22 of development and starts migrating caudally by day 26 to reach its definitive pretracheal position by day 48–50 of development. The parafollicular C cells arise from the paired lateral thyroid anlagen that develop from the fourth pharyngeal pouch by day 26 and give rise to two ultimobranchial bodies. Fusion of the two components occurs by day 44, following which terminal differentiation of thyroid follicular cells occurs. The histogenesis is complete by tenth week of gestation.Citation12 Expression of several genes that code for multiple thyroid (thyroid transcription factor-1 or TTF-1, also known as TITF1, NKX2.1, and T/EBP; TTF-2, also known as TITF2, FKHL15, and FOXE-1; PAX-8) and pituitary (PIT1, PROP1, LHX3, LHX4, HESX1) transcription factors are important for fetal gland development.Citation13 Recent data suggest that Tbx1–Fgf8 pathway in the pharyngeal mesoderm is a key size regulator of mammalian thyroid gland.Citation14
Triiodothyronine (T3) and thyroxine (T4) appear by 12 weeks of gestation and TSH by 10–12 weeks. Fetal thyroid axis does not function independently till midgestation, when transplacental T4 transfer is extremely important for fetal brain growth. For this reason, untreated hypothyroidism in the mother may result in poor neurodevelopmental outcome in the infant.Citation15 Maternal T4 is taken up by astrocytes and converted by type II monodeiodinase to T3 in brain, which is required for normal development of neocortex.Citation15
Etiology
CH may be permanent or transient. Permanent CH may result from primary or secondary causes. Primary causes include defects in thyroid gland development, deficiencies in thyroid hormone production, and defects of TSH binding or signal transduction. Secondary or central causes include defects of thyrotropin-releasing hormone (TRH) formation or binding, and TSH production. Peripheral hypothyroidism results from defects in thyroid hormone transport, metabolism, or resistance to thyroid hormone action.Citation16 CH patients having associated defects in other organ systems are classified as having syndromic hypothyroidism.
Data from NBS programs from developed countries have resulted in better understanding the etiology of CH.Citation17 An appropriate use of biochemistry, ultrasonography, and scintigraphy, supplemented with genetic analysis wherever available, allows an etiological diagnosis to be made in most cases. Establishing the exact etiology of CH is important in determining severity, clinical management, prognosis, and genetic counseling.Citation17 In the following sections, causes of permanent CH are discussed.
Thyroid dysgenesis
Thyroid dysgenesis (TD) is the most common cause of permanent CH and includes athyreosis, hypoplasia, hemiagenesis, and ectopic thyroid gland (ETG).Citation16 It accounts for approximately 85% of cases of permanent CH. Although most cases of TD are sporadic, higher prevalence in certain ethnic groups like Hispanics and Caucasians, in babies with Down syndrome, and familial occurrence in about 5% suggest the possibility of a role of genetic factors.Citation13,Citation18 A female preponderance in TD has been noted in previous studies.Citation19,Citation20 The sex differences have been suggested to modulate thyroid gland development probably by involving molecular mechanisms.Citation20 However, recent data do not indicate sexual dimorphism in TD.Citation21,Citation22 The observations that advanced maternal age is associated with TD need to be confirmed by further larger studies.Citation21,Citation22
The most common form (about two-third) of TD is ETG worldwide and occurs due to defective migration of the gland during development.Citation16 However, some recent reports indicate that the predominant form of TD is agenesis or hypoplasia.Citation22–Citation24 Ectopic gland may be located at sublingual, suprahyoid, or infrahyoid region in neck, locations other than midline of the neck, as well as aberrant locations in many other organs in the body.Citation25
Athyreosis or aplasia refers to the complete absence of thyroid tissue, usually indicated by absence of isotope uptake on technetium-99m pertechnetate scintigraphy. In some of these cases, high-resolution ultrasonography may identify small or normal-sized thyroid in orthotopic location indicating high TSH resistance. Combined use of thyroid scintigraphy and ultrasonography is therefore essential in correctly identifying true aplasia.Citation26 Also the thyroglobulin (TG) levels are found to be highly variable and suggest the presence of rudimentary thyroid tissue.Citation27 Hypoplasia refers to small-sized gland in normal location. This is identified by mild-to-moderate tracer uptake on scintiscan and a thyroid volume of less than the third percentiles of reference values on ultrasonography. Combined scanning is mandatory to determine hypoplasia, as several cases initially diagnosed as athyreosis by scintigraphy may have to be reclassified as hypoplasia.Citation26 Hemiagenesis is a rare form of TD that is more commonly reported from some countries.Citation22,Citation23 The higher prevalence is believed to be due to genetic factors resulting from frequent parental consanguinity.Citation23
Genes associated with TD include those causing syndromic CH (TTF-1, TTF-2, PAX-8, Gsα genes) and those causing nonsyndromic CH (TSHR gene).Citation13
Dyshormonogenesis
Dyshormonogenesis (DH) accounts for 10%–15% of patients with permanent CH.Citation16 The thyroid gland is eutopic and is often, but not always, enlarged. It is a heterogenic disorder caused by mutations in the genes that encode enzymes, membrane transporters, and a structural protein involved in thyroid hormone biosynthesis.Citation28 The sequential steps in thyroid hormone synthesis include trapping of iodine by sodium/iodide symporter (NIS), iodide efflux into follicular lumen by pendrin, organification of iodine and coupling by thyroperoxidase (TPO), hydrogen peroxide generation by dual oxidase 2, and intrathyroidal iodine recycling by dehalogenase.Citation28
Of all the biosynthetic defects, the most common is TPO deficiency that may result in total or partial iodide organification defect. A biallelic mutation in the TPO gene is present in majority of cases with permanent total iodide organification defects, while monoallelic defects are responsible for some cases.Citation29,Citation30 While virtually all patients with total iodide organification defects may have TPO gene mutations, the partial iodide organification defect is heterogeneous and includes defects in the hydrogen peroxide generator that drives TPO activity.Citation28
Mutations in the enzyme dual oxidase 2 (DUOX2 or THOX2) may cause deficient hydrogen peroxide generation and lead to total or partial organification defects.Citation16,Citation28 About 28 mutations have been reported after the first description in 2002.Citation28,Citation31 The majority of these are nonsense, frameshift, missense, or splice site mutations.Citation28 Recently, larger deletions of DUOX2 locus have been reported.Citation32 The expressivity of the DUOX2 gene defects depends on the genetic background and iodide intake, and may result in wide phenotypic variability.Citation28
Pendred syndrome (PDS) is characterized by a triad of hypothyroidism, goiter, and deafness caused by a defect in apical iodide efflux due to biallelic inactivating mutations in the SLC26A4 (PDS) gene.Citation33 About half of the patients with SLC26A4 defects do not have thyroid abnormalities.Citation28 Patients with PDS are often detected to have hypothyroidism when they present with deafness.Citation28
In TG deficiency, serum TG level is low and thyroid gland is enlarged.Citation28 About 40 inactivating mutations in the TG gene have been described since the first report.Citation34 Screening for TG gene mutations is usually undertaken after finding elevated TSH in a neonate with enlarged thyroid gland and absent or low serum TG level.Citation28
Iodide transport defect due to mutations in the NIS or SLC5A5 gene is a rare cause of DH. About 13 mutations of the SLC5A5 gene have been described since the molecular cloning of the gene in 1996.Citation35 Patients may be misdiagnosed as athyreosis if the goiter is absent or small, and TG concentrations are not measured, as the isotope uptake on scintigraphy is absent.Citation28
Defects of iodotyrosine deiodinase and TG action are extremely rare causes of DH.Citation16
TSH resistance
End-organ hormone resistance is usually caused by genetic defects of thyroid hormone receptors. Traditionally, the clinical phenotypes of TSH resistance due to inactivating TSHR mutations were thought to vary depending on the residual mutant receptor activity.Citation36 The two main phenotypes are classic (compensated or uncompensated resistance) and nonclassic TSH resistance.Citation36 Biallelic nonfunctional mutations produce thyroid hypoplasia with overt hypothyroidism, while hypomorphic mutations in at least one allele produce normal-sized thyroid gland with preserved thyroid functions.Citation36 Resistance may also occur in the absence of TSHR mutations. Rarer forms of TSH resistance include defects in TSH signaling (mutations in the α subunit of the stimulatory guanine nucleotide binding protein, Gsα that causes pseudohypoparathyroidism type 1a) and a dominantly inherited form with a locus on long arm of chromosome 15.Citation16,Citation36
Central hypothyroidism
Central CH is due to insufficient stimulation of thyroid gland by TSH, resulting from hypothalamic and/or pituitary dysfunction or defects in TSH production. This disorder is uncommon and usually occurs as a part of congenital hypopituitarism which in turn may be isolated or a component of other genetic syndromes. Familial hypopituitarism is caused by mutations in genes that regulate pituitary gland development which include HESX1, LHX3, LHX4, PIT1, and PROP1, and usually results in deficiencies of other pituitary hormones in addition to TSH deficiency. Specific gene defects causing isolated TSH deficiency are very rare and include mutations in the TSHβ subunit gene and TRH receptor gene.Citation16,Citation37
Defects of thyroid hormone transport, metabolism, or action
The peripheral defects of thyroid hormone metabolism are very rare causes of CH. These are associated with mutations of the gene encoding selenocysteine insertion sequence binding protein 2 (SBP2), the SBP2 gene.Citation13 A defect in the iodothyronine transporter, MCT8, results in CH and severe neurological deficits.Citation38 The defective transporter impairs the entry of iodothyronine into neurons resulting in elevation of T3, low T4, and normal TSH concentrations. First reported in boys with mental retardation, the disorder is also called Allan–Herndon–Dudley syndrome.Citation38 Permanent CH may also occur due to dominantly inherited mutations in genes encoding for thyroid hormone receptor beta, which results in resistance to thyroid hormone action.Citation39 The affected infants usually escape detection by NBS as thyroid hormone levels are mildly elevated without suppression of TSH.Citation39
Transient congenital hypothyroidism
Transient CH contributes significantly to the increasing incidence of CH as detected by NBS in many developed countries.Citation8,Citation40,Citation41 In a cohort of French children with CH, 38% were reported to have TH.Citation11 The following factors may result in TH:
Iodine deficiency: maternal iodine deficiency due to iodine-deficient diets, common in many European and Asian populations, puts infants, especially those born premature, at risk of developing TH.Citation11,Citation42 The condition usually lasts a few weeks after birth but may be prolonged. Some of these infants may require treatment.Citation42
Iodine excess: fetal or postnatal iodine excess may result in TH, especially in preterm babies, due to their inability to regulate the iodine uptake and reduced renal clearance.Citation42 Iodine excess may occur due to exposure to drugs like amiodarone, radiocontrast agents, and iodine-containing antiseptics in mother or infant.Citation42
Maternal TSHR-blocking antibodies: transfer of TSHR-blocking antibodies from mother to infant may result in neonatal hypothyroidism which lasts for 3–6 months and requires treatment in most cases.Citation42,Citation43 The scintigraphic findings suggest agenesis as there is no isotope uptake due to blocking of binding and action of TSH by antibodies.Citation43 However, the gland is normally visualized on ultrasonography. Persistence of antibodies in mother is a risk for recurrence of TH in future pregnancies.Citation44
Maternal ingestion of goitrogens: goitrogens are substances that interfere with the function of thyroid gland. Various maternal goitrogens include antithyroid drugs such as methimazole, carbimazole, and propylthiouracil, excessive consumption of vegetables in the Brassica genus (broccoli, brussel sprouts, cabbage, canola, cauliflower, mustard greens, radishes, and rapeseed) and other vegetables and fruits (spinach, cassava, peanuts, soybeans, strawberries, sweet potatoes, peaches, pears, and kale).Citation16 Recent data suggest that exposure to endocrine disrupters during pregnancy may also affect the fetal and neonatal thyroid function.Citation45
Mutations in DUOX2 and DUOXA2: both monoallelic and biallelic mutations in DUOX2 gene which is involved in production of hydrogen peroxide for thyroid peroxidation cause transient CH.Citation13
Isolated hyperthyrotropinemia: this is characterized by an elevated TSH but normal total and free T4. In some children, it is caused by transient immaturity of the hypothalamic-pituitary thyroid (HPT) axis. In others, inactivating mutations of TSHR may be associated with transient or permanent CH.Citation46 Follow-up of thyroid function is important as about one-third of such children can develop subclinical hypothyroidism.
Maternal hyperthyroidism: this may suppress the fetal TSH and lead to central hypothyroidism in the baby.Citation47 Free T4 concentrations in the serum are low, and TSH may be low or normal.
Prematurity and very low birth weight: these infants are at risk of developing hypothyroidism for a variety of reasons such as withdrawal of maternal–placental T4 transfer, HPT axis immaturity, developmental constraints on the synthesis and peripheral metabolism of iodothyronines, and iodine deficiency.Citation42
Steroids and dopamine: use of these drugs in critically ill neonates may cause central hypothyroidism by inhibition of TSH release.
Isolated hypothyroxinemia: this is characterized by low concentrations of free T4, but normal concentrations of TSH, and a normal response of TSH to TRH stimulation. The condition is common in preterm and very low birth weight babies and may also occur secondary to nonthyroidal illness and/or drug usage in these babies.Citation42 Similar thyroid dysfunction may also be found in T4-binding globulin (TBG) deficiency, undernutrition, liver dysfunction, or recovery from sick euthyroid syndrome.
Liver hemangiomas: congenital liver hemangiomas produce large amounts of type 3 iodothyronine deiodinase and result in severe consumptive hypothyroidism.Citation48 Serum T4 levels are low due to its conversion to inactive reverse T3 (rT3) by deiodinases. Large amounts of T4 are usually required, particularly during the proliferative phase of hemangioma during infancy. Some authors believe that a combination of liothyronine and T4 may work better as the exogenous T4 is also converted to inactive rT3.Citation49 Resolution of hypothyroidism occurs with treatment or spontaneous involution of hemangioma but may recur with regrowth.Citation48
Hypothyroidism with eutopic thyroid gland
Changes in screening strategies for CH in recent years have led to an increased detection of mild forms of CH associated with eutopic thyroid gland.Citation11 A normal thyroid gland is found in approximately 20% of the cases detected on neonatal screening. On reevaluation, an almost equal proportion of patients may show TH, permanent CH, or persistent hyperthyrotropinemia.Citation11,Citation50 Many of these cases may have iodine organification defects, DUOX2 mutations, or TSH resistance.Citation43 In the latter situation, there may be hypoplasia of thyroid gland. Other causes are iodine deficiency or overload and transplacentally transmitted antithyroid drugs or TSHR antibodies. To establish transient or permanent CH, a reevaluation is mandatory at about 3 years of age or earlier in those requiring lower T4 doses (<3.25 μg/kg).Citation50,Citation51
Sick euthyroid syndrome in critically ill neonates
Neonates with severe illness may show low T3 or T4 and elevated rT3 with normal TSH levels. Commonly known as “sick euthyroid syndrome”, it may be physiological or adaptive response to decrease the metabolic rate to divert energy expenditure to respond to acute illness.Citation52 It may be pathological as low T3 levels are due to inappropriately low TSH and TRH secretion.Citation52 The role of T4 replacement in such neonates is uncertain. Only when TSH elevation is more than 15–20 mU/L or TSH remains high for a month or longer, is treatment needed, while other cases may only need serial determinations of free T4 and TSH.Citation52
Special issues regarding preterm neonates
Thyroid dysfunction is common in premature neonates. It is due to immature HPT axis, postnatal depletion of T4 reserves, increased incidence of severe systemic illnesses, and exposure to drugs such as dopamine, steroids, topical iodine, commonly used in neonatal intensive care units (ICUs).Citation53 The HPT axis maturation continues through 35–40 weeks gestation.Citation12 The evolution of hypothyroid state in preterm babies remains difficult to predict. Approximately half of these babies may have TH, while the rest may either have permanent CH or persistent hyperthyrotropinemia.Citation53 Delayed TSH elevation in most preterm babies by early screening based on TSH-first approach often makes detection difficult. Additionally, premature babies tend to have elevated rT3, low normal T3, T4, and normal TSH concentrations mimicking the hormone profile of sick euthyroid syndrome.Citation53 For these reasons, a periodic reevaluation of thyroid functions should be done to avoid overtreatment.
Hypothyroidism in neonates with Down syndrome
Thyroid dysfunction is a well-known feature of Down syndrome, and its frequency increases with age.Citation54 Majority of the patients have subclinical hypothyroidism.Citation55 The etiology of CH in Down syndrome is still under debate. Absence of goiter indicates that DH is unlikely. Approximately one-third of the patients show mild abnormalities on scintigraphy.Citation55 A high prevalence of thyroid hypoplasia was found in a recent study, but these findings need to be confirmed on larger patient populations.Citation56 There is an underemphasis on diagnosis of hypothyroidism in children with Down syndrome in national programs. The decision to start treatment is often difficult as thyroid dysfunction is usually mild, and there are no clear recommendations for T4 supplementation.Citation54 In countries with well-established screening programs, treatment is usually begun in the neonatal period.Citation57 In other countries, delayed initiation of treatment is common due to absence of standardized national screening protocols and delayed diagnosis.Citation55 Recent data suggest that early T4 supplementation improves psychomotor and physical development in these children.Citation58,Citation59
Modes of inheritance in congenital hypothyroidism
Familial occurrence has been noted in TD and DH. However, mutations are detected in only about 2% of cases of TD, but in majority of patients with DH.Citation13,Citation28 The modes of inheritance are largely unknown. Recent studies suggest autosomal recessive inheritance and familial dominant occurrence of TD.Citation60,Citation61 An increased frequency of minor abnormalities of the thyroid development has been described in first-degree relatives of patients with TD.Citation18,Citation62 Although the available data are scarce, it has become apparent that TD, at least in a subset of patients (<5%), is caused by molecular genetic defects, which in principle are inheritable.Citation62 In majority of patients, however, a non-Mendelian genetic defect or somatic mutations have to be considered as possible mechanisms. The mode of inheritance of DH is considered autosomal recessive in most cases.Citation28 A high recurrence risk of CH is also seen in babies born to mothers with high levels of thyrotropin receptor blocking antibody (TRB-Ab).Citation16
Clinical features
The clinical features of CH are often subtle, and many newborn infants remain undiagnosed during the critical treatment-sensitive newborn period. The evolution of symptoms may also be very slow. Hence, early detection is only possible through NBS. Few of the common symptoms in infants are postmaturity, birth weight exceeding 90%, prolonged jaundice, feeding difficulties, abnormal cry, constipation, and lethargy. Examination may show macroglossia, cold or mottled skin, hypothermia, edema, wide fontanel, open sutures, flat nasal bridge, protuberant abdomen, umbilical hernia, delayed reflexes, and hypotonia. Presence of goiter indicates DH.
During childhood, patients may have short stature, decreased intelligence, gross/fine motor incoordination, speech disorders, attention deficit, and sensory neural deafness.Citation63 Patients with long-standing untreated CH may present with proximal muscle weakness associated with calf hypertrophy, also known as Kocher–Debre–Semelaigne syndrome.Citation64 Although delayed puberty is more common in CH, pseudoprecocious pubertal development, also called Van Wyk–Grumbach syndrome, may occur similar to children with untreated autoimmune thyroiditis.Citation65,Citation66 Other unusual features include prepubertal gynecomastiaCitation67 and menorrhagia.Citation68
Associated congenital anomalies
The prevalence of congenital malformations is increased in children with CH in comparison with the normal child population.Citation69 The prevalence is usually less than 20% of children with CH.Citation69,Citation70 An unusually high prevalence of 59% was noted in a study from India.Citation71 The extrathyroidal anomalies are more commonly seen in patients with TD than in other forms of CH. Majority of the malformations are cardiac, but malformations of genitourinary, gastrointestinal, skeletal, renal, and urogenital systems have also been described.Citation69–Citation71 Developmental dysplasia of hip, spina bifida occulta, spiky hair, cleft palate, and multiple malformations are also reported.Citation69–Citation71 The most common cardiac anomaly is atrial septal defect as opposed to ventricular septal defect in the general population.Citation69,Citation71 A close relationship of developing thyroid with developing heart has been proposed as the possible explanation for associated cardiac defects. Mutations in transcription factors and genes (NKX.2 and HEX) involved in the development of both cardiovascular system and thyroid may be responsible in some cases.Citation72,Citation73
Mutations of several genes that cause CH may also result in distinct clinical phenotypes. The presence of inner ear malformations is essential for diagnosis of PDS due to PDS gene mutations.Citation74 However, some patients may not have inner ear malformations, and mutations in the PDS gene.Citation75 This condition has been termed “pseudo-Pendred syndrome” and is considered to be of autoimmune origin. Additional mutational analysis for TPO gene has been suggested in these patients.Citation75,Citation76 Bamforth–Lazarus syndrome, characterized by TD, choanal atresia, cleft palate, and spiky hair, is caused by mutation of the gene for TTF-2, which is expressed in the tongue, epiglottis, and palate in addition to thyroid gland.Citation77 Mutations in NKX 2.1 cause CH along with respiratory distress and neurologic problems such as benign hereditary chorea and ataxia.Citation78 Another syndrome that features CH with sensorineural hearing loss and multiple other anomalies is known as Johanson–Blizzard syndrome.Citation79
Diagnosis and other evaluation
The diagnosis of CH is made after detection by NBS in most of the developed countries.Citation80 These regions include Japan, Australia, USA, Canada, Mexico, Western Europe, New Zealand, and Israel. Many countries in Eastern Europe, Asia, and Africa are in the process of developing NBS programs. Without neonatal screening, the diagnosis is usually delayed and made only after development of clinical features of CH. A complete diagnostic evaluation includes detection of CH by NBS, followed by confirmation by repeat thyroid function testing, and use of available modalities to determine the exact underlying etiology.
Neonatal screening and confirmatory tests
Blood from a heel prick (or cord) collected on special filter paper cards usually between 2 and 5 days of age is sent for either initial TSH or initial T4 test, with a follow-up TSH test according to the local NBS program.Citation80 With either approach, a diagnosis is made in most infants with primary CH. Some NBS programs measure both T4 and TSH so as to not miss any cases with central CH, mild hypothyroidism, and those with delayed TSH rise. Many programs employ age-related cutoffs for T4 and TSH and follow an algorithm as suggested in .Citation16 Each program must develop its own T4 and TSH cutoff for recall of infants with abnormal test results. Recent guidelines suggest immediate institution of therapy if venous TSH level is more than 20 mU/L or if venous free T4 levels are below norms for age.Citation81 If free T4 levels are normal and TSH values range from 6 mU/L to 20 mU/L, a repeat evaluation is mandated after 3 weeks of age to reestablish abnormal T4 and TSH levels and to obtain a definitive diagnosis and decide about initiation of therapy.Citation81 Most confirmatory serum tests are done after 2 weeks of life, when the upper TSH range falls to approximately 10 mU/L. The approximate normal reference ranges at 2–4 weeks of life are as follows: serum free T4 =10–26 pmol/L, total T4 =90–206 nmol/L, and TSH <10 mU/L.Citation16 The diagnosis of primary CH is confirmed by the finding of an elevated serum TSH level and a low free T4 or total T4. Subclinical primary hypothyroidism is diagnosed if free T4 or total T4 concentrations are normal, but TSH is elevated.Citation81 A low T4, with either a low or an “inappropriately normal” TSH level confirms central or secondary CH. Infants diagnosed to have central CH must undergo evaluation for other pituitary hormone deficiencies. Infants showing a low serum total T4, but a normal free T4 and normal TSH level may have TBG deficiency which can be confirmed by a low serum TBG level.Citation80,Citation81
Figure 1 Diagnostic algorithm for congenital hypothyroidism.
Adapted from Rastogi MV, LaFranchi SH. Congenital hypothyroidism. Orphanet J Rare Dis. 2010;5:17.Citation16
Abbreviations: T4, thyroxine; TSH, thyroid stimulating hormone; T3, triiodothyronine; CH, congenital hypothyroidism; HPT, hypothalamic-pituitary thyroid; TSHβ, TSH β subunit; MRI, magnetic resonance imaging; yr, years.
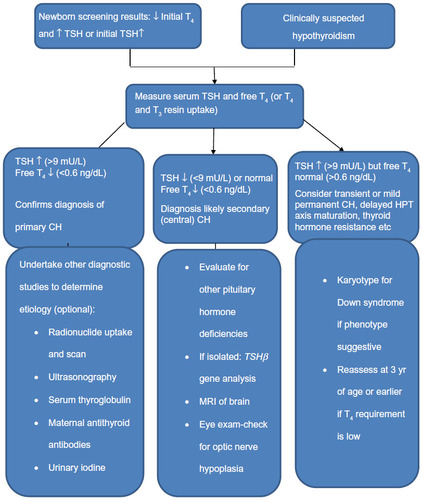
Evaluation for the underlying etiology
Several diagnostic studies may be undertaken to determine the precise etiology of CH, including thyroid ultrasonography, thyroid radionuclide uptake and scan, serum TG measurement, antithyroid antibody determinations, and measurement of urinary iodine.
All these studies are optional because their results do not influence treatment decisions, which are based on serum thyroid function tests. These studies help in differentiating transient from permanent CH, lead to performing specific genetic tests, especially in patients with DH, and guide genetic counseling in familial forms.
Thyroid ultrasonography
Due to its easy availability at most centers, ultrasound is usually the first imaging modality in infants with CH. It is rapid, noninvasive, cheap, and requires no special preparation. The disadvantages are operator dependency, lesser sensitivity for the most common variant, ie, TD, and inability to provide information on thyroid function.Citation82 Introduction of high-resolution and color Doppler ultrasonography has significantly increased the ability to pick ETG.Citation82 However, color Doppler ultrasonography is based on increased vascularity and may falsely identify vascular structures like thymus and lymph nodes as thyroid gland.Citation82
Thyroid scintigraphy
Thyroid radionuclide uptake and scan performed with either iodine-123 (I-123) or sodium pertechnetate 99m (Tc-99m) accurately defines all variants of TD.Citation83 If the scan shows no radionuclide uptake, it needs to be confirmed with ultrasonogarphy. Combined scanning using high-resolution ultrasonography and scintigraphy is considered more informative compared to single scanning in the diagnosis of CH.Citation26 In addition to thyroid aplasia, absent uptake is seen with TSH suppression from levothyroxine administration, cystic degeneration of thyroid gland, TSHβ gene mutations, TSHR mutations, iodide trapping defects, and with maternal TSHR- blocking antibodies. Ultrasonography and other additional studies such as estimation of serum TG or TSHR antibodies are carried out to differentiate these etiologies from aplasia.Citation16 Increased radionuclide uptake in an eutopic large gland is seen in biosynthetic defects beyond trapping. In these cases, a rapid discharge of iodide from the gland after high dose of perchlorate (perchlorate discharge test) suggests defective oxidation and organification of iodide. Subsequent genetic analysis may identify TPO gene mutations in these cases.
Thyroid scintigraphy is performed before initiation of levothyroxine during infancy, if this is readily available. Delaying initiation of therapy for carrying out scintigraphy is unjustified during infancy. In such cases, scan is performed after interruption of therapy after 3 years of age when brain growth has almost completed. Postponement of scintigraphy can be avoided with use of recombinant TSH which allows the scan to be performed without interruption of treatment.Citation84
Serum thyroglobulin determination
Serum TG concentration is an indirect marker of thyroid gland volume. The levels are lowest in thyroid aplasia, intermediate in ectopia, and high in large glands. Serum TG is absent in true aplasia if measured within few weeks after birth. However, some patients identified as having aplasia on scintigraphy may show detectable TG levels indicating the presence of functional thyroid tissue.Citation27 Also, patients with absent uptake on scintiscan and increased TG concentrations may have a trapping defect, maternal TSHR antibodies, or TSH receptor inactivating mutation.Citation60
Antithyroid antibodies
Determination of TSHR-blocking antibodies is recommended only if a mother with known autoimmune thyroid disease and with a previous child diagnosed with transient CH is pregnant again.Citation16 Transient CH caused by maternal transfer of TSHR-blocking antibodies is rare (approximately 1:100,000 newborns), despite common occurrence (about 5%) of maternal autoimmune thyroid disease.Citation16
24-hour urinary iodine
This determination is undertaken if iodine excess or deficiency is suspected to be the cause of CH, which is transient in almost all cases. The normal level in neonates is 50–100 μg.
Genetic tests
Genetic analysis is only undertaken if the other diagnostic studies indicate a specific defect. These are more useful in cases of DH because most of the biosynthetic defects are well characterized and associated with known gene mutations. In cases of TD, mutations are identified in only about 2%–5% of cases.Citation13,Citation59
Treatment
Timely initiation of T4 replacement in children with CH is crucial for neurological outcome. The goal of therapy is to achieve a growth and mental development close to the genetic potential of patients. A rapid normalization and maintenance of normal thyroid functions is important to achieve these goals.
Levothyroxine is the treatment of choice. Replacement with T3 is not needed as most of the T3 in brain is formed by local deiodination of T4. Treatment with only levothyroxine has been shown to normalize serum T3 and is considered sufficient.Citation85 Combination of T3 and T4 does not provide any additional benefits. In all countries except Europe, only levothyroxine tablets are approved for use, as suspensions may result in unreliable dosing.Citation16 The efficacy of liquid and tablet forms was, however, found to be similar in one study.Citation86 The absorption of medication is best on empty stomach and may be impaired by gastrointestinal disorders like celiac disease, inflammatory bowel disease, lactose intolerance, Helicobacter pylori infection, and atrophic gastritis as well as drugs such as proton pump inhibitors, aluminum-containing antacids, sucralfate, cholestyramine and other resins, ferrous sulfate, calcium, phosphate binders, fiber supplements, and soy protein formulas.Citation87
The dose and timing of levothyroxine therapy are crucial to the neurologic outcome in CH. Behavioral and cognitive testing scores are significantly lower when normalization of T4 takes more than 2 weeks.Citation88,Citation89 A high starting dose of 10–15 μg/kg/day is recommended by the American Academy of Pediatrics and the European Society for Paediatric Endocrinology (ESPE).Citation81 Additionally, the delay in TSH normalization is also related to neurodevelopmental outcome.Citation16 The T4 levels normalize in first 3 days of initiation of treatment, while TSH levels take up to 1 month for normalization even though a 50% decrease occurs in first 3–4 days.Citation90 The desired target levels for serum total T4, free T4, and TSH are 10–16 μg/dL (130–206 nmol/L), 1.4–2.3 ng/dL (18–30 pmol/L), and <5 mU/L, respectively.Citation81,Citation85 Care should be taken to avoid under- as well as overtreatment. Children having T4 levels <10 μg/dL and TSH >15 mU/L during the first year of life have been shown to have lower IQs than those with T4 levels >10 μg/dL.Citation91 Variations in TSH are also related to verbal IQs and other developmental indices.Citation91 Overtreatment may result in hyperactivity, delinquency, and aggression.Citation92
Traditionally, T4 administration is done as a once-daily dose.Citation16,Citation81,Citation88 The efficacy of this daily-dose regimen is proven. However, an alternate day dosing schedule which offers flexibility and choice of administration to caregivers has been used effectively to maintain euthyroidism in children with CH.Citation93
The ESPE guidelines, endorsed by many other endocrine societies of Asia-Pacific region, recommend first follow-up examination 1–2 weeks after start of treatment, every 2 weeks until complete normalization of TSH concentration, every 1–3 months till 12 months of age, every 2–4 months from 1 to 3 years of age, and every 3–12 months thereafter.Citation91 All children confirmed to have permanent CH need lifelong treatment. Serial TSH values and need for increasing levothyroxine dose with age are indicators of permanence of CH. In children suspected to have transient CH, a trial of discontinuation of therapy should be given at about 3 years of age or earlier to decide about further continuation of therapy.Citation42,Citation43,Citation51
Outcome
Factors known to influence the neurodevelopmental outcome in children with CH are the age at initiation of treatment, starting dose of levothyroxine, severity of hypothyroidism, serum T4 concentrations in the first 2 years of life, and compliance to therapy.Citation16 Early diagnosis and treatment of CH have significant impact on the long-term neurodevelopmental outcome of the affected children. Majority of the studies on outcome conducted during the pre-NBS era suggested an inverse relationship between the age at diagnosis and IQ outcome.Citation16 A study from Auckland on children detected through NBS reported normal intellectual and motor development after prompt normalization of thyroid function using initial high-dose T4 and frequent monitoring and dose alterations in first 2 years of life.Citation94 However, recent data from Netherlands found no correlations between starting day of treatment and developmental outcome in a cohort of toddlers who were started on T4 on day 9 of life.Citation95 As a group, the initial T4 concentration and initial T4 dose were found to be weak predictors for developmental outcome. However, the subgroup of children with severe CH was documented to be at risk for motor and cognitive problems, probably due to the consequence of prenatal hypothyroid state or thyroid hormone deficiency in early neonatal life.Citation95 After early initiation of therapy in neonatal life, careful monitoring of thyroid functions throughout childhood and adulthood has been suggested to avoid numerous problems related to sensorimotor, cognitive, behavior, socioeducational, growth, puberty, fertility, body composition, bone health, metabolic, and cardiovascular health in these patients.Citation96,Citation97
Conclusion
In children, CH is the most common congenital endocrine disorder and preventable cause of mental retardation. The incidence of CH has shown an increase over the past few decades. The etiology of TD, which is the most common cause of CH, is largely unknown. The optimum long-term outcome depends on an early detection through NBS, rapid normalization, and careful monitoring of thyroid functions throughout childhood and adulthood. There is an urgent need to implement NBS programs in all countries to prevent significant health burden due to CH.
Disclosure
The authors report no conflicts of interest in this work.
References
- Centers for Disease Control and Prevention (CDC). Economic costs associated with mental retardation, cerebral palsy, hearing loss, and vision impairment – United States, 2003. MMWR Morb Mortal Wkly Rep. 2004;53(3):57–59.
- Grosse SD, Van Vliet G. Prevention of intellectual disability through screening for congenital hypothyroidism: how much and at what level? Arch Dis Child. 2011;96(4):374–379.
- Van Vliet G, Grosse SD. The continuing health burden of congenital hypothyroidism in the era of neonatal screening. J Clin Endocrinol Metab. 2011;96(6):1671–1673.
- Alm J, Larsson A, Zetterström R. Congenital hypothyroidism in Sweden. Incidence and age at diagnosis. Acta Paediatr Scand. 1978;67(1):1–3.
- Klett M. Epidemiology of congenital hypothyroidism. Exp Clin Endocrinol Diabetes. 1997;105(Suppl 4):19–23.
- Prabhu SR, Mahadevan S, Jagadeesh S, Suresh S. Congenital hypothyroidism: recent Indian data. Indian J Endocrinol Metab. 2015;19:436–437.
- Sareen N, Pradhan R. Need for neonatal screening program in India: a national priority. Indian J Endocrinol Metab. 2015;19(2):204–220.
- Hinton CF, Harris KB, Borgfeld L, et al. Trends in incidence rates of congenital hypothyroidism related to select demographic factors: data from the United States, California, Massachusetts, New York, and Texas. Pediatrics. 2010;125(Suppl 2):S37–S47.
- Botler J, Camacho LA, Cruz MM. Phenylketonuria, congenital hypothyroidism and haemoglobinopathies: public health issues for a Brazilian newborn screening program. Cad Saude Publica. 2012;28(9):1623–1631.
- Skordis N, Toumba M, Savva SC, et al. High prevalence of congenital hypothyroidism in the Greek Cypriot population: results of the neonatal screening program 1990–2000. J Pediatr Endocrinol Metab. 2005;18(5):453–461.
- Gaudino R, Garel C, Czernichow P, Léger J. Proportion of various types of thyroid disorders among newborns with congenital hypothyroidism and normally located gland: a regional cohort study. Clin Endocrinol (Oxf). 2005;62(4):444–448.
- Hill MA. Embryology Endocrine – Thyroid Development. 2015, Available from: https://embryology.med.unsw.edu.au/embryology/index.php/Endocrine_-_Thyroid_Development. Accessed April 1, 2015.
- Park S, Chatterjee V. Genetics of congenital hypothyroidism. J Med Genet. 2005;42:379–389.
- Lania G, Zhang Z, Huynh T, et al. Early thyroid development requires a Tbx1-Fgf8 pathway. Dev Biol. 2009;328(1):109–117.
- Stenzel D, Huttner WB. Role of maternal thyroid hormones in the developing neocortex and during human evolution. Front Neuroanat. 2013;7:19.
- Rastogi MV, LaFranchi SH. Congenital hypothyroidism. Orphanet J Rare Dis. 2010;5:17.
- Dias VM, Campos AP, Chagas AJ, Silva RM. Congenital hypothyroidism: etiology. J Pediatr Endocrinol Metab. 2010;23(8):815–826.
- Sindhuja L, Dayal D, Sodhi KS, Sachdeva N, Bhattacharya A. Thyroid dysfunction and developmental anomalies in first degree relatives of children with thyroid dysgenesis: data from a developing country. World J Pediatr. In press 2015.
- Narumi S, Muroya K, Asakura Y, Adachi M, Hasegawa T. Transcription factor mutations and congenital hypothyroidism: systematic genetic screening of a population-based cohort of Japanese patients. J Clin Endocrinol Metab. 2010;95:1981–1985.
- Eugène D, Djemli A, Van Vliet G. Sexual dimorphism of thyroid function in newborns: with congenital hypothyroidism. J Clin Endocrinol Metab. 2005;90:2696–2700.
- Dayal D, Sindhuja L, Bhattacharya A, Bharti B. Advanced maternal age in Indian children with thyroid dysgenesis. Clin Pediatr Endocrinol. 2015;24(2):59–62.
- Kirmizibekmez H, Güven A, Yildiz M, Cebeci AN, Dursun F. Developmental defects of the thyroid gland: relationship with advanced maternal age. J Clin Res Pediatr Endocrinol. 2012;4(2):72–75.
- Hashemipour M, Ghasemi M, Hovsepian S, et al. Etiology of congenital hypothyroidism in Isfahan: Does it different? Adv Biomed Res. 2014;3:21.
- Dayal D, Sindhuja L, Bhattacharya A, Sodhi KS, Sachdeva N. Agenesis and not ectopia is common in North Indian children with thyroid dysgenesis. Indian J Endocrinol Metab. 2014;18(Suppl S1):97–99.
- Guerra G, Cinelli M, Mesolella M, et al. Morphological, diagnostic and surgical features of ectopic thyroid gland: a review of literature. Int J Surg. 2014;12(Suppl 1):S3–S11.
- Perry RJ, Maroo S, Maclennan AC, Jones JH, Donaldson MD. Combined ultrasound and isotope scanning is more informative in the diagnosis of congenital hypothyroidism than single scanning. Arch Dis Child. 2006;91:972–976.
- Djemli A, Fillion M, Belgoudi J, et al. Twenty years later: a reevaluation of the contribution of plasma thyroglobulin to the diagnosis of thyroid dysgenesis in infants with congenital hypothyroidism. Clin Biochem. 2004;37(9):818–822.
- Grasberger H, Refetoff S. Genetic causes of congenital hypothyroidism due to dyshormonogenesis. Curr Opin Pediatr. 2011;23(4):421–428.
- Abramowicz MJ, Targovnik HM, Varela V, et al. Identification of a mutation in the coding sequence of the human thyroid peroxidase gene causing congenital goiter. J Clin Invest. 1992;90(4):1200–1204.
- Fugazzola L, Cerutti N, Mannavola D, et al. Monoallelic expression of mutant thyroid peroxidase allele causing total iodide organification defect. J Clin Endocrinol Metab. 2003;88(7):3264–3271.
- Moreno JC, Bikker H, Kempers MJ, et al. Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. N Engl J Med. 2002;347(2):95–102.
- Hulur I, Hermanns P, Nestoris C, et al. A single copy of the recently identified dual oxidase maturation factor (DUOXA) 1 gene produces only mild transient hypothyroidism in a patient with a novel biallelic DUOXA2 mutation and monoallelic DUOXA1 deletion. J Clin Endocrinol Metab. 2011;96(5):E841–E845.
- Everett LA, Glaser B, Beck JC, et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet. 1997;17(4):411–422.
- Ieiri T, Cochaux P, Targovnik HM, et al. A 3′ splice site mutation in the thyroglobulin gene responsible for congenital goiter with hypothyroidism. J Clin Invest. 1991;88(6):1901–1905.
- Spitzweg C, Morris JC. Genetics and phenomics of hypothyroidism and goiter due to NIS mutations. Mol Cell Endocrinol. 2010;322(1–2):56–63.
- Narumi S, Hasegawa T. TSH resistance revisited (review). Endocr J. 2015;62(5):393–398.
- Gupta V, Lee M. Central hypothyroidism. Indian J Endocrinol Metab. 2011;15(Suppl 2):S99–S106.
- Friesema EC, Jansen J, Heuer H, Trajkovic M, Bauer K, Visser TJ. Mechanisms of disease: psychomotor retardation and high T3 levels caused by mutations in monocarboxylate transporter 8. Nat Clin Pract Endocrinol Metab. 2006;2(9):512–523.
- Olateju TO, Vanderpump MP. Thyroid hormone resistance. Ann Clin Biochem. 2006;43(Pt 6):431–440.
- Parks JS, Lin M, Grosse SD, et al. The impact of transient hypothyroidism on the increasing rate of congenital hypothyroidism in the United States. Pediatrics. 2010;125(Suppl 2):S54–S63.
- Oren A, Wang MK, Brnjac L, Mahmud FH, Palmert MR. Mild neonatal hyperthyrotrophinaemia: 10-year experience suggests the condition is increasingly common but often transient. Clin Endocrinol (Oxf). 2013;79(6):832–837.
- Bhavani N. Transient congenital hypothyroidism. Indian J Endocrinol Metab. 2011;15(Suppl 2):S117–S120.
- Yang RL, Zhu ZW, Zhou XL, Zhao ZY. Treatment and follow-up of children with transient congenital hypothyroidism. J Zhejiang Univ Sci B. 2005;6(12):1206–1209.
- Matsuura N, Yamada Y, Nohara Y, et al. Familial neonatal transient hypothyroidism due to maternal TSH-binding inhibitor immunoglobulins. N Engl J Med. 1980;303(13):738–741.
- Li C, Cheng Y, Tang Q, et al. The association between prenatal exposure to organochlorine pesticides and thyroid hormone levels in newborns in Yancheng, China. Environ Res. 2014;129:47–51.
- Zung A, Tenenbaum-Rakover Y, Barkan S, et al. Neonatal hyperthyrotropinemia: population characteristics, diagnosis, management and outcome after cessation of therapy. Clin Endocrinol (Oxf). 2010;72:264–271.
- Papendieck P, Chiesa A, Prieto L, Gruñeiro-Papendieck L. Thyroid disorders of neonates born to mothers with Graves’ disease. J Pediatr Endocrinol Metab. 2009;22(6):547–553.
- Bessho K, Etani Y, Ichimori H, et al. Increased type 3 iodothyronine deiodinase activity in a regrown hepatic hemangioma with consumptive hypothyroidism. Eur J Pediatr. 2010;169(2):215–221.
- Peters C, Langham S, Mullis PE, Dattani MT. Use of combined liothyronine and thyroxine therapy for consumptive hypothyroidism associated with hepatic haemangiomas in infancy. Horm Res Paediatr. 2010;74(2):149–152.
- Rabbiosi S, Vigone MC, Cortinovis F, et al. Congenital hypothyroidism with eutopic thyroid gland: analysis of clinical and biochemical features at diagnosis and after re-evaluation. J Clin Endocrinol Metab. 2013;98(4):1395–1402.
- Cho MS, Cho GS, Park SH, Jung MH, Suh BK, Koh DG. Earlier re-evaluation may be possible in pediatric patients with eutopic congenital hypothyroidism requiring lower L-thyroxine doses. Ann Pediatr Endocrinol Metab. 2014;19(3):141–145.
- Farwell AP. Nonthyroidal illness syndrome. Curr Opin Endocrinol Diabetes Obes. 2013;20(5):478–484.
- Vigone MC, Caiulo S, Di Frenna M, et al. Evolution of thyroid function in preterm infants detected by screening for congenital hypothyroidism. J Pediatr. 2014;164(6):1296–1302.
- King K, O’Gorman C, Gallagher S. Thyroid dysfunction in children with Down syndrome: a literature review. Ir J Med Sci. 2014;183(1):1–6.
- Dayal D, Jain P, Panigrahi I, Bhattacharya A, Sachdeva N. Thyroid dysfunction in Indian children with Down syndrome. Indian Pediatr. 2014;51(9):751–752.
- Cebeci AN, Güven A, Yildiz M. Profile of hypothyroidism in Down’s syndrome. J Clin Res Pediatr Endocrinol. 2013;5(2):116–120.
- Bull MJ; Committee on Genetics. Health supervision for children with Down syndrome. Pediatrics. 2011;128(2):393–406.
- van Trotsenburg AS, Vulsma T, van Rozenburg-Marres SL, et al. The effect of thyroxine treatment started in the neonatal period on development and growth of two-year-old Down syndrome children: a randomized clinical trial. J Clin Endocrinol Metab. 2005;90(6):3304–3311.
- Kowalczyk K, PukajŁo K, Malczewska A, Król-Chwastek A, Barg E. L-thyroxine therapy and growth processes in children with Down syndrome. Adv Clin Exp Med. 2013;22(1):85–92.
- Gagné N, Parma J, Deal C, Vassart G, Van Vliet G. Apparent congenital athyreosis contrasting with normal plasma thyroglobulin levels and associated with inactivating mutations in the thyrotropin receptor gene: are athyreosis and ectopic thyroid distinct entities? J Clin Endocrinol Metab. 1998;83(5):1771–1775.
- Castanet M, Polak M, Bonaïti-Pellié C, Lyonnet S, Czernichow P, Léger J. Nineteen years of national screening for congenital hypothyroidism: familial cases with thyroid dysgenesis suggest the involvement of genetic factors. J Clin Endocrinol Metab. 2001;86(5):2009–2014.
- Léger J, Marinovic D, Garel C, Bonaïti-Pellié C, Polak M, Czernichow P. Thyroid developmental anomalies in first degree relatives of children with congenital hypothyroidism. J Clin Endocrinol Metab. 2002;87(2):575–580.
- Nandi-Munshi D, Taplin CE. Thyroid-related neurological disorders and complications in children. Pediatr Neurol. 2015;52(4):373–382.
- Tashko V, Davachi F, Baboci R, Drishti G, Hoxha P. Kocher-Debré Sémélaigne syndrome. Clin Pediatr (Phila). 1999;38(2):113–115.
- Asami T, Kikuchi T, Kamimura T, Kinoshita S, Uchiyama M. Precocious puberty in a girl with congenital hypothyroidism receiving continuous L-thyroxine-replacement therapy. Pediatr Int. 2001;43(1):87–90.
- Sharma D, Dayal D, Gupta A, Saxena A. Premature menarche associated with primary hypothyroidism in a 5.5-year-old girl. Case Rep Endocrinol. 2011;2011:678305.
- Dayal D, Bhalla AK, Sachdeva N. A boy with prepubertal gynecomastia, hyperprolactinemia, and hypothyroidism. J Pediatr Endocr Metab 2013;26(3–4):357–360.
- Sukumar SP, Balachandran K, Jayakumar, et al. Congenital hypothyroidism – an usual suspect at an unusual age: a case series. Indian J Endocrinol Metab. 2013;17(Suppl 1):S184–S187.
- Kreisner E, Neto EC, Gross JL. High prevalence of extrathyroid malformations in a cohort of Brazilian patients with permanent primary congenital hypothyroidism. Thyroid. 2005;15(2):165–169.
- Kumar J, Gordillo R, Kaskel FJ, Druschel CM, Woroniecki RP. Increased prevalence of renal and urinary tract anomalies in children with congenital hypothyroidism. J Pediatr. 2009;154(2):263–266.
- Reddy PA, Rajagopal G, Harinarayan CV, et al. High prevalence of associated birth defects in congenital hypothyroidism. Int J Pediatr Endocrinol. 2010;2010:940980.
- Dentice M, Cordeddu V, Rosica A, et al. Missense mutation in the transcription factor NKX2-5: a novel molecular event in the pathogenesis of thyroid dysgenesis. J Clin Endocrinol Metab. 2006;91(4):1428–1433.
- Thomas PQ, Brown A, Beddington RS. Hex: a homeobox gene revealing peri-implantation asymmetry in the mouse embryo and an early transient marker of endothelial cell precursors. Development. 1998;125(1):85–94.
- Soh LM, Druce M, Grossman AB, et al. Evaluation of genotype-phenotype relationships in patients referred for endocrine assessment in suspected Pendred syndrome. Eur J Endocrinol. 2015;172(2):217–226.
- Kara C, Kiliç M, Uçaktürk A, Aydin M. Congenital goitrous hypothyroidism, deafness and iodide organification defect in four siblings: Pendred or pseudo-Pendred syndrome? J Clin Res Pediatr Endocrinol. 2010;2(2):81–84.
- Pfarr N, Borck G, Turk A, et al. Goitrous congenital hypothyroidism and hearing impairment associated with mutations in the TPO and SLC26A4/PDS genes. J Clin Endocrinol Metab. 2006;91(7):2678–2681.
- Clifton-Bligh RJ, Wentworth JM, Heinz P, et al. Mutation of the gene encoding human TTF-2 associated with thyroid agenesis, cleft palate and choanal atresia. Nat Genet. 1998;19(4):399–401.
- Ferrara AM, De Michele G, Salvatore E, et al. A novel NKX2.1 mutation in a family with hypothyroidism and benign hereditary chorea. Thyroid. 2008;18(9):1005–1009.
- Rezaei N, Sabbaghian M, Liu Z, Zenker M. Eponym: Johanson–Blizzard syndrome. Eur J Pediatr. 2011;170(2):179–183.
- Torresani T. Neonatal screening for congenital hypothyroidism. Endocr Dev. 2014;26:44–49.
- Léger J, Olivieri A, Donaldson M, et al; ESPE-PES-SLEP-JSPE-APEG-APPES-ISPAE; Congenital Hypothyroidism Consensus Conference Group. European Society for Paediatric Endocrinology consensus guidelines on screening, diagnosis, and management of congenital hypothyroidism. J Clin Endocrinol Metab. 2014;99(2):363–384.
- Clerc J. Imaging the thyroid in children. Best Pract Res Clin Endocrinol Metab. 2014;28(2):203–220.
- Clerc J, Monpeyssen H, Chevalier A, et al. Scintigraphic imaging of paediatric thyroid dysfunction. Horm Res. 2008;70(1):1–13.
- Fugazzola L, Persani L, Vannucchi G, et al. Thyroid scintigraphy and perchlorate test after recombinant human TSH: a new tool for the differential diagnosis of congenital hypothyroidism during infancy. Eur J Nucl Med Mol Imaging. 2007;34(9):1498–1503.
- Selva KA, Mandel SH, Rien L, et al. Initial treatment dose of L-thyroxine in congenital hypothyroidism. J Pediatr. 2002;141(6):786–792.
- Peroni E, Vigone MC, Mora S, et al. Congenital hypothyroidism treatment in infants: a comparative study between liquid and tablet formulations of levothyroxine. Horm Res Paediatr. 2014;81(1):50–54.
- Liwanpo L, Hershman JM. Conditions and drugs interfering with thyroxine absorption. Best Pract Res Clin Endocrinol Metab. 2009;23(6):781–792.
- LaFranchi SH, Austin J. How should we be treating children with congenital hypothyroidism? J Pediatr Endocrinol Metab. 2007;20(5):559–578.
- Selva KA, Harper A, Downs A, Blasco PA, Lafranchi SH. Neurodevelopmental outcomes in congenital hypothyroidism: comparison of initial T4 dose and time to reach target T4 and TSH. J Pediatr. 2005;147(6):775–780.
- Bakker B, Kempers MJ, De Vijlder JJ, et al. Dynamics of the plasma concentrations of TSH, FT4 and T3 following thyroxine supplementation in congenital hypothyroidism. Clin Endocrinol (Oxf). 2002;57(4):529–537.
- Heyerdahl S, Oerbeck B. Congenital hypothyroidism: developmental outcome in relation to levothyroxine treatment variables. Thyroid. 2003;13(11):1029–1038.
- Rovet JF, Ehrlich RM. Long-term effects of L-thyroxine therapy for congenital hypothyroidism. J Pediatr. 1995;126(3):380–386.
- Dayal D, Saini L, Attri SV, Singh B, Bhalla AK. Daily versus alternate day thyroxine therapy to maintain euthyroidism in children with congenital hypothyroidism. Int J Endocrinol Metab. 2013;11(4):e9499.
- Albert BB, Heather N, Derraik JG, et al. Neurodevelopmental and body composition outcomes in children with congenital hypothyroidism treated with high-dose initial replacement and close monitoring. J Clin Endocrinol Metab. 2013;98(9):3663–3670.
- van der Sluijs Veer L, Kempers MJ, Wiedijk BM, Last BF, Grootenhuis MA, Vulsma T. Evaluation of cognitive and motor development in toddlers with congenital hypothyroidism diagnosed by neonatal screening. J Dev Behav Pediatr. 2012;33(8):633–640.
- Donaldson M, Jones J. Optimising outcome in congenital hypothyroidism; current opinions on best practice in initial assessment and subsequent management. J Clin Res Pediatr Endocrinol. 2013;5(Suppl 1):13–22.
- Léger J. Congenital hypothyroidism: a clinical update of long-term outcome in young adults. Eur J Endocrinol. 2015;172(2):R67–R77.