
Abstract
Hereditary hemochromatosis (HH) is an inherited iron overload disorder due to a deficiency of hepcidin, or a failure of hepcidin to degrade ferroportin. The most common form of HH, Type 1 HH, is most commonly due to a homozygous C282Y mutation in HFE and is relatively well understood in significance and action; however, other rare forms of HH (Types 2–4) exist and are more difficult to identify and diagnose in clinical practice. In this review, we describe the clinical characteristics of HH Type 2–4 and the mutation patterns that have been described in these conditions. We also review the different methods for genetic testing available in clinical practice and a pragmatic approach to the patient with suspected non-HFE HH.
Introduction
Iron is an essential metal for its biological role in hemoglobin and myoglobin to deliver oxygen to tissues, and as a cofactor for a host of enzymes.Citation1,Citation2 Body iron stores are regulated at the level of intestinal absorption, and there is no physiologic mechanism other than blood loss via menses for elimination of excess iron.Citation3 Therefore, prevention of iron overload must occur at the level of absorption in the duodenum.
Hepcidin
Hepcidin is a 25-amino acid peptide that regulates systemic iron homeostasis by inhibition of iron absorption.Citation4 Hepcidin originates in the liver in response to elevated iron levels and binds to ferroportin (FPN) on intestinal cells, resulting in a reduction of cellular iron export.Citation5 Hepcidin has major effects on control of iron absorption in enterocytes and macrophages, and is crucial to the regulation of overall serum iron levels.Citation6
Iron Overload Disorders
Iron overload disorders may be classified as primary or secondary iron overload disorders. Primary iron overload disorders have a genetic basis, and can be attributed to either low hepcidin production or decreased binding interactions between hepcidin and FPN, the transmembrane cellular iron exporter.Citation7,Citation8 These primary iron overload disorders are classified as different types of hereditary hemochromatosis. These disorders have variable phenotypic expression but all share the central defect of decreased hepcidin activity due to different mutations. The most frequent form of hereditary hemochromatosis is one of the most common genetic disorders among Caucasians, with a homozygote frequency of approximately 1 in 250 individuals of Northern European descent.Citation9,Citation10 Type 1 or classical hereditary hemochromatosis, is due to mutations in HFE, the gene encoding the HFE protein.Citation11 Patients with HFE-associated HH may be asymptomatic; chronic fatigue and arthropathy may be early signs.Citation3 The most common HFE mutation is a guanine to alanine substitution at position 845 of the HFE gene, resulting in a cysteine to tyrosine change (p.C282Y).Citation11 This mutation is inherited in an autosomal recessive pattern, and p.C282Y heterozygotes generally do not express the hemochromatosis phenotype.Citation12 Clinical penetrance of p.C282Y hereditary hemochromatosis may be as low as 2%, and is influenced by other factors leading to iron overload such as excess alcohol intake or chronic hepatitis C13. The mutation responsible for p.C282Y was originally identified in 1996 by a full physical mapping of the 3-Mb genomic region around the HLA-A3 allele on chromosome 6 in diagnosed HH patients.Citation11 Along with p.C282Y, a second missense mutation in HFE was also identified in the same physical mapping, with a cytosine to guanine change resulting in a substitution from histidine to aspartic acid, p.H63D.Citation11 p.H63D heterozygotes may have an increased likelihood of iron overload if they are compound heterozygotes with the p.C282Y mutation; this form of HH is known as type 1B hereditary hemochromatosis. However, heterozygosity or homozygosity for H63D alone is unlikely to result in expression of the hemochromatosis phenotype.Citation13
The p.C282Y mutation is present in a large proportion of patients who present with the phenotype of hereditary hemochromatosis (80.6%).Citation14 Testing for HFE-associated hereditary hemochromatosis is readily available, as a multiplex-PCR can be performed to identify p.H63D and p.C282Y concurrently.Citation15
Other more rare forms of hereditary hemochromatosis relevant to the clinician may be caused by mutations in hemojuvelin (Type 2A HH), hepcidin antimicrobial peptide (Type 2B HH), transferrin receptor 2 (Type 3A HH), or ferroportin (Type 4 HH) ().Citation11,Citation16–Citation18 A recent publication by Sandhu et al is a comprehensive collection of cases of identified non-HFE hereditary hemochromatosisCitation19. It is important to note that iron overload may also be caused by inherited mutations in the H-ferritin gene (Type 5 HH); however, Type 5 HH has only been identified in one Japanese family.Citation20 Additionally, aceruloplasminemia can be the source of iron overload and present with increased serum ferritin, but is not generally considered a form of hereditary hemochromatosis.Citation21
Table 1 Hereditary Hemochromatosis Subtypes, Genes Affected and Most Common Mutations
Molecular Testing (General)
Molecular diagnosis of monogenic inherited diseases is based on identifying variants that may explain phenotypic patterns.Citation23 It is estimated that approximately 8% of all live births will be diagnosed with a genetic abnormality, with the majority due to monogenic autosomal variants.Citation24 Linkage mapping of recessive traits using restriction fragment length polymorphisms (RFLP) was first described as a method of diagnosis for recessive traits within consanguineous relatives in 1987,Citation25 and is particularly useful for autosomal recessive monogenic diseases such as cystic fibrosis.Citation26 However, linkage analysis limits resolution and may be unable to identify candidate genes.Citation23 In the late 20th century, identification efforts for pathogenic genes were typically carried out to identify monogenic diseases, and typically within single pedigrees.Citation27
The Human Genome Project (HGP) represented a watershed moment in sequencing and identification of genetic disease.Citation28 The HGP identified 19,000 protein-coding (exome) genes, representing approximately 1% of the genome; however, a majority of phenotypes and pathogenic variants occur in exomic loci.Citation29,Citation30 Massively parallel sequencing (MPS) or next-generation sequencing (NGS) drastically increased the data-to-price ratio of sequencing exomes, reducing genes of interest from 3 million base pairs of the genome to 20,000 single-nucleotide polymorphisms (SNPs) of exomes.Citation31 There are two approaches to exome sequencing: whole-exome sequencing and array-based sequencing.Citation32 NGS can be used as a diagnostic tool for rare diseases and can reveal novel mutations within known clinical genes of interest.Citation33 Whole-exome sequencing (WES) focuses screening on exomes of genes of interest, as interpretation of introns is limited.Citation34 This method has proved effective in postmortem diagnosis in early sudden death due to monogenic disease variants.Citation35 Exome sequencing is valuable for diagnosis of prenatal disease, and WES of parent and child can be used to identify de novo mutations of interest.Citation31 For example, WES has expanded genetic diagnosis of terminal prenatal conditions after ultrasound abnormalities, even when karyotyping appears normal.Citation34 Definitive diagnosis after WES has been estimated to be approximately 25 to 35% and is especially useful in determining autosomal dominant mutations.Citation36 Chromosomal microarray analysis (CMA) examines changes in genes within a known chromosomal region.Citation37 CMA has higher sensitivity for nucleotide additions and deletions and is typically used to determine copy number variants (CNVs) or SNPs of clinical significance.Citation38 Genetic testing for diagnosis of disease is a rapidly evolving field, with genotyping not only being used for diagnosis of diseases but may also to guide individualized treatment regimens.Citation39
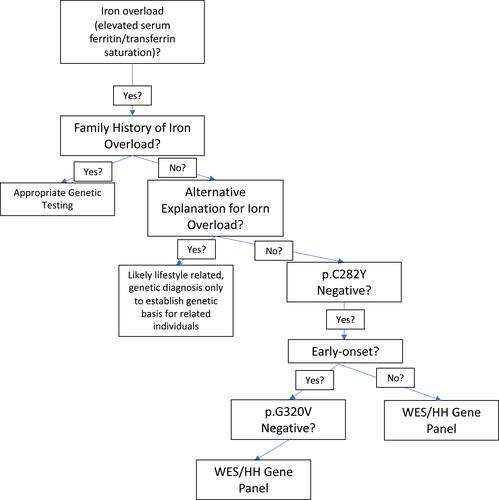
Hereditary hemochromatosis may also be identified through molecular testing but is not as straightforward as simply testing for p.C282Y. In a genotype analysis of African Americans, primary iron overload was found to be due to varying causes and not as straightforward as the relative frequency of p.C282Y in Caucasian patients with primary iron overload.Citation40 Since the clinical consequences and diagnosis of HH have been described in detail, in this review we discuss and elucidate features and diagnosis of non-HFE hemochromatosis. A variety of options exist for clinicians to test for non-HFE hemochromatosis (). Genetic testing for HH is usually ordered in patients with elevated serum transferrin-iron saturation and/or ferritin; the diagnosis of non-HFE hemochromatosis is usually only suspected in patients who test negative for the p.C282Y mutation.Citation41
Table 2 Available Testing for Non-HFE Related HH Subtypes, as of 6/22/21
Type 2A- HJV
Type 2 hereditary hemochromatosis, also known as juvenile hemochromatosis (JH), is an early-onset form of HH. Two subtypes of juvenile hemochromatosis have been identified: type 2A and type 2B. Type 2A hereditary hemochromatosis is caused by mutations in HJV, located on chromosome 1q which encodes hemojuvelin (HJV), which is expressed in the same tissues as hepcidin.Citation48 The median age of presentation for Type 2A is 25 years, and while Type 1 hemochromatosis shows a male predominance, HH Type 2A affects sexes equally.Citation2 Type 2A HH has a distinct set of clinical symptoms compared to Type 1 HH, and characteristically includes iron overload, hypogonadotropic hypogonadism, diabetes and most significantly, cardiomyopathy.Citation49 Type 2A HH may also result in amenorrhea in women, further increasing body iron stores.Citation50 If untreated, cardiomyopathy is a frequent cause of death among JH probands.Citation51,Citation52 Based on family members of one lineage, it was suggested that clinical penetrance of HJV mutations is higher than mutations in HFE.Citation50 The earliest age at which patients present with symptoms of iron overload associated with HJV mutations was age 4 in an African American patient with the homozygous nonsense mutation p.R54X.Citation53
Originally known as HFE2, HJV was first mapped by Papanikolaou et al, from a Greek family.Citation16 Six candidate mutations were identified based on polymorphisms compared to controls in ten Greek families, one Canadian family and one French family in juvenile hemochromatosis patients without p.C282Y mutations.Citation16 The most common mutation in HJV is a guanine to thymine transition originally discovered by Sanger sequencing, resulting in a glycine to valine substitution in position 320 of HJV (p.G320V).Citation16 WES also revealed a homozygous substitution in HJV from guanine to thymine in position 959, confirming WES validity in diagnosis of p.G320V hemochromatosis.Citation54 The second mutation in HJV identified by Papanikolaou et al was a substitution of thymine to adenine at position 665, known as p.I222N.Citation16 The p.I281T mutation has been detected both as a homozygous mutation in a Greek patient and as compound heterozygous with the nonsense mutation p.C321X in Chinese patients.Citation16,Citation55 Mutations in HJV are not always identified in patients with juvenile hemochromatosis; HJV mutations in Japan (p.D249H, p.Q312X) have resulted in HH phenotypes with onset in middle age.Citation56
In a PCR-RFLP study in Alabama of Caucasians and African Americans there were no p.G320V homozygotes and only one heterozygote for p.I222N was found, indicating the relative rarity of these mutations in the population.Citation57 It has been proposed that WES should be considered for a possible diagnosis of Type 2A HH only after demonstration of elevated ferritin and transferrin saturation, and negative HFE and p.G320V testing.Citation54 Classical symptoms of Type 2A HH accompanied by secondary hypothyroidism and iron overload in the pituitary may be due to a frameshift mutation from a cytosine deletion at position 697 of the HJV gene, resulting in a stop codon at amino acid 245 of HJV.Citation58 Commercially available testing for Type 2A HH includes single gene panels for p.G320V available from FulgentCitation43 and Invitae;Citation44 exome sequencing available from Blueprint Genetics,Citation42 Prevention Genetics,Citation45 the Valencian Institute of Microbiology (Ivami)Citation46 and Invitae.Citation44 Companies identified for testing were identified by the use of search engine, using keywords “non-HFE hemochromatosis testing, HJV testing”.
Type 2B- HAMP
The HAMP gene located on chromosome 19 directly encodes HAMP, a propeptide that is cleaved into the 25 amino acid hepcidin. Clinical features of HAMP hemochromatosis are similar to those of Type 2A HH.Citation13 HAMP mutations are relatively rare and heterozygosity in HAMP can be observed as a modifier of the p.C282Y HFE phenotype.Citation59 Microsatellite analysis of chromosome 19q13 in two individuals of Italian and Greek descent with juvenile hemochromatosis (JH) but with no HJV mutations revealed two HAMP mutations: a frameshift mutation resulting from a guanine deletion at position 3 of HAMP (93delG) and a cytosine to guanine transversion resulting in a nonsense mutation (p.R56X).Citation60 This led HAMP-associated HH to be classified as a subtype of JH. PCR screening of the HAMP gene in 21 unrelated JH patients identified a missense from cysteine to arginine (p.C70R), removing a stabilizing disulfide in the structure of hepcidin.Citation61 Mutations in HAMP have also been observed in both Russia and Portugal, and the overall geographic range is not completely defined.Citation61,Citation62
A mutation in the 5ʹUTR of HAMP mRNA has been identified in Portuguese patients, creating a mutant initiation codon resulting in iron overload phenotypes from mistranslation of hepcidin.Citation63
As in Type 2A HH, testing for Type 2B HH includes exome sequencing available from Blueprint GeneticsCitation42 Prevention Genetics,Citation45 Valencian Institute of Microbiology (Ivami)Citation46 and Invitae,Citation44 and next-generation sequencing from Fulgent.Citation43 Companies identified for testing were identified by the use of search engine, using keywords “non-HFE hemochromatosis testing, HAMP testing”.
Type 3- TFR2
Type 3 hereditary hemochromatosis is caused by mutations in the transferrin receptor 2 (TFR2) gene on chromosome 7; TFR2 is relatively long with a length of 20 kB and many mutations and polymorphisms have been identified.Citation64 It was initially thought that HFE formed dimers in the duodenum with transferrin receptor 2 (TFR2), but TFR2 acts as an independent regulator of hepcidin in hepatocytes.Citation65 TFR2-associated hemochromatosis is described as intermediate in clinical presentation between HFE-HH and HJV-HH, and mutations in TFR2 can produce a form of HH resembling juvenile hemochromatosis.Citation66,Citation67
TFR2-associated hereditary hemochromatosis is rare, with approximately 65 cases in 44 family lineages across a wide geographic area.Citation68 Although TFR2-HH was initially discovered in Italy, subsequent cases were found in Scotland, Spain, Japan and Taiwan.Citation68–Citation71 However, TFR2-associated HH is still most commonly found in Italy and Japan.
Mutations in TFR2 were first discovered upon analysis of two unrelated families of Sicilian origin (one heavily inbred) presenting with hemochromatosis but with no mutations in p.C282Y.Citation17 Linkage mapping excluding HFE and HJV revealed homozygosity in the region of TFR2 on chromosome 7; microsatellite analysis confirmed a cytosine to guanine substitution in exon 6 of TFR2, creating a nonsense mutation (p.Y250X) in TFR2.Citation17 The second mutation in TFR2 to be discovered was within a consanguineous family, and was an insertion of a cytosine into exon 2, resulting in a nonsense mutation at amino acid 60 (p.E60X).Citation72
A homozygous deletion in exon 16 of TFR2 resulting in deletion of amino acids 594–597 was found in a proband of Northern Italian descent with known iron overload, and intrafamilial haplotyping of affected siblings detected early-onset HH phenotype.Citation69 Three potential mutations of TFR2 were proposed in a Scottish man in 2006, including p.R396X, and p.G792R, but could not be directly attributed to the observed HH phenotype.Citation70 Despite the relative rarity of HH in Asian populations, WES of TFR2 in a Taiwanese woman found a mutation of guanosine to adenosine in exon 11 (p.R481H) resulting in extreme iron overload and diabetes mellitus.Citation71 The first described change in TFR2 mRNA levels was a splice site mutation resulting in skipping of exon 4 in TFR2 reported in a woman of Southern Italian descent.Citation73 In a study of four unrelated Spanish patients with HH symptoms and no mutations in HFE, HJV, HAMP and SLC40A1, homozygous mutations in TFR2 were discovered, including a missense mutation p.G792R, and nonsense mutations p.Q306X and p.Q672X.Citation68
TFR2 sequencing as a single gene is available from InvitaeCitation44 and Fulgent,Citation43 and whole exome sequencing is available from Blueprint Genetics,Citation42 IvamiCitation46 and Prevention Genetics.Citation45 Additionally, Baylor Genetics Laboratories has created a sequence-specific test designed to detect private mutations.Citation47 Companies identified for testing were identified by the use of search engine, using keywords “non-HFE hemochromatosis testing, TFR2 testing”.
Type 4A- FPN Disease
Type 4A hereditary hemochromatosis is also known as classic ferroportin disease (FD), and can be categorized as its own disease associated with iron overload. Outside of HFE-HH, FD is the most prevalent form of genetic iron overload regardless of race.Citation18,Citation74 FD is caused by pathogenic mutations in Fpn or SLC40A1, which encodes ferroportin (FPN), also known as Solute Carrier Family 40 Member 1 protein. FD is distinct from hereditary hemochromatosis because it is not associated with high transferrin-iron saturation or low hepcidin concentrations, and unlike other forms of HH, FD is typically inherited in an autosomal dominant pattern.Citation74,Citation75 In FD, the hepcidin-FPN interaction marking FPN for degradation is disturbed, resulting in a heterogeneity of clinical symptoms.Citation22
The most common FD genotypes are an alanine to aspartic acid at residue 77 (p.A77D) substitution in exon 3 of the FPN gene and a deletion of valine at residue 162 (p.V162del).Citation8,Citation22 p.A77D was detected by PCR amplification of exons in the FPN gene comparing probands and control groups.Citation8 Two missense mutations causing FD (p.I152F and p.L233P) were identified in probands by denaturing high-performance liquid chromatography (DHPLC), where Fpn mutations were transfected in vitro into kidney cell lines and then injected into zebrafish embryos, using immunofluorescence to detect FPN localization and degradation in the cell.Citation76
Molecular testing for SLC40A1 includes whole exome sequencing is available from Blueprint Genetics,Citation42 IvamiCitation46 and Prevention Genetics,Citation45 and next-generation sequencing from Fulgent.Citation43 Companies identified for testing were identified by the use of search engine, using keywords “non-HFE hemochromatosis testing, FPN testing”.
Type 4B
Type 4B hereditary hemochromatosis also results from a mutation in SLC40A1; however, Type 4B HH is not caused by a change in expression of SLC40A1, but rather is a gain-of-function mutation in FPN. This “resistant” form of FPN is no longer susceptible to degradation by hepcidin binding and remains hyperactive on the cell membrane.Citation77 Types 4A and 4B HH can be distinguished clinically by the finding of a low to normal transferrin saturation (TS) in type 4A and high TS in type 4B.Citation22 MRI has been suggested as a non-invasive tool to distinguish between the gain-of-function and loss-of-function forms of Type 4 HH.Citation78
The first form of Type 4B HH was discovered by RFLP analysis of Thai and Vietnamese patients by an autosomal dominant mutation p.C326Y creating a resistant FPN with no effect on protein localization.Citation79 The cysteine residue at position 326 is involved in a disulfide interaction between hepcidin and FPN, and any change in the cysteine at position 326 of FPN results in minimal hepcidin internalization.Citation80,Citation81 An Australian proband with cirrhosis and parenchymal iron overload onset was identified with relatively early onset of disease at 32 years old, with a change in the asparagine residue to aspartic acid at position 144.Citation82 A splice mutation in SLC40A1 was detected by PCR analysis of HFE, HJV, HAMP, TFR2 compared against a cDNA library in a middle-aged Chinese woman with unexplained iron overload.Citation83
Conclusion
Primary iron overload not due to mutations in HFE can be due to a variety of mutations in HJV, HAMP, TFR2 and/or FPN. These rare forms of non-HFE HH and difficult to diagnose with certainty, given the variety of mutations, many of which are private. Moreover, compound heterozygosity can add challenges to the diagnosis when HFE mutations accompany HJV or HAMP mutations (). It is also important to note that the majority of suspected HH cases are not attributable to any genetic cause, so adequate clinical suspicion is necessary for diagnosis of genetic HH and it is important to eliminate secondary causes of iron overload. Clinicians should always seek alternative explanations for iron overload in patients with iron overload in the absence of HFE mutations before considering a diagnosis of non-HFE hereditary hemochromatosis given the rarity of these disorders. Patient genetic history and ancestry should be taken into account before diagnosis and treatment with therapeutic phlebotomy.Citation13 Genetic testing for non-HFE HH should be considered in patients with documented iron overload after other causes if iron overload have been excluded, especially among patients with a family history of iron overload. Several clinical laboratories offer genetic testing for these forms of HH and appropriate use of genetic testing may provide confirmation of the diagnosis and prognosis.
Figure 1 Flowchart for clinician approach to patients with unexplained iron overload.
Disclosure
Dr Kris V Kowdley reports grants, personal fees from La Jolla, personal fees from Protagonist, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
- Wang C-Y, Babitt JL. Liver iron sensing and body iron homeostasis. Blood. 2019;133:18–29.
- Gerhard GS, Paynton BV, DiStefano JK. Identification of genes for hereditary hemochromatosis. Methods Mol Biol. 2018;1706:353–365.
- Brissot P, Pietrangelo A, Adams PC, et al. Haemochromatosis. Nat Rev Dis Primers. 2018;4(1):18016. doi:10.1038/nrdp.2018.16
- Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276(11):7806–7810. doi:10.1074/jbc.M008922200
- Anderson GJ, Frazer DM. Current understanding of iron homeostasis. Am J Clin Nutr. 2017;106(Supplement 6):1559S–1566S. doi:10.3945/ajcn.117.155804
- Kowdley KV, Gochanour EM, Sundaram V, Shah RA, Handa P. Hepcidin signaling in health and disease: ironing out the details. Hepatol Commun. 2021;5(5):723–735. doi:10.1002/hep4.1717
- Siddique A, Kowdley KV. Review article: the iron overload syndromes. Aliment Pharmacol Ther. 2012;35(8):876–893. doi:10.1111/j.1365-2036.2012.05051.x
- Montosi G, Donovan A, Totaro A, et al. Autosomal-dominant hemochrom-atosis is associated with a mutation in the ferroportin (SLC11A3) gene. J Clin Invest. 2001;108(4):619–623. doi:10.1172/JCI200113468
- Adams PC, Reboussin DM, Barton JC, et al. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med. 2005;352(17):1769–1778. doi:10.1056/NEJMoa041534
- Gochee PA, Powell LW. What’s new in hemochromatosis. Curr Opin Hematol. 2001;8(2):98–104. doi:10.1097/00062752-200103000-00007
- Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I–like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13(4):399–408. doi:10.1038/ng0896-399
- Zaloumis SG, Allen KJ, Bertalli NA, et al. Natural history of HFE simple heterozygosity for C282Y and H63D: a prospective 12-year study. J Gastroenterol Hepatol. 2015;30(4):719–725. doi:10.1111/jgh.12804
- Kowdley KV, Brown KE, Ahn J, Sundaram V. ACG clinical guideline: hereditary hemochromatosis. Am J Gastroenterol. 2019;114(8):1202–1218. doi:10.14309/ajg.0000000000000315
- European Association For The Study Of The Liver. EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol. 2010;53:3–22.
- Arts HH, Eng B, Waye JS. Multiplex allele-specific PCR for simultaneous detection of H63D and C282Y HFE mutations in hereditary hemochromatosis. J Appl Lab Med. 2018;3(1):10–17. doi:10.1373/jalm.2017.024984
- Papanikolaou G, Samuels ME, Ludwig EH, et al. Mutations in HFE2 cause iron overload in chromosome 1q–linked juvenile hemochromatosis. Nat Genet. 2004;36(1):77–82. doi:10.1038/ng1274
- Camaschella C, Roetto A, Calì A, et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet. 2000;25(1):14–15. doi:10.1038/75534
- Pietrangelo A. The ferroportin disease. Blood Cells Mol Dis. 2004;32(1):131–138. doi:10.1016/j.bcmd.2003.08.003
- Sandhu K, Flintoff K, Chatfield MD, et al. Phenotypic analysis of hemochromatosis subtypes reveals variations in severity of iron overload and clinical disease. Blood. 2018 Jul 5;132(1):101–110. doi:10.1182/blood-2018-02-830562
- Kato J, Fujikawa K, Kanda M, et al. A mutation, in the iron-responsive element of H ferritin mRNA, causing autosomal dominant iron overload. Am J Hum Genet. 2001;69(1):191–197. doi:10.1086/321261
- Roberti MDRF, Borges Filho HM, Gonçalves CH, Lima FL. Aceruloplasminemia: a rare disease - diagnosis and treatment of two cases. Rev Bras Hematol Hemoter. 2011;33(5):389–392. doi:10.5581/1516-8484.20110104
- Mayr R, Janecke AR, Schranz M, et al. Ferroportin disease: a systematic meta-analysis of clinical and molecular findings. J Hepatol. 2010;53(5):941–949. doi:10.1016/j.jhep.2010.05.016
- Botstein D, Risch N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat Genet. 2003;33(S3):228–237. doi:10.1038/ng1090
- Baird PA, Anderson TW, Newcombe HB, Lowry RB. Genetic disorders in children and young adults: a population study. Am J Hum Genet. 1988;42:677–693.
- Lander ES, Botstein D. Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science. 1987;236(4808):1567–1570. doi:10.1126/science.2884728
- Kerem B, Rommens J, Buchanan J, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245(4922):1073–1080. doi:10.1126/science.2570460
- Claussnitzer M, Cho JH, Collins R, et al. A brief history of human disease genetics. Nature. 2020;577(7789):179–189. doi:10.1038/s41586-019-1879-7
- Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921.
- Chong JX, Buckingham K, Jhangiani S, et al. The genetic basis of mendelian phenotypes: discoveries, challenges, and opportunities. Am J Hum Genet. 2015;97(2):199–215. doi:10.1016/j.ajhg.2015.06.009
- Makrythanasis P, Antonarakis SE. Pathogenic variants in non-protein-coding sequences. Clin Genet. 2013;84(5):422–428. doi:10.1111/cge.12272
- Stranneheim H, Wedell A. Exome and genome sequencing: a revolution for the discovery and diagnosis of monogenic disorders. J Intern Med. 2016;279(1):3–15. doi:10.1111/joim.12399
- Schwarze K, Buchanan J, Taylor JC, Wordsworth S. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet Med. 2018;20(10):1122–1130. doi:10.1038/gim.2017.247
- Gilissen C, Hoischen A, Brunner HG, Veltman JA. Unlocking mendelian disease using exome sequencing. Genome Biol. 2011;12(9):228. doi:10.1186/gb-2011-12-9-228
- Jelin AC, Vora N. Whole exome sequencing: applications in prenatal genetics. Obstet Gynecol Clin North Am. 2018;45(1):69–81. doi:10.1016/j.ogc.2017.10.003
- Salfati EL, Spencer EG, Topol SE, et al. Re-analysis of whole-exome sequencing data uncovers novel diagnostic variants and improves molecular diagnostic yields for sudden death and idiopathic diseases. Genome Med. 2019;11(1):83. doi:10.1186/s13073-019-0702-2
- Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369(16):1502–1511. doi:10.1056/NEJMoa1306555
- Chang Y-S, Lin C-Y, Huang H-Y, Chang J-G, Kuo H-T. Chromosomal microarray and whole-exome sequence analysis in Taiwanese patients with autism spectrum disorder. Mol Genet Genomic Med. 2019;7(12):e996. doi:10.1002/mgg3.996
- Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–764. doi:10.1016/j.ajhg.2010.04.006
- Franceschini N, Frick A, Kopp JB. Genetic testing in clinical settings. Am J Kidney Dis. 2018;72(4):569–581. doi:10.1053/j.ajkd.2018.02.351
- Barton JC, Acton RT, Rivers CA, et al. Genotypic and phenotypic heterogeneity of African Americans with primary iron overload. Blood Cells Mol Dis. 2003;31(3):310–319. doi:10.1016/S1079-9796(03)00166-9
- Santos PCJL, Krieger JE, Pereira AC. Molecular diagnostic and pathogenesis of hereditary hemochromatosis. Int J Mol Sci. 2012;13(2):1497–1511. doi:10.3390/ijms13021497
- Blueprint Genetics. Available from: https://blueprintgenetics.com/tests/panels/metabolic-disorders/hereditary-hemochromatosis-panel/. Accessed July 20, 2021.
- Fulgent Genetics. Available from: https://www.fulgentgenetics.com/products/disease/raredisease.html. Accessed July 20, 2021.
- Invitae. Available from: https://www.invitae.com/en/physician/tests/05201/. Accessed July 20, 2021.
- Prevention Genetics. Available from: https://www.preventiongenetics.com/testInfo?val=Hereditary+Hemochromatosis+Panel. Accessed July 20, 2021.
- Valencian Institute of Microbiology (Ivami). Available from: https://www.ivami.com/en/genetic-testing-human-gene-mutations-diseases-neoplasias-and-pharmacogenetics/4301-genetic-testing-hereditary-hemochromatosis-i-hamp-hfe-hfe2-slca40a1-i-and-i-tfr2-i. Accessed July 20, 2021.
- Baylor Genetics. Available from: https://baylorgenetics.com/. Accessed July 20, 2021.
- Pagani A, Silvestri L, Nai A, Camaschella C. Hemojuvelin N-terminal mutants reach the plasma membrane but do not activate the hepcidin response. Haematologica. 2008;93(10):1466–1472. doi:10.3324/haematol.12508
- Li C-X, Zhang L, Wang P, Sun L. Clinicopathological diagnosis and treatment of juvenile hemochromatosis. Chin Med J. 2019;132(24):3018–3020. doi:10.1097/CM9.0000000000000547
- Lee PL, Beutler E, Rao SV, Barton JC. Genetic abnormalities and juvenile hemochromatosis: mutations of the HJV gene encoding hemojuvelin. Blood. 2004;103(12):4669–4671. doi:10.1182/blood-2004-01-0072
- Cherfane C, Lee P, Guerin L, Brown K. A late presentation of a fatal disease: juvenile hemochromatosis. Case Rep Med. 2013;2013:875093. doi:10.1155/2013/875093
- Filali M, Le Jeunne C, Durand E, et al. Juvenile hemochromatosis HJV-related revealed by cardiogenic shock. Blood Cells Mol Dis. 2004;33(2):120–124. doi:10.1016/j.bcmd.2004.05.001
- Murugan RC, Lee PL, Kalavar MR, Barton JC. Early age-of-onset iron overload and homozygosity for the novel hemojuvelin mutation HJV R54X (exon 3; c.160A→T) in an African American male of West Indies descent. Clin Genet. 2008;74(1):88–92. doi:10.1111/j.1399-0004.2008.01017.x
- Farrell CP, Parker CJ, Phillips JD. Exome sequencing for molecular characterization of non-HFE hereditary hemochromatosis. Blood Cells Mol Dis. 2015;55(2):101–103. doi:10.1016/j.bcmd.2015.04.002
- Lv T, Zhang W, Xu A, et al. Non- HFE mutations in haemochromatosis in China: combination of heterozygous mutations involving HJV signal peptide variants. J Med Genet. 2018;55(10):650–660. doi:10.1136/jmedgenet-2018-105348
- Koyama C, Hayashi H, Wakusawa S, et al. Three patients with middle-age-onset hemochromatosis caused by novel mutations in the hemojuvelin gene. J Hepatol. 2005;43(4):740–742. doi:10.1016/j.jhep.2005.06.024
- Barton JC, Rivers CA, Niyongere S, Bohannon SB, Acton RT. Allele frequencies of hemojuvelin gene (HJV) I222N and G320V missense mutations in white and African American subjects from the general Alabama population. BMC Med Genet. 2004;5(1):29. doi:10.1186/1471-2350-5-29
- Santiago de Sousa Azulay R, Magalhães M, Tavares MDG, et al. Novel mutation in the Hemojuvelin Gene (HJV) in a patient with juvenile hemochromatosis presenting with insulin-dependent diabetes mellitus, secondary hypothyroidism and hypogonadism. Am J Case Rep. 2020;21:e923108. doi:10.12659/AJCR.923108
- Jacolot S, Le Gac G, Scotet V, et al. HAMP as a modifier gene that increases the phenotypic expression of the HFE pC282Y homozygous genotype. Blood. 2004;103(7):2835–2840. doi:10.1182/blood-2003-10-3366
- Roetto A, Papanikolaou G, Politou M, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. 2003;33(1):21–22. doi:10.1038/ng1053
- Roetto A, Daraio F, Porporato P, et al. Screening hepcidin for mutations in juvenile hemochromatosis: identification of a new mutation (C70R). Blood. 2004;103(6):2407–2409. doi:10.1182/blood-2003-10-3390
- Potekhina ES, Lavrov AV, Samokhodskaya LM, et al. Unique genetic profile of hereditary hemochromatosis in Russians: high frequency of C282Y mutation in population, but not in patients. Blood Cells Mol Dis. 2005;35(2):182–188. doi:10.1016/j.bcmd.2005.06.012
- Matthes T, Aguilar-Martinez P, Pizzi-Bosman L, et al. Severe hemochromatosis in a Portuguese family associated with a new mutation in the 5′-UTR of the HAMP gene. Blood. 2004;104(7):2181–2183. doi:10.1182/blood-2004-01-0332
- Glöckner G, Scherer S, Schattevoy R, et al. Large-scale sequencing of two regions in human chromosome 7q22: analysis of 650 kb of genomic sequence around the EPO and CUTL1 loci reveals 17 genes. Genome Res. 1998;8(10):1060–1073. doi:10.1101/gr.8.10.1060
- Rishi G, Crampton EM, Wallace DF, Subramaniam VN, Avila M. In situ proximity ligation assays indicate that hemochromatosis proteins Hfe and transferrin receptor 2 (Tfr2) do not interact. PLoS One. 2013;8(10):e77267. doi:10.1371/journal.pone.0077267
- Radio FC, Majore S, Binni F, et al. TFR2-related hereditary hemochromatosis as a frequent cause of primary iron overload in patients from Central-Southern Italy. Blood Cells Mol Dis. 2014;52(2–3):83–87. doi:10.1016/j.bcmd.2013.08.003
- Pietrangelo A, Caleffi A, Henrion J, et al. Juvenile hemochromatosis associated with pathogenic mutations of adult hemochromatosis genes. Gastroenterology. 2005;128(2):470–479. doi:10.1053/j.gastro.2004.11.057
- Joshi R, Shvartsman M, Morán E, et al. Functional consequences of transferrin receptor-2 mutations causing hereditary hemochromatosis type 3. Mol Genet Genomic Med. 2015;3(3):221–232. doi:10.1002/mgg3.136
- Girelli D, Bozzini C, Roetto A, et al. Clinical and pathologic findings in hemochromatosis type 3 due to a novel mutation in transferrin receptor 2 gene. Gastroenterology. 2002;122(5):1295–1302. doi:10.1053/gast.2002.32984
- Lee PL, Barton JC. Hemochromatosis and severe iron overload associated with compound heterozygosity for TFR2 R455Q and two novel mutations TFR2 R396X and G792R. Acta Haematol. 2006;115(1–2):102–105. doi:10.1159/000089474
- Hsiao P-J, Tsai K-B, Shin S-J, et al. A novel mutation of transferrin receptor 2 in a Taiwanese woman with type 3 hemochromatosis. J Hepatol. 2007;47(2):303–306. doi:10.1016/j.jhep.2007.04.014
- Roetto A, Totaro A, Piperno A, et al. New mutations inactivating transferrin receptor 2 in hemochromatosis type 3. Blood. 2001;97(9):2555–2560. doi:10.1182/blood.V97.9.2555
- Pelucchi S, Mariani R, Trombini P, et al. Expression of hepcidin and other iron-related genes in type 3 hemochromatosis due to a novel mutation in transferrin receptor-2. Haematologica. 2009;94(2):276–279. doi:10.3324/haematol.13576
- Pietrangelo A. Ferroportin disease: pathogenesis, diagnosis and treatment. Haematologica. 2017;102(12):1972–1984. doi:10.3324/haematol.2017.170720
- Sham RL, Phatak PD, Nemeth E, Ganz T. Hereditary hemochromatosis due to resistance to hepcidin: high hepcidin concentrations in a family with C326S ferroportin mutation. Blood. 2009;114(2):493–494. doi:10.1182/blood-2009-04-216226
- Girelli D, De Domenico I, Bozzini C, et al. Clinical, pathological, and molecular correlates in ferroportin disease: a study of two novel mutations. J Hepatol. 2008;49(4):664–671. doi:10.1016/j.jhep.2008.05.028
- Majore S, Bonaccorsi Di Patti MC, Valiante M, et al. Characterization of three novel pathogenic SLC40A1 mutations and genotype/phenotype correlations in 7 Italian families with type 4 hereditary hemochromatosis. Biochim Biophys Acta. 2018;1864(2):464–470. doi:10.1016/j.bbadis.2017.11.006
- Pietrangelo A, Corradini E, Ferrara F, et al. Magnetic resonance imaging to identify classic and nonclassic forms of ferroportin disease. Blood Cells Mol Dis. 2006;37:192–196. doi:10.1016/j.bcmd.2006.08.007
- Viprakasit V, Merryweather-Clarke AT, Chinthammitr Y, et al. Molecular diagnosis of the first ferroportin mutation (C326Y) in the far east causing a dominant form of inherited iron overload. Blood. 2004;104(11):3204. doi:10.1182/blood.V104.11.3204.3204
- Preza GC, Ruchala P, Pinon R, et al. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J Clin Invest. 2011;121(12):4880–4888. doi:10.1172/JCI57693
- Fernandes A, Preza GC, Phung Y, et al. The molecular basis of hepcidin-resistant hereditary hemochromatosis. Blood. 2009;114(2):437–443. doi:10.1182/blood-2008-03-146134
- Wallace DF, Clark RM, Harley HAJ, Subramaniam VN. Autosomal dominant iron overload due to a novel mutation of ferroportin1 associated with parenchymal iron loading and cirrhosis. J Hepatol. 2004;40(4):710–713. doi:10.1016/j.jhep.2003.12.008
- Zhang W, Lv T, Huang J, Ou X. Type 4B hereditary hemochromatosis associated with a novel mutation in the SLC40A1 gene: a case report and a review of the literature. Medicine. 2017;96:e8064.