
Abstract
Background
β-Thalassemia is the most prevalent genetic disorder in India. Its traits and coinheritance vary from mild to severe conditions, resulting in thalassemia minor, intermediate, and major, depending upon many factors.
Purpose
The objective of this study was to identify the incidence of β-thalassemia traits, their coinheritance, and mutations, as well as to support the patients already diagnosed with β-thalassemia in East-Western Indian population for better management.
Patients and methods
Seventy-five referral cases for β-thalassemia were analyzed for various β-thalassemia traits, heterozygosity, and homozygosity conditions. Blood phenotypic parameters using cell counter and capillary electrophoresis were investigated. Analyses of eight common mutations of thalassemia in India were carried out using polymerase chain reaction-amplification refractory mutation system, end point polymerase chain reaction, and DNA sequencing methods.
Results
Of these (75) referral cases from East-Western Indian region, 68 were positive for β-thalassemia (90.67%). The majority of case types were of β-thalassemia minor (49, 65.33%), followed by HbE traits (6, 8.0%) and β-thalassemia major, including heterozygous and homozygous (5, 6.66%; 4, 5.33%) types and then HbE homozygous (2, 2.66%), as well as one each of the HbE/β-thalassemia and HbD/β-thalassemia (1, 1.34%) combination. Mutation analysis also revealed that the highest frequency of mutation was c.92+5G>C (41, 60.29%) followed by deletion 619bp (9, 13.23%) and c.79G>A (8, 11.76%) in our study group. Five cases (nos. 24, 27, 33, 58, and 71) exhibited coinheritance between β0/β+ (2), β0/β D (1), and c.124_127delTTCT/β+ or β0(2) affecting the Rajasthani and Gujarati populations in our study of the Western region of India.
Conclusion
We strongly recommend these Western populations for genetic screening before adopting reproductive technologies and interracial marital relations.
Introduction
β-Thalassemia is one of the hemoglobinopathies belonging to a class of genetic disorders. It occurs due to mutation in β-gene of autosomal chromosome 11.Citation1 The incidence of β-thalassemia trait in India is 3.3% with 1%–7% of couples being affected annually.Citation2 Approximately 300 mutations would occur in this type, affecting β-chain globin synthesis. If the synthesis of two β-chains is absent (β0/β0), the person has β-thalassemia major (Cooley’s anemia). This condition follows severe microcytic and hyochromic anemia. The person requires lifelong transfusion. β-Thalassemia minor is asymptomatic and results in microcytosis and mild anemia and HbA2 level increases, designated as β+/β or β0/β. Usually thalassemia intermedia is a condition between the major and minor forms depending on the severity of the anemic condition (β+/β+/or β0/β+) among other cases.Citation3,Citation4 Others are HbE trait, HbE homozygous, HbD/β-thalassemia, and HbE/β-thalassemia hemoglobinopathies. The latter one, HbE/β-thalassemia, is maximum in Thailand.Citation5 The gene mutation takes place in another one of β-gene only in addition to β-thalassemia minor/allele (β0/β or β+/β), leading to coin-heritance. In India, such coexisting HbE/β-thalassemia and HbD/β-thalassemia are less debated and occur in some parts of India, Pakistan, and Iran.Citation6,Citation7 Recently, a report was published in Eastern Indian population about the status of thalassemia and hemoglobinopathies and suggested that more such studies are necessary in other regions of India.Citation8 Prevalence of common hemoglobinopathies in Gujarati population was documented by Patel et alCitation9 in screening programs, where β-thalassemia minor cases were maximum comparatively. Hence, we report β-thalassemia, HbE, HbD traits, and their coinheritance as well as the mutation analysis of β-thalassemia distribution in East-Western Indian population using electrophoresis, polymerase chain reaction-amplification refractory mutation system (PCR-ARMS), end point PCR, and gene sequencing technology in our study.
Patients and methods
Patient selection
Blood samples of 75 referral cases of both sexes varying in age from 6 months to 38 years were collected from Gujarat (17), Rajasthan (40), Maharashtra (7), Assam (3), and West Bengal (6) in India for β-thalassemia testing after duly filling the patient consent form in our Supratech Micropath Research Institute, Ahmedabad. These patients were referred at random. This project was approved by Human Ethical Committee (HEC) of Gujarat University, Ahmedabad (GU/HEC-001/15), in 2015 for investigation.
Hematological analysis and DNA extraction
Hematogram report was carried out on CELL DYN RUBY automated cell counter. Hemoglobin (Hb) levels were estimated by Sebia Capillary 2 Flex piercing electrophoresis. The DNA was extracted from 2 mL of EDTA blood using PerkinElmer Prepito DNA Blood 250 Kit automatic machine. The kit was used according to the manufacturer’s instructions. The extracted genomic DNA was used as a template and was kept at 4°C until further use after routine DNA check.
Amplification, purification, and cycle sequencing
The primers were synthesized from the Eurofins, India. The amplification reaction was performed in Veriti Thermal Cycler. The PCR products were loaded on a 2.5% agarose gel, and the amplicons were visualized under ultraviolet transillumination after staining with ethidium bromide. PCR product cleanup using USB ExoSAP-IT kit (Affymetrix, Santa Clara, CA, USA) and cycle sequencing using BigDye Termi-natorV3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA, USA) were used for further mutation identification.
Mutation analysis
The β-globin gene mutations were first characterized using two sets of allele-specific PCR-ARMS to detect eight common mutations in India including c.92+5G>C, deletion 619 bp, c.79G>A (p.E27K), c.47G>A (p.Trp16Ter), c.364G>C (p.E122Q), c.27_28insG, c.51delC, and c.124_127delTTCT. Unknown β-thalassemia genes were further characterized by direct DNA sequencing using 3500 Genetic Analyzer Applied Biosystems (ABI) for all coding regions and exon–intron boundaries to detect uncommon point mutations and small rearrangements in the β-globin gene. The c.92+5G>C mutation was detected by Sanger sequencing and PCR-ARMS, and deletion 619 bp was done by end point PCR (gel electrophoresis). Other mutations were analyzed only by Sanger sequencer. The data were analyzed using CodonCode Aligner v5.0.2 (CodonCode Corporation, Centerville, MA, USA) and Mutation Surveyor v5.0 (Softgenetics, State College, PA, USA). Mean and percentage were calculated wherever necessary.
Results
β-Thalassemia and other traits
Referral cases of 75 at our Supratech Micropath Research Institute, Ahmedabad, from 2015 to 2016 were analyzed for β-thalassemia and other traits based on Hb levels blood indices and mutation analysis from different parts of India. The affected (68) contributed 90.67% of the referral cases. High percentage (65.33%) had β-thalassemia followed by HbE trait (8%) and β-thalassemia major (heterozygous 6.66%; homozygous 5.33%). Others were HbE homozygous (2.66%), HbE/β-thalassemia, and HbD/β-thalassemia contributed only 1.34% each. Thus, 49 cases (65.33%) had β-thalassemia minor followed by HbE trait (8%) and β-thalassemia major (compound heterozygous 6.66% and homozygous 5.33%). Two were HbE homozygous (2.66%) and HbE/β-thalassemia and HbD/β-thalassemia contributed only 1.34% each. These hemoglobinopathies are well supported by increased levels of mean HbA2 (08.66%) with decreased mean corpuscular hemoglobin (MCH), mean corpuscular volume values, and altered mean HbD (1.25%), HbF (7.21%), and HbE (2.73%) levels, measured by capillary electrophoresis ( and and ).
Figure 1 Percentage (%) distribution of β-thalassemia traits.
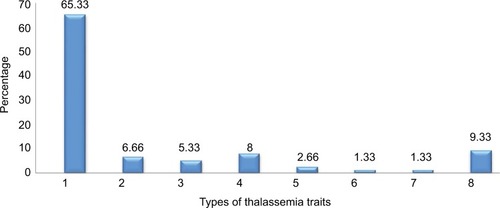
Table 1 Classification of thalassemia traits in our study
Table 2 Sex wise distribution of Hb variants and mutations in our study
Mutation analysis
We have analyzed conventional mutations of eight in Indian population using PCR-ARMS and end point PCR allayed with using Codon code Aligner V6.0.2 and mutation survey 5.0 software for exact specific mutation nomenclature from 68 affected cases. The data showed that c.92+5 G>C was higher (41, 60.29%), followed by nine cases of deletion 619 bp (13.23%), eight cases of c.79G>A (p.E27K) (11.76%), and five cases each of c.27_28insG (7.35%) and c.47G>A (p.Trp16Ter) (7.35%) with two cases each of c.124_127delTTCT (2.94%) and c.51delC (2.94%) and one case each of c.364G>C (p.E122Q) and c.47G>A (p.Trp16Ter) (1.47%), respectively, with no sex difference as female and male ratio was (1:1.13) (). Deletion 619 bp was only detected by gel electrophoresis, and c.92+5G>C was identified using PCR-ARMS and also gene sequencing as that of others (). In Rajasthan and Gujarat, where more are accumulated (40 and 17) respectively, in both cases, the most frequent mutation is c.92+G>C (26 and 10) followed by 619 bp (3 and 4) and 619 bp deletions ( and ).
Figure 2 Seven common mutations identified by direct DNA sequencing.
Abbreviations: S, G/C; R, G/A; K, G/T; M, C/A; Y, C/T.
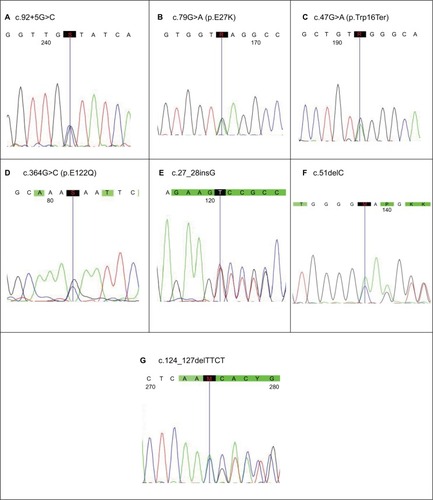
Figure 3 Region wise percentage distribution of mutations in β-thalassemia and numbers in parentheses indicate cases.
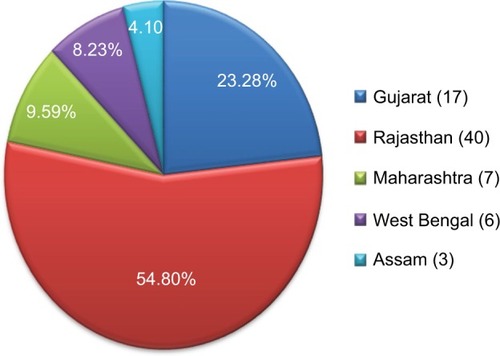
Table 3 Percentage of mutation types in our thalassemic cases (68) of different regions
Discussion
β-Thalassemia is one of the heterozygous inheritable disorders in India. It causes reduced or absence of β-chain synthesis of Hb. Its variants in addition to carrier identification and prenatal analysis are necessary for its management and to avoid marriages between carrier of mutated genes including consanguineous types.Citation4,Citation10–Citation12 Hence, from 75 referral cases of Western and Eastern India, the blood was collected to identify various traits and mutations accurately using electrophoretic and molecular diagnostic techniques in our laboratory including coinheritance with β-thalassemia. Of the total referral patients, 68 cases were affected having 90.67% in this study. Of these, 65.33% of β-thalassemia traits (49) were detected followed by HbE trait and β-thalassemia major with HbE homozygous and their HbE/β-thalassemia and HbD/β-thalassemia coinherited cases depending on altered Hb, MCH, red blood cell distribution width, and MCV values. It indicated that β-thalassemia cases (carriers) are maximum followed by others and support the data of previous workers in India.Citation13–Citation16 Similarly, Hb patterns were measured and presented in the study of Mondal and Mandal,Citation8 who obtained few cases of HbE, HbD traits, and β-thalassemia major comparatively. This could be due to changing lifestyles, environmental and genetic factors, and coinheritance of HbE, HbD, and/or α-thalassemia with β-thalassemia carriers.Citation17,Citation18 Further, these factors may also be the cause of β-thalassemia major with heterozygosity/homozygosity who are less in number requiring blood transfusion. Similarly, coinheritance of HbE/β-thalassemia and HbD/β-thalassemia and HbE homozygous cases were also reduced in number in our report. HbE trait had six cases having less severity of clinical condition. However, Olivieri et alCitation17 mentioned that these conditions may vary from severe to mild depending upon genetic and environmental factors, and such patients are also less frequent to support our data. We detected one each of HbD/β-thalassemia and HbE/β-thalassemia cases in addition to HbE patients with variable phenotypic indices expressing mild heterozygous state.Citation19–Citation22 These patients may require transfusion in severe condition only, due to coinheritance of the disease.Citation23
Further, we extended our investigation on molecular analysis of mutations of β-thalassemia and systematically using latest molecular biology tools such as PCR-ARMS, end point PCR, and Sanger Gene Sequencing. Data revealed 92+5 G>C (IVS-1–5) is the maximum in cases (60.29%), of Rajasthan and Gujarat followed by deletion 619 bp and is conformed with others documented earlier in Gujarat, Maharashtra, and Rajasthan.Citation2,Citation9,Citation12,Citation15,Citation24–Citation26 But Hassan et al,Citation27 from Thailand, found cd26 (A–G) HbE and cd41/42 (−TTCT) were higher in their studies. Thong et alCitation28 presented cd41/42 (−TTCT) and IVS-2 654 (C–T) were maximum in Chinese population. Similarly, second highest mutation was 619 bp in (9, 13.23%) this study similar to that of other studies in Western India conducted by Sheth et al,Citation10 Grow et al,Citation13 Colah et al,Citation29 and Nigam et al.Citation16 The third largest mutation in our study was c.79G>A (p.E27K) followed by c.47G>A (p.Trp16Ter) and c.27_28insG different from other investigators,Citation2 followed by other mutations, ie, c.51delC, c.124_127delTTCT(novel), and c.364G>C (p.E122Q). The incidence of these mutations does not seem to be related to sex, as our sex ratio was 1:1.13 (M:F). The variation in occurrence of these mutations is related to regional, ethical, migration, interracial marriages, study plan, and other factors as mentioned by others.Citation12,Citation29,Citation30
Conclusion
Our study showed that 68 cases were affected by β-thalassemia in our referral cases (75), from East-Western Indian region. β-Thalassemia carriers were 49 (65.33%) followed by HbE trait and β-thalassemia major with heterozygous and homozygous condition using hematological profiles. Detection of molecular analysis of mutations using PCR-ARMS, end point PCR, and gene sequencing methods revealed c.92+5G>C mutation exhibited higher incidence (26+10+2) followed by deletion 619 bp (3+4+2) in Rajasthan, Gujarat, and Maharashtra (Western India) as compared to West Bengal and Assam (3:0; 0:0) of Eastern India, respectively. This requires further elucidation. The variation in incidence of these mutations is dependent on ethnic diversity, migration, genetic factors, and other lifestyles. We, hence, recommend the mass screening, Prenatal Diagnostic Techniques, genetic counseling, transfusion programs and clinical management made available to these populations before adopting assisted reproductive and preimplantation technologies in India.
Author contributions
Parth S Shah and Nidhi D Shah have contributed to writing results and discussion in the manuscript preparation when they visit India. Hari P Ray, Nikunj B Khatri, Ketan K Vaghasia, and Rutvik J Raval are involved in collection of blood from the patients after duly filled consent forms, blood analysis, DNA extraction, DNA sequencing, PCR-ARMS, end point PCR, and data analysis of 75 patients. Dr Sandip C Shah and Dr Mandava V Rao have contributed to preparation of reports after finalization of the results and preparation of the manuscript finally for submission to the journal. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Acknowledgments
The authors are thankful to all the staff including Clinicians of Supratech Micropath Laboratory, Ahmedabad, for their continuous assistance in this work. Parth S Shah, Nidhi D Shah, and Sandip C Shah are son, daughter-in-law, and father, respectively, who run this research institute.
Disclosure
The authors report no conflicts of interest in this work.
References
- WeatherallDJCleggJBInherited haemoglobin disorders: an increasing global health problemBull World Health Organ200179870471211545326
- AnsariMIPatelNGCharacterization of β-thalassemia mutations from north Maharashtra regionJ Pharm Biol Sci20151031316
- CooleyTBLeePA series of cases of splenomegaly in children with anemia and peculiar bone changesTrans Am Pediatr Soc1925372930
- RaoMVShahSRPatelAPβ-thalassemiaGuptaPDSrivastavaLMEssentials of Inborn Metabolic and Genetic DisordersChennaiPug Publication Pvt Ltd2015169179
- BoonyawatBMonsereenusornCTraivareeCMolecular analysis of beta-globin gene mutations among thai beta-thalassemia children: results from a single center studyAppl Clin Genet2014725325825525381
- TaghaviBMKarimipoorMAmirianACo-inheritance of hemoglobin D and B-thalassemia trait in three Iranian families: clinical relevanceArch Iran Med2011141616321194265
- AbolghasemiHAmidAZeinaliSThalassemia in Iran: epidemiology, prevention and managementJ Pediatr Hematol Oncol200729423323817414565
- MondalSKMandalSPrevalence of thalassemia and hemoglobinopathy in eastern India: a 10-year high-performance liquid chromatography study of 119,336 casesAsian J Transfus Sci201610110511027011683
- PatelAPNaikMRShahNMSharmaNPParmarPHPrevalence of common hemoglobinopathies in Gujarat: an analysis of a large population screening programNatl J Comm Med201231112116
- ShethJJShethFJPandyaPBeta thalassaemia mutations in western IndiaInd J Pediatr20086567570
- MishraAKTiwariAScreening and molecular characterization of β-thalassaemia mutations in parents and siblings of β-thalassaemia major patientsInd J Basic Appl Medl Res201452481486
- CaoAGalanelloRBeta-thalassemiaGenet Med2010122617620098328
- GrowKVashistMAbrolPSharmaSYadavRBeta thalassemia in India: current status and the challenges aheadInt J Pharm Pharm Sci2014642833
- BalgirRSThe burden of haemoglobinopathies in India and the challenges aheadCurr Sci2000791115361547
- SatputeSBBankarMPMominAAThe prevalence of β-thalassemia mutations in south western MaharashtraInd J Clin Biochem2012274389393
- NigamNMunshiMPatelMSoniADistribution of beta thalassemia mutation and its correlation with alpha thalassemia in Gujarati familiesInt J Hum Genet200334221224
- OlivieriNFPakbazZVichinskyEHb E/beta-thalassaemia: a common & clinically diverse disorder IndianJ Med Res20111344522531
- ColahRGorashekarAPhanasgaonkarSEpidemiology of beta thalassemia in western India: mapping the frequencies & mutations in subversion of Maharashtra and GujaratBr J Haematol2010149573974720230396
- BurtisCAAshwoodERBrunsDETietz Textbook of Clinical Chemistry and Molecular Diagnostics5th edIndiaElsevier2012
- WilliamsonMASnyderLMWallach’s Interpretation of Diagnostic Tests9th edNew York, NYLippincott Pub2011
- TheodoridouSAlemayechouMPerperidouPSinopoulouCKarafoulidouTKiriakopoulouGCompound heterozygosity for Hb D-Punjab/b-thalassemia and blood donation: case reportTurk J Hematol2009262100101
- RahimiZAkramipourRKoraniSNagelRLHb D-Punjab [beta 121 (GH4) Glu→Gln]/beta0-thalassemia [IVSII.1(G→A)] in two cases from an Iranian family: first reportAm J Hematol2006814302303
- MenzelSGarnerCGutIA QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15Nat Genet200739101197119917767159
- PanjaAGhoshTKBasuAGenetics of thalassemia in Indian populationJ Community Nutr Health2012113946
- BhukhanvalaDSItaliaKSawantPColahRGhoshKGupteSCMolecular characterization of β-thalassemia in four communities in South Gujarat-codon 30 (G→A) a predominant mutation in the Kachhiya Patel communityAnn Hematol201392111473147623665927
- ColahRBGorakshakarACNadkarniAHInvasive & noninvasive approaches for prenatal diagnosis of haemoglobinopathies: experiences from IndiaInd J Med Res20111344552560
- HassanSAhmadRZakariaZZulkafliZAbdullahWZDetection of β-globin gene mutations among β-thalassaemia carriers and patients in Malaysia: application of multiplex amplification refractory mutation system–polymerase chain reactionMalays J Med Sci20132011320
- ThongMKTanJATanKLYapSFCharacterisation of beta-globin gene mutation in Malaysian children: a strategy for the control of beta-thalassemia in a develop countryJ Trop Pediatr200551632833315967770
- ColahRGorakshakarANadkarniARegional heterogeneity of beta-thalassemia mutations in the multi ethnic Indian populationBlood Cells Mol Dis200942324124619254853
- NadkarniAHNairSBItaliaKYMolecular diversity of hemoglobin H disease in IndiaAm J Clin Pathol2010133349149420154289