
Abstract
Citrin deficiency is a recessively inherited metabolic disorder with age-dependent clinical manifestations. It causes neonatal intrahepatic cholestasis (NICCD) and adult-onset type II citrullinemia (CTLN2). Patients with NICCD present with intrahepatic cholestasis in the neonatal period and usually respond to the treatment with medium-chain triglyceride (MCT) supplement and lactose-restricted formula. In adulthood, CTLN2 develops in <10 % of the patients showing hyperammonemic encephalopathy. Patients with CTLN2 required liver transplantation for the most promising prognosis; however, they were successfully treated with MCT supplement with a low carbohydrate formula. Citrin deficiency is caused by mutations in SLC25A13 on chromosome 7q21.3, with a high frequency in East Asia, including Japan. Citrin is aspartate/glutamate transporter in mitochondria, a component of malate-aspartate nicotinamide adenine dinucleotide hydrogen shuttle, and is essential for the hepatic glycolysis. Although the precise pathophysiology of citrin deficiency remains unclear, recent reports for the effective MCT supplement therapy and downregulation of peroxisome proliferator-activated receptor α suggest that citrin deficiency impairs hepatic de novo lipogenesis coupled with glycolysis leading to the energy deficit of hepatocytes. Herein, we review the current therapeutic and pathological understanding of CTLN2.
Introduction
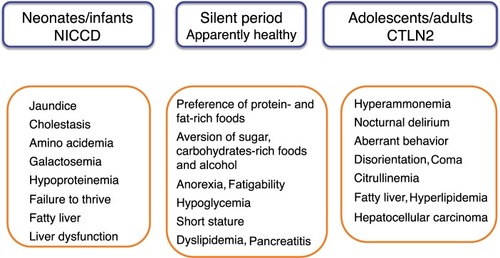
Citrin deficiency is an autosomal recessive disorder caused by mutations in SLC25A13 and is prevalent in East Asia, including Japan.Citation1 There are common mutations in the Japanese population; the carrier rate is 1/42, and an estimated frequency of patients with citrin deficiency is 1/7,100.Citation2 Citrin deficiency causes neonatal intrahepatic cholestasis (NICCD; Online Mendelian Inheritance in Man [OMIM] 603471) and adult-onset type II citrullinemia (CTLN2; OMIM 605814). In 2006, Kobayashi et alCitation3 identified SLC25A13 on chromosome 7q21.3 as a disease-causing gene and contributed to clarify the age-dependent clinical phenotypes of citrin deficiency (). Patients with NICCD present with intrahepatic cholestasis in the neonatal period and usually respond to the treatment with medium-chain triglyceride (MCT) supplement and lactose-restricted formula.Citation4 After recovery of NICCD, some patients complain of fatigue, recurrent episode of hypoglycemia, and short stature. In adulthood, CTLN2 develops in <10% of patients showing hyperammonemic encephalopathy. Liver transplantation has been considered as the only definitive therapy for CTLN2. However, we recently found that MCT supplement with low carbohydrate formula was very effective for the management of CTLN2.Citation5,Citation6 MCT effectiveness provided the opportunity to consider the basic pathophysiology of citrin deficiency.
Figure 1 Age-dependent clinical phenotypes of citrin deficiency.
Abbreviations: CTLN2, adult-onset type II citrullinemia; NICCD, neonatal intrahepatic cholestasis due to citrin deficiency.
Clinical features of CTLN2
After recovery of NICCD, the patients show a dietary preference for protein- and lipid-rich foods and an aversion to sugar, carbohydrate-rich foods, and alcohol. Hyperammonemic encephalopathy develops in some patients aged 20–70 years. A male preponderance was observed; the male-to-female ratio is 2.4:1.Citation1 Disease onset is often associated with surgery (fasting), alcohol intake, and medication. They usually present with nocturnal delirium, aberrant behavior (aggression, irritability, and restlessness), disorientation, drowsiness, loss of memory, flapping tremor, convulsion, and coma. The patients usually experience increases in blood ammonia levels after dinner and exhibit delirium and aberrant behavior in the evening. In the morning, they exhibit normal behavior with no hyperammonemia at an early stage. Without treatment, cerebral edema develops, resulting in brain death. More than 90% have a body mass index of <20 kg/m2; ~10% of the patients have complications such as hyperlipidemia, pancreatitis, and hepatocellular carcinoma.
With regard to the diagnosis, food preference is useful information for CTLN2 in addition to hyperammonemia and citrullinemia. A liver biopsy and ultrasonography, computed tomography scan, and magnetic resonance imaging studies reveal fatty liver. Some patients have a history of NICCD. Definitive diagnosis is dependent on the mutation analysis of SLC25A13.
The recommended management of CTLN2 is MCT supplement therapy with low carbohydrate formula.Citation5,Citation6 Sodium pyruvate administration with a low-carbohydrate diet was reported to decrease the frequency of hyperammonemic encephalopathy.Citation7 However, this treatment did not prevent the relapse of encephalopathy nor improve the Fischer ratio and citrullinemia.
Pathophysiology of CTLN2
Function of citrin
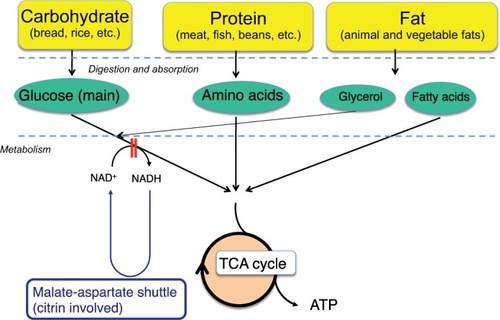
Citrin consists of a polypeptide of 675 amino acids and has a bipartite structure: a C-terminal half with a mitochondrial carrier motif and an N-terminal extension harboring four EF-hand calcium-binding domains.Citation3 Citrin and isoform protein aralar (encoded by SLC25A12) are the proteins that function in the transport of aspartate from mitochondria to cytosol in exchange for glutamate.Citation3,Citation8,Citation9 They play a role in the transfer of cytosolic nicotinamide adenine dinucleotide hydrogen (NADH)-reducing equivalents into mitochondria as a member of the malate-aspartate NADH shuttle (). CaCitation2 signaling in mitochondria is important for regulating mitochondrial function in response to intracellular and extracellular cues. CaCitation2 activation of the malate-aspartate NADH shuttle was demonstrated; however, its physiological involvement is not elucidated. Citrin is expressed in the liver, kidney, heart, and small intestine; aralar is found in the brain, skeletal muscle, kidney, and heart.Citation3,Citation10 Citrin and aralar play a substantial role in the liver and brain, respectively, probably due to the compensatory effects of each isoform. As a result, citrin deficiency predominantly impairs the NADH shuttle in the liver, leading to the impairment of hepatic glycolysis.
Figure 2 Glycolysis and lipogenesis in the liver.
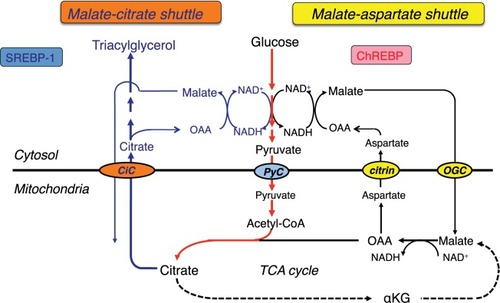
Glucose metabolism and energy source of hepatocytes and the effect of MCT supplement
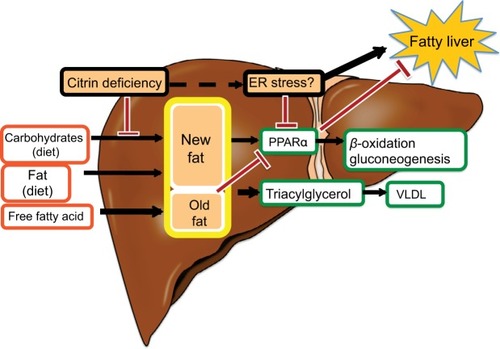
The major energy source of hepatocytes is fatty acids.Citation11 Hepatic glycolysis is coupled with lipogenesis. Hepato-cytes uptake glucose only during the fed state, use it as an energy source in part, and synthesize glycogen and fatty acids (de novo lipogenesis) from the remainder of the glucose.Citation12,Citation13 Citrin deficiency is predicted to impair glycolysis and de novo lipogenesis. De novo lipogenesis in the liver is a minor pathway in adults under conditions of a normal Western (fat-rich) diet.Citation14 However, Chakravarthy et alCitation15 reported that new fat synthesized in the liver from fat and carbohydrates in the diet activates the peroxisome proliferator-activated receptor α (PPARα), which controls β-oxidation of fatty acids. Komatsu et alCitation16 demonstrated the downregulation of PPARα in the liver of CTLN2 patients. Thus, the hepatocytes in citrin deficiency have difficulty using glucose as an energy source because of a defect in glycolysis and also have difficulty using fatty acids as a major energy source because of the downregulation of PPARα. The basic pathophysiology of CTLN2 is likely the persistent and cumulative energy deficit of the hepatocytes ().
Figure 3 Fatty liver in citrin deficiency.
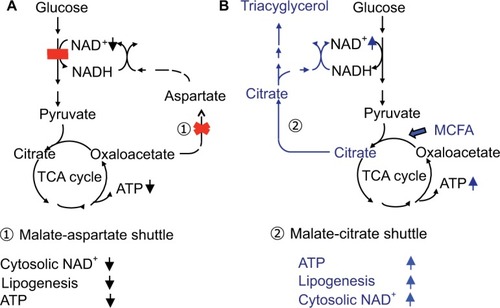
MCT is rapidly hydrolyzed and is absorbed mainly as medium-chain fatty acids (MCFA), which reach the liver via the portal vein and are metabolized to acetyl-CoA by β-oxidation, enhance tricarboxylic acid cycle activity, and increase adenosine triphosphate (ATP) levels in hepatocytes ().Citation17 MCT supplementation can promote lipogenesis, resulting in an increase in the nicotinamide adenine dinucleotide-oxidized (NAD+)/NADH ratio through the malate-citrate shuttle and reduction of oxidative stress.Citation5,Citation18 Intake of MCT with each meal is important to provide energy promptly to hepatocytes and promote hepatic glycolysis, glycogenesis, and lipogenesis, because insulin secretion after meal is expected to suppress the mobilization of fatty acids from adipocytes and decrease the utilization of fatty acids in the hepatocytes resulting in the energy deficit. The patients treated a few months after CTLN2 onset had no relapse of hyperammonemic encephalopathy, but did not have a complete recovery of biochemical findings and fatty liver probably due to irreversible change. On the other hand, the patients treated soon after CTLN2 onset showed a complete recovery of biochemical findings and fatty liver. Our studies suggest that MCT supplement can prevent the CTLN2 onset and is recommended for the children with citrin deficiency.
Figure 4 (A) Glucose metabolism in citrin deficiency. (B) Metabolic change by MCT treatment.
Food preference
The patients show peculiar preference for high-protein and high-fat foods, such as bean products, eggs, fish, and meat. In contrast, these patients dislike carbohydrates, such as cereals, sweets, and cooked rice. They also dislike alcohol drinks. Nutritional assessment revealed low-carbohydrate daily diet: protein: fat: carbohydrate ratio of 15%–25%: 40%–50%: 30%–40%, compared with 10% to 15%: 25% to 35%: 50% to 60% of healthy Japanese controls.Citation19 The patients cannot metabolize carbohydrates effectively in the liver because of the impairment of NADH shuttle. As shown in , protein (amino acids) and fat (fatty acids) can produce energy independent on NADH shuttle. The metabolic sensing function of the liver may be present, by which the metabolic information from the liver affects the brain.Citation20 Their preference may be a compensatory behavior to increase hepatic energy. With regard to the sugar aversion, fructose loading in patients with citrin deficiency is similar to the fructose loading in patients with fructose intolerance.Citation21 Fructose is quickly metabolized in the liver, but its metabolism would be blocked at the step of glyceride 3-phosphate dehydrogenase in CTLN2 because of the impairment of NADH shuttle. The administration of fructose-added glycerol for brain edema is fatal and is contraindicated in the patients with CTLN2.Citation22 Fructose loading would result in a decrease or depletion of inorganic phosphate and ATP in liver. The patients may prefer the foods that can increase ATP in the liver and have an aversion to the foods that may decrease ATP and/or accumulate the toxic metabolites in the liver.
Figure 5 Energy conversion of food. Food preferenceAbbreviations: ATP, adenosine triphosphate; NADH, nicotinamide adenine dinucleotide hydrogen; NAD+, nicotinamide adenine dinucleotide-oxidized; TCA, tricarboxylic acid.
Fatty liver and hyperlipidemia
Liver histology of CTLN2 patients shows macrovesicular and microvesicular steatosis, infiltration of inflammatory cells in the lobular and portal areas, and changes identical to nonalcoholic steatohepatitis.Citation1 Hepatic steatosis develops when the influx of fatty acids and lipogenesis exceed hepatic β-oxidation and triglycerides export. Hepatic steatosis is frequently associated with obesity and/or diabetes, but is also caused by starvation.Citation23 Hepatic steatosis in CTLN2 is likely caused by the mechanism similar to starvation. During periods of starvation, the triglycerides stored in adipose tissue are hydrolyzed to free fatty acids and mobilized into plasma to reach the liver. However, free fatty acids cannot be effectively oxidized because of the downregulation of PPARα and thus accumulate in the liver.Citation24 PPARα is activated by new fat synthesized in liver.Citation15 Patients with citrin deficiency have defects in hepatic glycolysis and de novo lipogenesis and have a downregulation of PPARα ().Citation16 The downregulation of PPARα also causes hypertriglyceridemia. Augmented oxidative stress was confirmed and would further deteriorate the steatosis in citrin deficiency.Citation25 Improvement in fatty liver was observed after long-term MCT supplement therapy in one patient.Citation6
Hyperammonemia
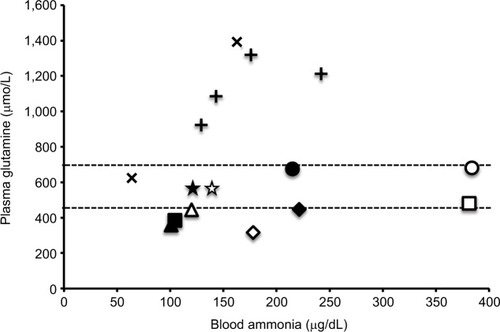
There are two major ammonia-detoxification systems, ureagenesis and glutamine synthesis in the liver ().Citation26 A system of ureagenesis in periportal hepatocytes has a low affinity, but high detoxicating capacity for ammonia. The glutamine synthesis system in the perivenous hepatocytes is a high-affinity scavenger for ammonia; it catches the ammonia escaped from ureagenesis and plays a significant role in the maintenance of physiologically low ammonia concentrations. Glutamine synthase (GS) catalyzes the ATP-dependent synthesis of glutamine from glutamate with ammonia. Impairment of either system causes hyperam-monemia.Citation27–Citation29 Citrullinemia (521±290 µmol/L) in CTLN2 reflects an impairment of the argininosuccinate synthase 1 (ASS1) step of the urea cycle. The ASS1 catalyzes the ATP-dependent synthesis of argininosuccinate in cytosol from citrulline and aspartate (). Hepatic ASS1 enzyme activity in CTLN2 is mostly reduced to <20% of control due to a decrease in the amount of enzyme protein.Citation30 Decrease in the ASS1 enzyme protein has been considered a main reason for hyperammonemia in CTLN2. However, a few patients had 50%–80% of control ASS1 enzyme activities,Citation30 and one patient had fluctuating serum citrulline levels during episodic manifesting periods.Citation31 In contrast, citrullinemia type 1 (CTLN1; OMIM 215700) is caused by ASS1 mutation, and these patients show hyperammonemia and usually increased plasma citrulline levels >1,000 µmol/L. Recently, there were reports of patients with mild and asymptomatic CTLN1 carrying ASS1 mutations, called benign citrullinemia. These patients had 7%–26% of control ASS1 enzyme activity and showed increased plasma citrulline levels (<1,000 µmol/L) similar to the levels of plasma citrulline in CTLN2.Citation32–Citation34 These findings suggest that additional factors are required for the development of hyperammonemia in CTLN2. We found that plasma glutamine levels were never increased to more than the normal range in hyperammonemia and rather decreased to lower than the normal range in some patients ().Citation6 These findings were different from carbamoyl phosphate transferase or ornithine transcarbamylase deficiency, and MCT therapy promptly decreased blood ammonia accompanying with an increase in plasma glutamine and a mild decline in plasma citrulline levels. These data indicated that the GS ammonia-detoxification system was also impaired in CTLN2 in addition to the impairment of ASS1. However, we did not find a decrease in the GS activity in the liver specimens from the CTLN2 patients obtained at transplantation.Citation6 Hepatic GS activity is impaired in CTLN2 possibly due to the decrease in the contents of ATP and/or substrates of hepatocytes. MCT supplement therapy can supply ATP and/or substrates and enhance the GS ammonia-detoxification system.
Figure 6 Ammonia metabolism in liver.
Abbreviations: ASS1, argininosuccinate synthase 1; ATP, adenosine triphosphate; CPS, carbamoyl phosphate synthetase; GDH, glutamate dehydrogenase; GLNase, glutaminase; GS, glutamine synthase.
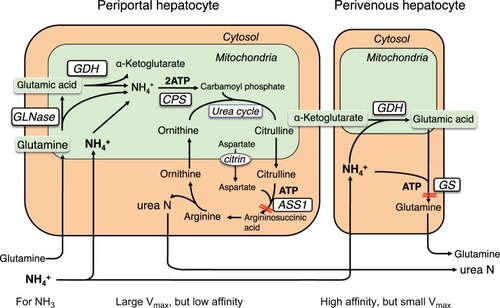
Figure 7 Relationship between blood ammonia and plasma glutamine.
Citrin transports aspartate to cytosol, and citrin deficiency has been considered to limit the supply of aspartate for hepatic ASS1. However, the individuals post-NICCD have neither citrullinemia nor hyperammonemia. NICCD patients show mild citrullinemia but have no hyperammonemia with normal hepatic ASS1 activity.Citation35 Citrullinemia in NICCD is promptly improved after MCT supplement therapy.Citation4 These suggest that the amounts of aspartate and/or ATP in cytosol are sufficient for hepatic ASS1 reaction in individuals post-NICCD but are not enough in NICCD patients. Aspartate is not an essential amino acid and can be synthesized through a transamination reaction of oxaloacetate in the ordinary state. In CTLN2, the level of plasma citrulline was mildly decreased by MCT supplement therapy as a short-term effect. Citrullinemia in CTLN2 is likely caused mainly by a decrease in hepatic ASS1 enzyme protein and partly by a deficiency of aspartate and/or ATP. The mechanism of a decrease in hepatic ASS1 enzyme protein is still unknown; however, the recovery of ASS1 expression was observed after a long-term MCT supplement therapy in one patient.Citation6
Prospective aspects of citrin deficiency
The pathophysiology of clinical features including low birth weight, male preponderance, and the presence of hepatocellular carcinoma is still unknown. Impairment of hepatic de novo lipogenesis, energy deficit of the hepatocytes, and augmented oxidative stress may be associated with those symptoms. On the basis of further pathophysiological information, more effective therapy can be established.
Disclosure
The authors report no conflicts of interest in this work.
References
- SahekiTSongYZCitrin deficiencyAdamMPArdingerHHPagonRAGeneReviews® [Internet]Seattle, WAUniversity of Washington200519932017
- KikuchiAArai-IchinoiNSakamotoOSimple and rapid genetic testing for citrin deficiency by screening 11 prevalent mutations in SLC25A13Mol Genet Metab2012105455355822277121
- KobayashiKSinasacDSIijimaMThe gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier proteinNat Genet199922215916310369257
- HayasakaKNumakuraCToyotaKKimuraTTreatment with lactose (galactose)-restricted and medium-chain triglyceride-supplemented formula for neonatal intrahepatic cholestasis caused by citrin deficiencyJIMD Rep20122374423430852
- HayasakaKNumakuraCToyotaKMedium-chain triglyceride supplementation under a low-carbohydrate formula is a promising therapy for adult-onset type II citrullinemiaMol Genet Metab Rep20141425027896073
- HayasakaKNumakuraCYamakawaMMedium-chain triglycerides supplement therapy with a low-carbohydrate formula can supply energy and enhance ammonia detoxification in the hepatocytes of patients with adult-onset type II citrullinemiaJ Inherit Metab Dis201841577778429651749
- YazakiMFukushimaKSahekiTIkedaSTherapeutic strategy for patients with adult onset type II citrullinemia (CTLN2)Program and abstracts for the 3rd Asian Congress for Inherited Metabolic Diseases/the 55th Annual Meeting of the Japanese Society for Inherited Metabolic Diseases2013Chiba101
- del ArcoASatrústeguiJMolecular cloning of Aralar, a new member of the mitochondrial carrier superfamily that binds calcium and is present in human muscle and brainJ Biol Chem19982733623327233349722566
- PalmieriLPardoBLasorsaFMCitrin and aralar1 are Ca(2+)-stimulated aspartate/glutamate transporters in mitochondriaEMBO J200120185060506911566871
- BegumLJalilMAKobayashiKExpression of three mitochondrial solute carriers, citrin, aralar1 and ornithine transporter, in relation to urea cycle in miceBiochim Biophys Acta20021574328329211997094
- SeifterSEnglardSEnergy metabolismAriasIWolkoffABoyerJThe Liver: Biology and Pathobiology5th edNew YorkRaven2009142
- UyedaKRepaJJCarbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesisCell Metab20064210711016890538
- WangYViscarraJKimSJSulHSTranscriptional regulation of hepatic lipogenesisNat Rev Mol Cell Biol2015161167868926490400
- HellersteinMKDe novo lipogenesis in humans: metabolic and regulatory aspectsEur J Clin Nutr199953Suppl 1S53S6510365981
- ChakravarthyMVPanZZhuY“New” hepatic fat activates PPARalpha to maintain glucose, lipid, and cholesterol homeostasisCell Metab20051530932216054078
- KomatsuMKimuraTYazakiMSteatogenesis in adult-onset type II citrullinemia is associated with down-regulation of PPARαBiochim Biophys Acta20151852347348125533124
- BachACBabayanVKMedium-chain triglycerides: an updateAm J Clin Nutr19823659509626814231
- MartenBPfeufferMSchrezenmeirJMedium-chain triglyceridesInt Dairy J2006161113741382
- SahekiTKobayashiKTerashiMReduced carbohydrate intake in citrin-deficient subjectsJ Inherit Metab Dis200831338639418415701
- YiCXla FleurSEFliersEKalsbeekAThe role of the autonomic nervous liver innervation in the control of energy metabolismBiochim Biophys Acta20101802441643120060897
- SteinmannBGitzelmannRVan den BergheGDisorders of fructose metabolismScriverCRBeaudetALSlyWSThe Metabolic and Molecular Bases of Inherited Disease7th edNew YorkMcGraw-Hill1995905934
- YazakiMTakeiYKobayashiKSahekiTIkedaSRisk of worsened encephalopathy after intravenous glycerol therapy in patients with adult-onset type II citrullinemia (CTLN2)Intern Med200544318819515805705
- AnguloPNonalcoholic fatty liver diseaseN Engl J Med2002346161221123111961152
- HashimotoTCookWSQiCYeldandiAVReddyJKRaoMSDefect in peroxisome proliferator-activated receptor alpha-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fastingJ Biol Chem200027537289182892810844002
- NagasakaHOkanoYTsukaharaHSustaining hypercitrullinemia, hypercholesterolemia and augmented oxidative stress in Japanese children with aspartate/glutamate carrier isoform 2-citrin-deficiency even during the silent periodMol Genet Metab2009971212619232506
- HäussingerDSchliessFGlutamine metabolism and signaling in the liverFront Biosci20071237139117127305
- HäussingerDHepatocyte heterogeneity in glutamine and ammonia metabolism and the role of an intercellular glutamine cycle during ureogenesis in perfused rat liverEur J Biochem198313322692756852039
- HäberleJGörgBRutschFCongenital glutamine deficiency with glutamine synthetase mutationsN Engl J Med2005353181926193316267323
- QvartskhavaNLangPAGörgBHyperammonemia in gene-targeted mice lacking functional hepatic glutamine synthetaseProc Natl Acad Sci U S A2015112175521552625870278
- YasudaTYamaguchiNKobayashiKIdentification of two novel mutations in the SLC25A13 gene and detection of seven mutations in 102 patients with adult-onset type II citrullinemiaHum Genet2000107653754511153906
- MaruyamaHOgawaMNishioTKobayashiKSahekiTSunoharaNCitrullinemia type II in a 64-year-old man with fluctuating serum citrulline levelsJ Neurol Sci2001182216717011137523
- HäberleJPauliSLinnebankMStructure of the human arginino-succinate synthetase gene and an improved system for molecular diagnostics in patients with classical and mild citrullinemiaHum Genet2002110432733311941481
- RuitenbeekWKobayashiKIijimaMModerate citrullinaemia without hyperammonaemia in a child with mutated and deficient argininosuccinate synthetaseAnn Clin Biochem200340Pt 110210712542919
- HäberleJPauliSSchmidtESchulze-EilfingBBerningCKochHGMild citrullinemia in Caucasians is an allelic variant of argininosuc-cinate synthetase deficiency (citrullinemia type 1)Mol Genet Metab200380330230614680976
- TazawaYKobayashiKOhuraTInfantile cholestatic jaundice associated with adult-onset type II citrullinemiaJ Pediatr2001138573574011343052