Abstract
Acute hyperammonemia is a medical emergency requiring rapid recognition and treatment to prevent devastating neurologic sequelae. Its varying etiologies include primary hepatic failure, drug toxicity, infection, and inherited disorders of metabolism. Ornithine transcarbamylase (OTC) deficiency is the most common inherited urea cycle disorder and can result in hyperammonemic encephalopathy and coma, often presenting in the newborn or early childhood. Partial deficiencies of the enzyme can present later in adulthood with protean neuropsychiatric signs and symptoms. Early recognition and management of metabolic encephalopathy is crucial to avoid neurologic damage, and may require hemodialysis for rapid removal of ammonia, with adjunctive medications and dietary modifications to decrease endogenous nitrogen production and activate alternate pathways of nitrogen excretion. We present the case of an adult patient with partial OTC deficiency who presented with encephalopathy, coma, and seizures, accompanied by hyperammonemia and treated acutely with hemodialysis.
Introduction
Ornithine transcarbamylase (OTC) deficiency is the most common inherited disorder of the urea cycle and is a well-known, but infrequent cause of hyperammonemia. Severe or complete OTC deficiency manifests as encephalopathy, coma, and seizures, resulting in death, usually in newborns and infants. Partial OTC deficiency may present more variably especially in adults, progressing from behavioral and mood changes, nausea and emesis (particularly after consuming meat and other high-protein foods), encephalopathy with irritability, confusion, seizures, and/or coma (CitationHawkes et al 2001; CitationSummar et al 2005). Unfortunately, these early indistinct clinical features lead to delays in recognizing the diagnosis and initiating treatment, leading to poor neurologic outcomes and overall survival. As such, the diagnosis of an underlying inherited disorder of the urea cycle is often missed, often until late acute neurologic deterioration occurs, or sometimes the diagnosis remains undiscovered until post-mortem investigation.
OTC deficiency is the most common of the urea cycle enzyme deficiencies (estimated incidence of 1/14,000 (CitationSmith et al 2005) and has X-linked inheritance (CitationNassogne et al 2005). The other urea cycle enzyme deficiencies have an autosomal recessive inheritance pattern (CitationNassogne et al 2005). The urea cycle functions to offload nitrogenous wastes (derived from normal protein catabolism) – ammonia in particular – in the form of urea, facilitating its excretion via the kidneys into the urine. Disruption of urea synthesis by inborn errors of metabolism leads to hyperammonemia and accumulation of other metabolic intermediaries (CitationSummar et al 2005).
Elevated ammonia in the central nervous system (CNS) leads to neurotoxicity not via a direct mechanism but rather due to the accumulation of glutamate as a result of interruptions to the normal breakdown of ammonia via the urea cycle. Hyperammonemia shifts the equilibrium of the glutamate dehydrogenase reaction toward the direction of glutamate formation, depleting alpha-ketoglutarate, and resulting in decreased cellular oxidation and ATP production, leaving the brain vulnerable due to its high energy consumption. Additionally, glutamate is osmotically active, and accumulates intracellularly in astrocytes. Accompanied by this is activation of the N-methyl-D-aspartate (NMDA) receptor, which enhances the formation of nitric oxide and causes cerebral vasodilatation. These osmotic and vasodilatory actions can chronically result in cerebral atrophy, ventricular enlargement, and delayed myelination; acutely the effect is cerebral edema, resulting in increased intracranial pressure and uncal herniation (CitationMathias et al 2001), manifesting clinically with depression in consciousness, loss of higher CNS functions, and seizures.
Radiologic modalities, such as computed tomography (CT) and magnetic resonance imaging (MRI) can demonstrate cerebral edema. Cerebral swelling with symmetric cortical and subcortical damage that does not match a vascular distribution, effectively excludes focal ischemic and hemorrhagic injury. In patients with cerebral edema secondary to OTC deficiency, MRI commonly shows symmetric involvement of both cerebral hemispheres and pallidal nuclei, with specific involvement of the insula, frontal, and temporal lobes, and sparing the brainstem and cerebellum. The absence of atherosclerotic vascular disease or drug abuse, coupled with the above imaging, should draw suspicion towards an acute metabolic process (CitationGaspari et al 2003).
Urea cycle disorders, such as OTC deficiency, that manifest during adulthood are more often discovered after common etiologies for encephalopathy are ruled out. Often, unexplained elevated serum ammonia levels in the setting of normal liver-associated-enzymes without evidence of primary hepatic failure, are the first clue. A laboratory profi le of respiratory alkalosis, anion gap metabolic acidosis, hypoglycemia, ketonuria, elevated transaminase level, or bone marrow suppression, should raise suspicion for an inherited metabolic disorder (urea cycle defect, organic acidemia, fatty acid oxidation disorder) (CitationGreene et al 1988; CitationMathias et al 2001). Respiratory alkalosis is a classic feature of urea cycle defects in patients with cerebral edema, and occurs due to hyperventilation, resulting from stimulation of central respiratory drive. Metabolic acidosis can be present in severely ill patients with urea cycle defects, due to perfusion deficits (CitationSmith et al 2005). Once a clinical diagnosis of hyperammonemia due to a urea cycle disorder is suspected, further laboratory studies, to include urine amino acid and organic acid analysis, with quantitative orotic acid analysis, can be helpful. In OTC deficiency elevations of urine orotic acid, serum glutamine, and decreased serum arginine and citrulline are often seen.
Case report
A 34-year-old otherwise healthy Caucasian male was found unconscious in his quarters by members of his unit. He had been deployed to Iraq in support of Operation Iraqi Freedom for 9 months. He had recently been home on mid-tour leave, noticed he had lost weight, and began taking protein supplements. He had done well medically during the first part of the deployment, but began experiencing post-prandial nausea and vomiting, inability to eat meat, and unintentional weight loss. After being found unconscious, he was taken to a nearby Military Treatment Facility, and expeditiously transported to a Combat Support Hospital in theater, with limited laboratory capabilities.
Evaluation at that time was significant for a GCS of 9 with intact cranial nerve exam, normal head CT and normal serum laboratories to include electrolytes and blood counts. His serum ammonia level was 125 μmol/L (normal 9–33 μmol/L), but in the combat medical facility the specimen had to be sent out for processing, and a result was not available at time of evaluation. He was transferred to Germany within 24 hours and the diagnosis of hyperammonemia was made, after an extensive neurologic work-up to include lumbar puncture, infectious serologies, and imaging. In Germany, his examination was significant for worsening neurologic exam with inability to protect his airway, requiring endotracheal intubation and mechanical ventilation. Laboratory evaluation at that time was remarkable for respiratory alkalosis and a serum ammonia ranging to 233 μmol/L. CT of the head showed effacement of the gyri, cerebral edema, and no midline shift. He was then transferred to our medical center for definitive care.
En route, his serum ammonia rose to 250 μmol/L. He developed seizures that progressed to status epilepticus while in-flight, necessitating treatment with pentobarbital and propofol coma.
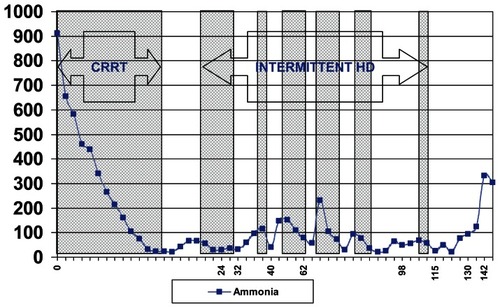
At our hospital, the patient’s initial serum ammonia was 956 μmol/L. Hemodialysis was instituted with the goal of reducing his serum ammonia level to less than 125 μmol/L. Further studies at that time revealed an elevated urinary orotic acid level of 609 μmol/molCr (normal 0–3 μmol/molCr), with a urine creatinine of 0.80 mmol/L (normal 2.38–26.55 μmol/L). His urine amino acid analysis showed an elevated level of glutamine (227.6 μmol/molCr, normal 21.5–57.6), and low arginine (0.0 μmol/molCr, normal 1.1–10.2 μmol/molCr) (). These lab findings were felt to be consistent with a diagnosis of partial OTC deficiency. Treatment with lactulose and neomycin was used to decontaminate the intestinal tract, and antibiotics were given to cover an aspiration pneumonia of the right lower lobe. Hemodialysis was unsuccessful in effectively reducing the ammonia level rapidly enough to facilitate clinical improvement, and serial CT exams of the brain showed progressive cerebral edema. The patient continued to deteriorate, and ultimately, brain death was diagnosed and care withdrawn. Orthotopic liver transplantation was discussed but this was predicated on return of neurologic function with correction of hyperammonemia.
Figure 1 Ammonia clearance during dialysis.
This patient had no prior significant medical problems. He was a self-selected vegetarian due to experiencing headaches, somnolence, and malaise with high protein-content meals, and his brother had a history of “liver problems,” and, “comas,” in the past. He was later tested and diagnosed with partial OTC deficiency.
Discussion
This case provides a classic presentation of an uncommon cause of acute mental status deterioration and coma. Partial OTC deficiency can present later in adulthood, sometimes as old as 62 (CitationRohininath et al 2004), and commonly after significant stress, such as starvation, weight loss, dehydration, trauma, surgery, infection (even mild, such as pharyngitis; CitationWeng et al 2004) and post-partum (CitationPeterson 2003).
Our patient likely had manifestations of OTC deficiency for two reasons. He was in a catabolic state as evidenced by his weight loss in the face of the stress of deployment and he was also taking protein supplements, a known precipitant of OTC deficiency (CitationWeng et al 2004; CitationSummar et al 2005).
The most important step in managing patients with OTC deficiency is suspecting and identifying the diagnosis, with expeditious transfer to a facility experienced in managing inherited metabolic disorders. Acute management strategies, once hyperammonemia secondary to an inherited metabolic disorder is diagnosed, are three-fold. First, endogenous ammonia production and protein catabolism are reduced through restricting exogenous nitrogenous intake (CitationMathias et al 2001). Second substrates of the urea cycle that are absent, secondary to enzymatic defect (such as the supplementation of arginine in OTC deficiency) are administered. And third, ammonia removal is facilitated.
Maximizing nutrition with high-calorie intake to prevent further catabolism, is critical. Protein restriction, coupled with hyper-caloric supplementation (high in carbohydrates and lipids (CitationMathias et al 2001), helps prevent further increases in protein (created by the catabolic state inherent in critically ill patients). The clinical relevance of protein restriction is highlighted in cases of severely ill patients who present with an encephalopathic picture and deteriorate following enteric nutritional supplementation (CitationChan et al 2002). Practically, this can be accomplished with high dextrose intravenous fluids and intralipid infusion.
Alternative pathway therapy typically encompasses arginine, to incorporate nitrogen in unaffected portions of the urea cycle and to replenish circulating amino acids, and sodium phenylacetate/sodium benzoate (Ammonul®, recently FDA-approved), to scavenge nitrogen. Benzoate conjugates to glycine, and phenylacetate conjugates to glutamine. These function to direct nitrogen away from defective urea cycle pathways towards hippurate and phenylacetylglutamine, which can be rapidly excreted in the urine (CitationMathias et al 2001).
For cases with severe hyperammonemia, dialysis is needed to rapidly clear serum ammonia to nontoxic levels (reported in the literature as <100 mg/dl) (CitationGaspari et al 2003). Hyperammonemia duration greater than 24 hours and higher serum levels (ammonia >800 mg/dl), are associated with worse neurologic outcomes and overall survival (CitationMsall et al 1984; CitationYoshino et al 1991; CitationPicca et al 2001; CitationLeonard and Morris 2002; CitationRajpoot and Gargus 2004). Therefore, immediate rapid removal of ammonia with dialysis favors a better prognosis. Ammonia is a small molecule (MW 17.03), with a molecular weight less than urea, and as such its clearance by a dialyzer membrane is greater than the clearance rate for urea. Various dialysis modalities – peritoneal dialysis (PD), intermittentent hemodialysis (IHD), continuous venovenous hemodialysis (CVVHD), and continuous arteriovenous hemodialysis (CAVH) – have all been used with varying degrees of efficacy. There is a paucity of adult experience with using extracorporeal modalities to treat the acute hyperammonemia associated with OTC deficiency, and treatment strategies, though effective, are largely derived or inferred from experience with treating neonates. CAVH has been described in the treatment of an adult female heterozygous for OTC deficiency with a late hyperammonemic coma (CitationHa et al 1997). For patients with severe encephalopathy, continuous renal replacement therapy may be superior because of decreased changes in intracranial pressure (CitationDavenport et al 1990; CitationChang et al 1999). Peritoneal dialysis, while having the potential to reduce the risk of elevated intracranial pressure, has shown to be repeatedly inferior in the pediatric literature, requiring longer treatment duration (>24 hours, and continuation for 1–5 days) to reach adequate ammonia clearance (CitationSchaefer et al 1999). IHD is superior to both PD and exchange transfusion in anuric newborns (CitationDonn et al 1979). The clearance of ammonia (CitationWiegand et al 1980) with HD is up to 10 times greater than in PD (19.4 ml/min in CVVH and up to 20–84 ml/min with IHD, versus 2.15 ml/min in PD) (CitationChan et al 2002) and is therefore the modality of choice for rapid removal of ammonia via extracorporeal therapy.
The goals of therapy with dialysis are to clear ammonia levels to a nontoxic level (accepted in the literature as below 100 mg/dl), improve neurologic status, and to remove other potentially-confounding CNS-acting substances such as anticonvulsants and sedatives. Because ammonia is not osmotically active, the risk for dialysis disequilibrium syndrome is considered to be low (CitationWiegand et al 1980). Dialysis in the hyperammonemic patient with normal renal function can lead to hypokalemia, hypomagnesemia, and hypophosphatemia, therefore renal replacement therapy should be conducted in a tertiary care facility with nephrologists experienced in the care of dialysis patients. Limitations of dialysis technique include hemodynamic instability, dialysis catheter restrictions on blood-flow rates and potential for access recirculation, and the potential for rebound ammonia regeneration due to ongoing catabolism if hemodialysis is discontinued before adjunctive medical therapies are in place to decrease the endogenous nitrogen load and to activate alternate pathways for nitrogenous waste removal. Prompt referral to a tertiary care center and treatment in conjunction with physicians knowledgeable in the diagnosis and management of metabolic disease is crucial (CitationWilcken 2004).
Orthotopic liver transplantion (OLT) has been performed in patients with OTC deficiency, primarily in the pediatric population. Indications for OLT include severe or progressive liver disease or life-threatening complications not amenable to medical and dietary management (CitationLeonard and McKiernan 2004). Short- and long-term outcomes are excellent; however neurologic damage pretransplant is typically irreversible. As our patient was newly diagnosed and had not “failed medical management”, it is unclear if OLT would even have been appropriate.
This case highlights both the difficulty and the importance of early diagnosis and treatment in preventing lasting neurologic damage. As a caveat, this case further demonstrates the importance of patient and doctor knowledge of family history of genetic diseases. Unfortunately, our patient presented in a war zone, to a combat medical facility with limited laboratory support. Perhaps prior knowledge of his family history may have prevented his deployment and subsequent decompensation.
Disclosure
The views expressed in this paper are those of the authors and do not reflect the official policy of the Department of the Army, Department of Defense, or the US Government. The authors report no conflicts of interest in this work.
References
- ChanWKButWMLawCW2002Ammonia detoxification by continuous venovenous haemofiltration in an infant with urea cycle defectHong Kong Med J82071012055368
- ChangMYFangJTChenY-C1999Continuous venovenous haemofiltration in hyperammonaemic coma of an adult with non-diagnosed partial ornithine transcarbamylase deficiencyNephrol Dial Transplant141282410344381
- DavenportAWillEJDavisonAM1990Early changes in intracranial pressure during haemofiltration treatment in patients with grade 4 hepatic encephalopathy and acute oliguric renal failureNephrol Dial Transplant519282113646
- DonnSMSwartzRDThoeneJG1979Comparison of exchange transfusion, peritoneal dialysis, and hemodialysis for the treatment of hyperammonemia in an anuric newborn infantJ Pediatr956770480015
- GaspariRArcangeliAMensiS2003Late-onset presentation of ornithine transcarbamylase deficiency in a young woman with hyperammonemic comaAnn Emerg Med41104912514690
- GreeneCLBlitzerMGShapiraE1988Inborn errors of metabolism and Reye syndrome: differential diagnosisJ Pediatr1131 Pt 115693290414
- HaYMcCannMTTuchmanM1997Substrate-induced conformational change in a trimeric ornithine transcarbamoylaseProc Natl Acad Sci USA94955059275160
- HawkesNDThomasGAJurewiczA2001Non-hepatic hyperammonaemia: an important, potentially reversible cause of encephalopathyPostgrad Med J777172211677282
- LeonardJVMcKiernanPJ2004The role of liver transplantation in urea cycle disordersMol Genet Metab81Suppl 1S74815050978
- LeonardJVMorrisAA2002Urea cycle disordersSemin Neonatol7273512069536
- MathiasRSKostinerDPackmanS2001Hyperammonemia in urea cycle disorders: role of the nephrologistAm J Kidney Dis3710698011325692
- MsallMBatshawMLSussR1984Neurologic outcome in children with inborn errors of urea synthesis. Outcome of urea-cycle enzymopathiesN Engl J Med310150056717540
- NassogneMCHéronBTouatiG2005Urea cycle defects: 1 and outcomeJ Inherit Metab Dis284071415868473
- PetersonDE2003Acute postpartum mental status change and coma caused by previously undiagnosed ornithine transcarbamylase deficiencyObstet Gynecol1025 Pt 212121514607061
- PiccaSDionisi-ViciCAbeniD2001Extracorporeal dialysis in neonatal hyperammonemia: modalities and prognostic indicatorsPediatr Nephrol16862711685590
- RajpootDKGargusJJ2004Acute hemodialysis for hyperammonemia in small neonatesPediatr Nephrol19390514997371
- RohininathTCostelloDJLynchT2004Fatal presentation of ornithine transcarbamylase deficiency in a 62-year-old man and family studiesJ Inherit Metab Dis27285815243986
- SchaeferFStraubeEOhJ1999Dialysis in neonates with inborn errors of metabolismNephrol Dial Transplant149101810328469
- SmithWKishnaniPSLeeB2005Urea cycle disorders: clinical presentation outside the newborn periodCrit Care Clin214 SupplS91716227115
- SummarMLBarrFDawlingS2005Unmasked adult-onset urea cycle disorders in the critical care settingCrit Care Clin214 SupplS1816227111
- WengTIShihFFChenWJ2004Unusual causes of hyperammonemia in the EDAm J Emerg Med22105715011224
- WiegandCThompsonTBockGH1980The management of life-threatening hyperammonemia: a comparison of several therapeutic modalitiesJ Pediatr9614247350296
- WilckenB2004Problems in the management of urea cycle disordersMol Genet Metab81Suppl 1S869115050980
- YoshinoMSakaguchiYKuriyaN1991A nationwide survey on transient hyperammonemia in newborn infants in Japan: prognosis of life and neurological outcomeNeuropediatrics221982021775216