
Abstract
Holt-Oram syndrome (HOS) is an autosomal dominant disorder characterized by congenital cardiac defects and congenital deformities of the upper limbs. Herein, we report the case of a 2-year-old patient presenting with clinical diagnostic criteria of HOS with interatrial and interventricular communication associated with hip dysplasia and upper limb reduction composed of radial ray anomaly. A novel de novo, potentially pathogenic variant in the TBX5 gene at NM_181486.2:c.243-1G>C was identified.
Introduction
Holt-Oram syndrome (HOS) is an autosomal dominant disorder (MIM: 142900) described for the first time by Holt and Oram in 1959.Citation1 The incidence is 1:100,000 live births, and more than 300 cases have been reported with diverse ethnic and geographic distribution.Citation2,Citation3 HOS is characterized by congenital deformities of the superior limbs (CDSL) associated with congenital heart defects.Citation4 The clinical diagnostic criteria include the presence of a radial ray preaxial anomaly in one of the superior limbs and personal or family history of cardiac defects.Citation5 Skeletal defects of the superior limbs can be from bone carpal abnormalities or the triphalangeal thumb to the bilateral phocomelia.Citation6 The most common congenital heart defects are the ostium secundum-type atrial septal defects and ventricular septal defects. However, more severe cardiac defects, such as Tetralogy of Fallot or a hypoplastic left heart, have been documented.Citation6,Citation7
This syndrome is caused by a TBX5 heterozygous mutation localized in the long arm of chromosome 12 (12q24.1). Forty percent of the cases occur due to new mutations, and the remaining cases occur due to familial inheritance.Citation1 TBX5 proteins are members of a conserved family of transcription factors that share a common DNA-binding domain called the T-box.Citation8 This transcriptional factor is associated with heart and limb development.Citation9 TBX5 is expressed in the epicardium, myocardium, and endocardium of embryonic and adult hearts.Citation10 In early cardiac development, this transcriptional factor activates genes associated with morphological septation and cardiomyocytes’ maturation; later it is involved in the development of the cardiac conduction system.Citation11 Studies in animal models indicate that TBX5 is expressed in the forelimb field and is implicated in the initiation of limb outgrowth.Citation12
Almost 90 different variants in this gene have been reported, and most of them are results of missense, frameshift, or splice-site mutations, which create truncated proteins and generate haploinsufficiency. In addition, these variants may alter the nuclear localization of the protein and its ability to bind to DNA.Citation13–Citation15 Some cases have also been reported with chromosomal alterations, such as translocations, deletions, and duplications.Citation13,Citation15 A mutation in TBX5 has been identified in 70% of patients with HOS,Citation5 with a variable phenotype associated with the mutation type and the localization.Citation17,Citation18 Furthermore, other genes, such as SALL4, have also been identified to be associated with disease development.Citation16
Herein, we report a novel de novo, potentially pathogenic mutation in the TBX5 gene as a cause of HOS in one patient with clear clinical diagnosis.
Case presentation
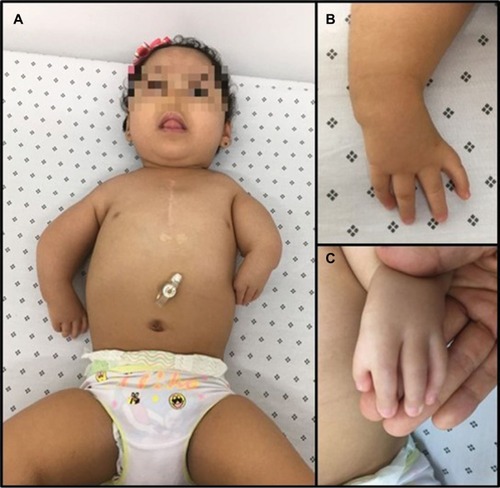
The patient was a Colombian 2-year-old female infant who was a product of non-consanguineous pregnancy and the first and only child of the family. She was born when her father was 34 years old and her mother was 30 years old. She had a family history of seizure syndrome and febrile crisis from the maternal lineage without other important family histories, such as genetic or neurological disorders. No complications were reported during the perinatal care and prenatal studies. She was born through vaginal delivery at 39.5 weeks gesta tion without any complications, weighed 3.170 g (50th–75th percentile), had a height of 51 cm (50th–75th percentile), with an Apgar score of 9–10. She was born with multiple CDSL and bilateral phocomelia, shortening of the left arm mainly, syndactyly of the second and third fingers of the left hand, and agenesis of both thumbs (). She underwent a period of hospitalization for these multiple malformations.
Figure 1 Morphological abnormalities.
The patient presented with ostium secundum-type atrial septal defect, muscular ventricular septal defect, and pulmonary hypertension that worsened when she was 6 months old. Due to this worsening, she was placed under pediatric critical care again. The computerized tomography scan showed diffuse cerebral edema with a partial collapse of the ventricular system without other focal lesions such as embolism and bleeding. Magnetic resonance imaging of the brain revealed restriction of generalized diffusion predominantly in the posterior and right areas without ventricular collapse and subdural efflux without compressive effect associated with adjacent cortical changes ascribable to necrosis. The electroencephalogram showed the absence of deep rhythms, severe slowing of the wave registers, and high-voltage bursts. Surgical intervention was provided to close the aortopulmonary and the pulmonary cerclage. After the first surgery, she presented with cerebral edema with encephalopathy due to axonal sprouting after the cortical lesion and generalized hypertonia as a consequence of the motor descending tract lesion, which was treated with clonazepam and phenobarbital without new episodes.
At 14 months of age, she presented with oropharyngeal dysphagia with moderate oral motor skills. For that reason, she received enteral feeding by gastrostomy. An intestinal transit study showed a duodenal corkscrew sign in the second intestinal portion at the Treitz angle level on the right side close to the right pedicle (L1). The image showed that the first jejunal handles were at the right center. All of these findings were related to intestinal malrotation changes. These changes included normal cecum position and gastroesophageal reflux grade III, which reached the barium column up to the proximal region of the esophagus. The follow-up electrocardiogram showed increased signs of sinus rhythm and right ventricular function. In addition, there was evidence of successful modification of the aortopulmonary window and pulmonary cerclage, muscular restrictive interventricular communication with a few left to right shunts, properly biventricular function without overflow, and pulmonary hypertension signs without clots and vegetations.
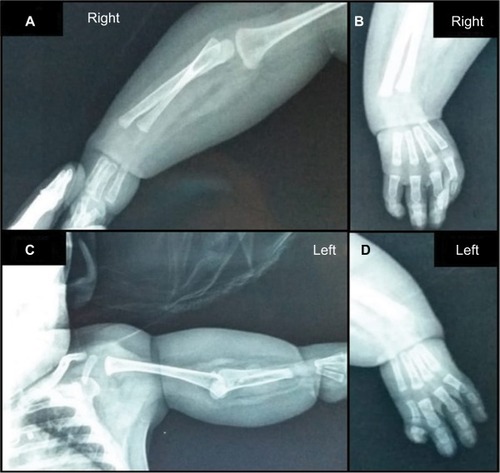
Radiological studies of the upper limbs revealed absence of bilateral metacarpal bones and phalanges of the first finger, bilateral radioulnar hypoplasia, absence of the ulna in the left arm (), hip dysplasia, and femoral–tibial varum angle (genu varum). No radiological changes in the spinal column X-ray were detected.
Figure 2 X-ray images of the upper limbs.
She was referred for genetic consultation at 2 years of age with a weight of 11.8 kg (50th–75th percentile), height 88 cm (75th–90th percentile). She presented with forehead hemangioma and hypoplasia of the nasal bones without appropriate visual follow-up, cephalic support was not successful, trunk and upper limbs were hypotonic, superior limbs with spasticity were more notable on the left side, and nipple hypertelorism.
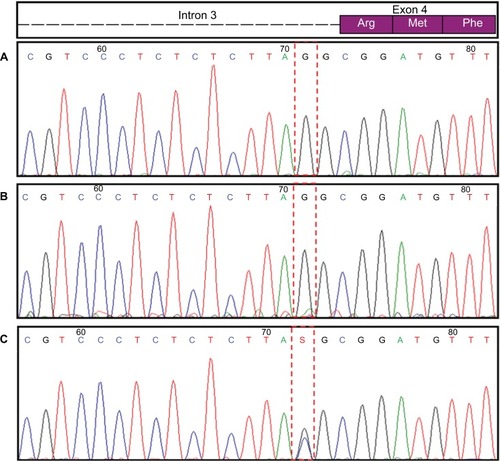
A complete sequence of the TBX5 gene was generated and compared to the NG_007373.1 (GeneBank) reference sequence which identified a novel potentially pathogenic variant in the heterozygous status NM_181486.2:c.243-1G>C. This splice-site mutation alters the acceptor site of the third intron, which is predicted to affect the splicing and lead to a nonfunctional protein. Using Sanger sequencing, this novel variant was confirmed using an ABI 3500 sequencer (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA). A carrier study in the parents was negative for this mutation (). For that reason, this mutation was considered to be a novel de novo mutation, which has not been previously reported.
Figure 3 TBX5 gene sequence.
Abbreviation: HOS, Holt-Oram syndrome.
Discussion
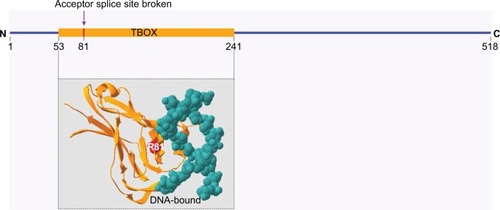
HOS is an autosomal dominant disorder, and in the majority of cases, it is caused by TBX5 mutations. This gene is located in the long arm of chromosome 12 (12q24.1), which codes for TBX5 protein and has a fundamental role in tissue and organ formation during embryonic development. It is involved in the normal development of the upper limbs and heart (structure and electric system) by activating different genes.Citation9,Citation19,Citation20 In the present case report, a point mutation in the TBX5 gene was identified in the acceptor site of the third intron, NM_181486.2:c.243-1G>C, which has not been previously reported. Bioinformatic analyses predicted an alteration in the acceptor site, which affects splicing (HSF v 3.1) and is classified as a pathogenic variant according to the ACMG Standards and Guidelines.Citation21 Furthermore, this mutation is located in a site of T-box domain that may affect the binding with DNA (). In our patient, because none of the family members had morphological anomalies in the upper limbs or heart defects and the carrier studies were negative, a de novo mutation was confirmed.
Figure 4 Affected site due to splicing mutation localization of the splice site mutation (NM_181486.2:c.243-1G>C) in a site of T-box domain (amino acid 81, arginine) near DNA binding site.
Our patient meets the HOS criteria described in the literature,Citation5 which includes upper limb malformations and heart defects. The patient presented with severe asymmetric radial malformations combined with mild cardiac abnormalities which were corrected by surgical procedures. Superior limb abnormalities can be unilateral or bilateral and asymmetric. They are usually worse on the left side compared to the right side,Citation2 which was clearly seen in this case wherein the patient had bilateral malformation predominantly on the left side of the body with the absence of the left ulna and a shortened upper left limb. The absence of the thumb is the most frequent limb abnormality, whereas ulnar abnormalities are less frequent than radius alterations.Citation6
Lower limb, craniofacial, pulmonary, genitourinary, and gastrointestinal malformations have been occasionally reported.Citation6,Citation22–Citation24 However, they are not the main characteristics of this syndrome.Citation1,Citation17 Hip dysplasia is a congenital malformation previously reported in only one patient with HOS,Citation6 due to hip malformation being an isolated sign of HOS, it is proposed as a random association and does not eliminate the diagnosis to HOS, as some authors have suggested in previous studies.Citation2,Citation25
The radius and ulna can easily be observed between the 13th and 16th week of gestation, and a majority of congenital defects are visible during routine prenatal care between the 18th and 20th week of pregnancy. However, more than 60% of the cases were not suspected during the prenatal follow-up,Citation6 which was the case in our patient. The different congenital malformations were not detected during pregnancy.
Conclusion
We identified and reported a novel, potentially pathogenic mutation as a cause of HOS in one patient with clear clinical diagnosis. We also presented an atypical sign of HOS (hip dysplasia), which might not be directly related to the clinical spectrum of this syndrome, but did not exclude HOS diagnosis in this case.
Consent for publication
Written informed consent was obtained from the patient’s parents for publication of her images and clinical data for scientific purposes. Data were collected in the context of studies performed in accordance with the Declaration of Helsinki Good Clinical Guidelines and protocol no 509 “registry of surveillance and survival of congenital defects of the Colombian South-West” approved by the Ethics Committee of Universidad Icesi (Act 192/2011).
Acknowledgments
We thank the family for being a part of this study.
Disclosure
The authors report no conflicts of interest in this work.
References
- HoltMOramSFamilial heart disease with skeletal malformationsBr Heart J196022Case 123624214402857
- HuangTCurrent advances in Holt-Oram syndromeCurr Opin Pediatr200214669169512436037
- orpha.net [homepage on the Internet]BossertTWaltherTGummertJHubaldRKostelkaMMohrFWHolt-Oram syndrome. Orphanet Encycl2003 Available from: http://www.orpha.net/data/patho/GB/uk-HOS.pdfAccessed November 1, 2018
- HurstAHallCMBaraitserMThe Holt-Oram syndromeJ Med Genet19912864064101870097
- McdermottDABressanMCHeJTBX5 genetic testing validates strict clinical criteria for Holt-Oram syndromePediatr Res200558598198616183809
- BarisicIBobanLGreenleesRHolt Oram syndrome: a registry-based study in EuropeOrphanet J Rare Dis2014915625344219
- SlettenLJPierpontMEVariation in severity of cardiac disease in Holt-Oram syndromeAm J Med Genet19966521281328911604
- Weizmann Institute of ScienceGeneCards Human Gene Database. TBX5 Gene Available from: http://www.genecards.org/cgi-bin/card-disp.pl?gene=TBX5Accessed November 1, 2018
- SimonHT-box genes and the formation of vertebrate forelimb- and hindlimb specific patternCell Tissue Res19992961576610199965
- HatcherCJGoldsteinMMMahCSDeliaCSBassonCTIdentification and localization of TBX5 transcription factor during human cardiac morphogenesisDev Dyn20002191909510974675
- SteimleJDMoskowitzIPTBX5: a key regulator of heart developmentCurr Top Dev Biol201712219522128057264
- AgarwalPWylieJNGalceranJTbx5 is essential for forelimb bud initiation following patterning of the limb field in the mouse embryoDevelopment2003130362363312490567
- KimuraMKikuchiAIchinoiNKureSNovel TBX5 duplication in a Japanese family with Holt-Oram syndromePediatr Cardiol201536124424725274398
- MoriADBruneauBGTBX5 mutations and congenital heart disease: Holt-Oram syndrome revealedCurr Opin Cardiol200419321121515096952
- FanCLiuMWangQFunctional analysis of TBX5 missense mutations associated with Holt-Oram syndromeJ Biol Chem2003278108780878512499378
- ChenLi BSunSXuKWuRYGenetic Analyses Identified a SALL4 Gene Mutation Associated with Holt–Oram SyndromeDNA Cell Biol201837439840429461882
- BassonCTCowleyGSSolomonSDThe clinical and genetic spectrum of the Holt-Oram syndrome (heart-hand syndromeN Engl J Med1994330138858918114858
- SmemoSCamposLCMoskowitzIPKriegerJEPereiraACNobregaMARegulatory variation in a TBX5 enhancer leads to isolated congenital heart diseaseHum Mol Genet201221143255326322543974
- StennardFAHarveyRPT-box transcription factors and their roles in regulatory hierarchies in the developing heartDevelopment2005132224897491016258075
- HatcherCJMcdermottDAUsing the TBX5 transcription factor to grow and sculpt the heartAm J Med Genet part A2006140131414141816691575
- ACMGGenetics Evaluation Guidelines for the Etiologic Diagnosis of Congenital Hearing Loss. Genetic Evaluation of Congenital Hearing Loss Expert Panel. ACMG statementGenet Med20024316217112180152
- BrassingtonAMSungSSToydemirRMExpressivity of Holt-Oram syndrome is not predicted by TBX5 genotypeAm J Hum Genet2003731748512789647
- TsengYRYnSFlLHolt-Oram syndrome with right lung agenesis caused by a de novo mutation in the TBX5 geneAm J Med Genet Part A2007143910121014
- GaravelliLde BrasiDVerriRHolt-Oram syndrome associated with anomalies of the feetAm J Med Genet Part A2008146A91185118918351627
- ChinJPereiraSCamachoAHolt-Oram syndrome: a case reportRev Port Cardiol20143311737.e1525455949