
Abstract
Genomic imprinting is an epigenetically regulated mechanism leading to parental-origin allele-specific expression. Beckwith–Wiedemann syndrome (BWS) is an imprinting disease related to 11p15.5 genetic and epigenetic alterations, among them loss-of-function CDKN1C mutations. Intriguing is that CDKN1C gain-of-function variations were recently found in patients with IMAGe syndrome (intrauterine growth restriction, metaphyseal dysplasia, congenital adrenal hypoplasia, and genital anomalies). BWS and IMAGe share an imprinted mode of inheritance; familial analysis demonstrated the presence of the phenotype exclusively when the mutant CDKN1C allele is inherited from the mother. Interestingly, both IMAGe and BWS are characterized by growth disturbances, although with opposite clinical phenotypes; IMAGe patients display growth restriction whereas BWS patients display overgrowth. CDKN1C codifies for CDKN1C/KIP2, a nuclear protein and potent tight-binding inhibitor of several cyclin/Cdk complexes, playing a role in maintenance of the nonproliferative state of cells. The mirror phenotype of BWS and IMAGe can be, at least in part, explained by the effect of mutations on protein functions. All the IMAGe-associated mutations are clustered in the proliferating cell nuclear antigen-binding domain of CDKN1C and cause a dramatic increase in the stability of the protein, which probably results in a functional gain of growth inhibition properties. In contrast, BWS mutations are not clustered within a single domain, are loss-of-function, and promote cell proliferation. CDKN1C is an example of allelic heterogeneity associated with opposite syndromes.
Video abstract
Point your SmartPhone at the code above. If you have a QR code reader the video abstract will appear. Or use:
Introduction
Genomic imprinting is an epigenetically regulated process determining parental-origin allele-specific expression. Aberrant expression of imprinted genes characterizes imprinting diseases, including Beckwith–Wiedemann syndrome (BWS, OMIM 130650, chromosome 11p15.5). To date, eight imprinting diseases have been reported: Prader-Willi (OMIM 176270) and Angelman (OMIM 105830) syndromes (chromosome 15q11–q13), transient diabetes mellitus (OMIM 601410; chromosome 6q24), maternal/paternal uniparental disomy 14 syndromes (OMIM 608149; chromosome 14), Silver-Russell syndrome (SRS, OMIM 180860; chromosome 7 and 11p15), pseudohypoparathyroidism type IB syndrome (OMIM 603233; 20q13) and BWS. Generally, the chromosomal regions subjected to imprinting contain genes that are involved in growth and development (eg, IGF2 and H19).Citation1,Citation2 This statement is in accordance with the model of the “struggle of the sexes”, based on the observation that paternally expressed genes are often growth promoting, while maternally expressed ones are growth inhibiting.Citation3
Genetic and epigenetic alterations in BWS involve the imprinted chromosomal region 11p15.5 containing genes with expression controlled by two neighboring imprinted domains, ie, ICR1 (imprinting control region 1, regulating IGF2/H19 genes) and ICR2 (regulating KCNQ1/CDKN1C genes). ICR1 is imprinted in the male germline and operates as an insulator; ICR2 is imprinted in the female germline and acts as a promoter for the regulatory noncoding RNA KCNQ1OT1.Citation4 The pathogenetic alterations recognized in BWS () comprise CDKN1C loss-of-function mutations present in the maternally derived allele.
Table 1 Molecular heterogeneity of BWS
The 11p15.5 region is also involved in another two syndromes with imprinting defects: SRS and the IMAGe (intrauterine growth retardation, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital anomalies syndrome, OMIM 614732). IMAGe syndrome is not due to primary imprinting defects such as BWS and SRS, and is exclusively related to mutations of CDKN1C.Citation5 This gene contains three exons and encodes p57(KIP2), a potent tight-binding inhibitor of several G1 cyclin/Cdk complexes (cyclin E-CDK2, cyclin D2-CDK4, and cyclin A-CDK2).Citation6 It is a negative regulator of cell proliferation, playing a role in the maintenance of the nonproliferative state throughout life, probably acting as a tumor suppressor gene. CDKN1C is expressed in the placenta, heart, brain, lung, skeletal muscle, kidney, pancreas, and testis, in the eye, and in the subcapsular or developing definitive zone of the adrenal gland. It is paternally imprinted, with a preferential expression of the maternal allele;Citation7 however, the imprint is not absolute, as the paternal allele is also expressed at levels comparable with the maternal one in the fetal brain.Citation8 The codified protein consists of three distinct domains, ie, a cyclin-dependent kinase inhibitory domain, a proline and alanine repeat domain, and a QT domain (). The cyclin-dependent kinase inhibitory domain contains a cyclin-binding region; the proline and alanine repeats interact with the LIM domain kinase 1 and regulate actin dynamics; the proliferating cell nuclear antigen (PCNA) binding domain, able to prevent DNA replication in vitro and S-phase entry in vivo.Citation6 As in the RET gene,Citation9 different mutations in CDKN1C can give rise to different phenotypes.
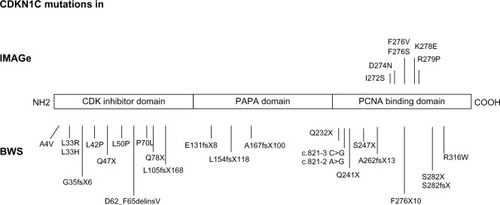
Figure 1 Mutations of CDKN1C in IMAGe syndrome (upper part) and BWS (lower part). The mutations in IMAGe syndrome are clustered within the PCNA-binding domain and are considered gain-of-function. The pathogenetic variations in BWS are spread throughout the gene and considered loss-of-function. The mutations reported here were previously described by Romanelli et alCitation29 and by Hamajima et al.Citation48
Beckwith–Wiedemann syndrome
The Beckwith–Wiedemann syndrome, described for the first time in 1963 by Beckwith, and again in 1964 by Wiedemann, is the most common overgrowth syndrome, with an incidence of about one in 13,700 live births.Citation10,Citation11 This occurrence is probably underestimated as milder phenotypes may not be ascertained.Citation12 Males and females are affected equally, with the exception of monozygotic twins who show a female predominance.Citation13 BWS occurs sporadically in most cases (85%), with the remaining cases having autosomal dominant inheritance.Citation14
Clinical diagnosis
Clinical findings of BWS include macrosomia, macroglossia, abdominal wall defects (diastasis recti, omphalocele, or umbilical hernia), visceromegaly, hemihyperplasia, anterior ear creases and posterior helical pits, kidney abnormalities (medullary dysplasia, later development of medullary sponge kidney), cytomegaly of the adrenal fetal cortex, a positive family history of BWS, and, rarely, cleft palate. Height and weight are typically around the 97th percentile in children, with head size closer to the 50th percentile, while adult height generally settles in the normal range.Citation11,Citation15,Citation16 Additional findings may include neonatal hypoglycemia, nevus flammeus, cardiomegaly, structural cardiac defects, cardiomyopathy, advanced bone age, and a characteristic facial appearance. Most BWS patients have normal psychomotor development, but mental retardation has been noted in cases with chromosomal abnormalities and/or perinatal complications.Citation17
Pregnancies with fetuses affected with BWS may be complicated by polyhydramnios, large/dysplastic placenta,Citation16,Citation18 a long and thickened umbilical cord, and an increased risk for premature delivery.Citation16 Children conceived by in vitro fertilization are at increased risk of developing BWS, and the overall risk of BWS with in vitro fertilization is about one in 4,000.Citation19–Citation21
Although consensus diagnostic criteria for BWS have not been defined, the presence of three major features (eg, prenatal and postnatal overgrowth, macroglossia, and abdominal wall defects) or two major features and one minor feature (eg, ear anomalies, neonatal hypoglycemia, nephromegaly, and hemihyperplasia) is required for the postnatal clinical diagnosis of BWS.Citation4,Citation22
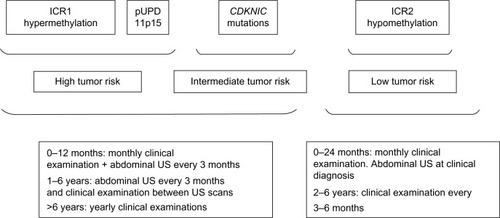
Early diagnosis of BWS is crucial because of the well documented increased risk of cancer, most commonly Wilms’ tumor and hepatoblastoma, but also adrenocortical carcinoma, rhabdomyosarcoma, and neuroblastoma.Citation11,Citation14,Citation15,Citation23–Citation25 Isolated hemihyperplasia (OMIM 235000) is a congenital overgrowth disorder related to BWS that displays asymmetric involvement of the body. It may manifest few of the abnormal features associated with BWS; however, patients with isolated hemihyperplasia have an increased risk for embryonal tumors, primarily Wilms’ tumor and hepatoblastoma, which are usually diagnosed before 10 years of age.Citation26 Furthermore, BWS patients with hemihyperplasia have a four-fold increased tumor risk compared with BWS patients without hemihyperplasia, emphasizing the increased risk of tumors in isolated hemihyperplasia.Citation25 Isolated hemihyperplasia cases should also be included in the specific follow-up created for BWS patients ().Citation27
Figure 2 Suggested follow-up for BWS according to risk classes.
Molecular genetic testing
Molecular diagnosis is important to confirm the provisional BWS clinical diagnosis and to identify BWS patients with cancer susceptibility. An etiologic molecular heterogeneity is clearly evident in BWS, due to various genetic and/or epigenetic alterations in growth regulatory genes located on the chromosome 11p15 region.
The genetic and epigenetic abnormalities associated with BWS are listed in and include loss of methylation on the maternal chromosome at IC2, gain of methylation on the maternal chromosome at IC1, paternal uniparental disomy for chromosome 11p15 (usually as mosaicism), mutations in the CDKN1C gene, and cytogenetically detectable or submicroscopic genomic alteration within 11p15.5.Citation22,Citation28 CDKN1C mutations are found in about 5%–10% of sporadic cases, whereas dominant maternal transmission of germline mutations are found in 40% of familial BWS cases.Citation4,Citation11
It is largely accepted that the variability of oncologic risk in BWS mostly depends on genetic heterogeneity () and duration of the observation. It is now well established that patients with a molecular abnormality of the telomeric domain (ICR1 hypermethylation) and 11p15 paternal uniparental disomy have an increased risk of embryonal tumors (especially Wilms’ tumor), whereas patients with defects of the centromeric domain only (ICR2 hypomethylation and CDKN1C mutations) are at lower risk of such tumors.Citation22
CDKN1C mutations in BWS: genotype-phenotype correlations
Focusing on CDKN1C mutations, subfertility in males, predisposition to aneurysmal arterial dilation, renal abnormalities, hearing loss, and, perhaps, increased risk for adult-onset malignancy are known.Citation12 Romanelli et alCitation29 reviewed the CDKN1C-related phenotypic features in BWS patients, and suggested that these patients show a peculiar pattern of clinical malformations in comparison with those with other molecular defects, and therefore the peculiar clinical presentation of the patients can be useful to address the molecular analysis. Genotype-phenotype studies indicate that mutations in CDKN1C are more frequently associated with omphalocele,Citation30 clefting,Citation31 and genital anomaliesCitation29,Citation32 and less frequently with development of tumors,Citation30 with the exception of rare cases with neuroblastoma.Citation33–Citation35 Focusing on cleft palate, although this finding has been rarely reported in patients affected by BWS (2.5%),Citation15,Citation29,Citation36–Citation39 it has usually been associated with CDKN1C alterations; instead, cleft lip has never been documented in these patients.
In addition, CDKN1C seems to be highly expressed in the placenta, and therefore may have importance in the pathophysiology of pre-eclampsia/HELLP syndrome during pregnancy.Citation40 Furthermore, a few patients with other rare malformations such as polydactyly and supernumerary nipples have been reported.Citation29
IMAGe syndrome
IMAGe syndrome is a rare developmental disorder, mainly characterized by growth restriction and described for the first time in 1999 by Vilain et al.Citation41 To date, there are only 12 familial or isolated cases reported in the literature, but this condition could be underestimated as the causative gene and the mode of inheritance have only recently been identified by Arboleda et al.Citation5
Clinical features
The acronym IMAGe indicates the presence of Intrauterine growth restriction, Metaphyseal dysplasia, congenital Adrenal hypoplasia, and Genital anomalies. The first sign that becomes evident after birth is adrenal insufficiency, which can be severe and life-threatening if not recognized and treated early; it usually manifests in the first days of life with adrenal crises, but a case of late onset has been reported.Citation42 Radiologic identification of metaphyseal dysplasia is often crucial for the diagnosis, but this could be very mild and identifiable only in late infancy or in childhood and then progress with age. A more precocious sign, ie, delayed endochondral ossification associated with osteopenia, hypercalcemia, and/or hypercalciuria, can be present at birth and normalizes later in infancy.Citation43,Citation44 Growth hormone deficiency has also been described in some patients,Citation40 and it has been shown that an early substitutive therapy assessment could help to improve the linear growth.
Dysmorphic craniofacial features in IMAGe syndrome include nonspecific signs, such as prominent forehead, low-set ears, flat nasal bridge, and short nose, while genital abnormalities seem to be confined to males and include micropenis, undescended testes, and varying severity of hypospadias.Citation42
Molecular genetic testing
The IMAGe causative gene and inheritance model have been recently identified.Citation5,Citation45–Citation47 To date, six missense mutations, all of them occurring in the PCNA-binding domain in the carboxy-terminal region of CDKN1C, were reported ().Citation5,Citation45 Recently, Hamajima et alCitation48 demonstrated that the IMAGe-associated mutations cause a dramatically increased instability of the CDKN1C proteins that probably results in a functional gain. This hypothesis is also supported by evidence of CDKN1C truncation mutants lacking PCNA binding (ie, F276X10; ) identified in BWS patients showing the loss-of-function phenotype of CDKN1C.Citation49
For molecular diagnosis of IMAGe syndrome, CDKN1C sequencing is recommended. Notably, familial analysis demonstrated de novo mutations or an imprinted mode of inheritance, exclusively with maternal transmission of the mutation. This means that each child of a woman heterozygous for the CDKN1C mutation has a 50% chance of inheriting the pathogenic variant and being affected, whereas each child of a man with the mutation has a 50% chance of inheriting the CDKN1C pathogenic variant but is expected to be unaffected.Citation50 This pattern of transmission suggests that IMAGe can be considered as an imprinting disease, albeit characterized by point gene mutations.
Same gene, mirror phenotypes
The mechanisms underlying genomic imprinting are very complex, in particular regarding the 11p15.5 imprinted region. Indeed, different mutations (or epimutations) in the same gene (CDKN1C) or imprinted region can result in opposite phenotypes. CDKN1C can be associated with BWS, SRS, and IMAGe syndrome, and IC1 opposite imprinting defects with BWS (IC1 hypermethylation) and SRS (IC1 hypomethylation). Among these syndromes, BWS is mainly characterized by overgrowth, and IMAGe and SRS by growth restriction.Citation46 A detailed description of the mirror phenotype in BWS and IMAGe is reported in .
Table 2 Mirror phenotypes in BWS and IMAGe syndrome
BWS-associated CDKN1C mutations are most often truncating mutations distributed throughout the gene, or missense mutations in the aminoterminal CDK inhibitor domain, and those associated with IMAGe are missense variants in the PCNA-binding domain (), suggesting that the mutations causing these two imprinting disorders have different biologic consequences.
CDKN1C mutations in BWS act as a “double-negative”, promoting proliferation due to loss of cell cycle inhibition, and then predisposition to cancer. In contrast, IMAGe syndrome is caused by mutations leading to a gain-of-function of CDKN1C protein, most probably related to increased stability of the protein.Citation5 IMAGe-associated mutations inhibit binding to PCNA and ubiquitination of CDKN1C. Evidence for a gain-of-function effect was detected in Drosophila melanogaster, in which expression of the mutant but not the wild-type human protein reduced eye and wing size.Citation5 It is conceivable that CDKN1C mutations in IMAGe patients can trigger an excess of inhibition of growth and differentiation.
A good example of the effects of these mutations on the single organ has been provided by Arboleda et al,Citation5 who showed how CDKN1C was strongly expressed during the embryonic development of the adrenal glands in mice. In BWS, the decreased inhibition of G1 CDKs due to loss-of-function mutations causes adrenal cytomegaly (one of the most common signs in BWS fetuses) and adrenocortical tumors (also sporadic adrenocortical tumors can display CDKN1C inactivation), while in IMAGe syndrome the increased inhibition of the cell cycle due to augmented stability of CDKN1C results in congenital adrenal hypoplasia ().
Finally, in the fetal brain, as mentioned before, CDKN1C is biparentally expressed. This observation could explain why the occipitofrontal circumference in IMAGe syndrome is relatively well preserved in comparison with length/height and weight that are severely compromised.Citation45
The link between the nature of CDKN1C mutation and the mirror phenotype of BWS and IMAGe can be explained by the importance of p57Kip2 in embryogenesis, which is also highlighted by the finding that knockout mice exhibit gastrointestinal tract defects, omphalocele, abnormal endochondral ossification, cleft palate, adrenal cortex enlargement, renal medullary dysplasia, increased body weight, and placental abnormalities. p57Kip2 excess in mice increases embryonic lethality and decreases body size, suggesting that embryonic growth requires accurate control of p57Kip2 dosage.Citation51
In conclusion, after the discovery that SRS is characterized by the opposite growth defect compared with BWS, IMAGe syndrome represents an additional imprinting disorder with a mirror phenotype of BWS, strengthening the primary role of the 11p15.5 imprinted region in prenatal and postnatal growth regulation.
Disclosure
The authors report no conflicts of interest in this work.
References
- MiozzoMSimoniGThe role of imprinted genes in fetal growthBiol Neonate20028121722812011565
- TabanoSColapietroPCetinIEpigenetic modulation of the IGF2/H19 imprinted domain in human embryonic and extra-embryonic compartments and its possible role in fetal growth restrictionEpigenetics2010165313324
- JurkowskaRZJeltschAGenomic imprinting – the struggle of the genders at the molecular levelAngew Chem Int Ed Engl201352135241353624346936
- ChoufaniSShumanCWeksbergRBeckwith–Wiedemann syndromeAm J Med Genet C Semin Med Genet2010154C34335420803657
- ArboledaVALeeHParnaikRMutations in the PCNA-binding domain of CDKN1C cause IMAGe syndromeNat Genet20124478878922634751
- LeeMHReynisdottirIMassagueJCloning of p57(KIP2), a cyclin-dependent kinase inhibitor with unique domain structure and tissue distributionGenes Dev199596396497729683
- HatadaIMukaiTGenomic imprinting of p57KIP2, a cyclin-dependent kinase inhibitor, in mouseNat Genet1995112042067550351
- MatsuokaSThompsonJSEdwardsMCImprinting of the gene encoding a human cyclin-dependent kinase inhibitor, p57KIP2, on chromosome 11p15Proc Natl Acad Sci U S A199693302630308610162
- MarxSJMolecular genetics of multiple endocrine neoplasia types 1 and 2Nat Rev Cancer2005536737515864278
- EngströmWLindhamSSchofieldPWiedemann-Beckwith syndromeEur J Pediatr19881474504573044795
- WeksbergRShumanCBeckwithJBBeckwith–Wiedemann syndromeEur J Hum Genet20101881419550435
- GreerKJKirkpatrickSJWeksbergRPauliRMBeckwith–Wiedemann syndrome in adults: observations from one family and recommendations for careAm J Med Genet Part A2008146A1707171218546283
- BliekJAldersMMaasSMLessons from BWS twins: complex maternal and paternal hypomethylation and a common source of haematopoietic stem cellsEur J Hum Genet2009171625163419513094
- CohenMMJrBeckwith–Wiedemann syndrome: historical, clinicopathological, and etiopathogenetic perspectivesPediatr Dev Pathol2005828730416010495
- PettenatiMJHainesJLHigginsRRWappnerRSPalmerCGWeaverDDWiedemann–Beckwith syndrome: presentation of clinical and cytogenetic data on 22 new cases and review of the literatureHum Genet1986741431543770742
- WengEYMoeschlerJBJrGrahamJMLongitudinal observations on 15 children with Wiedemann-Beckwith syndromeAm J Med Genet1995563663737541608
- MunnsCBatchJHyperinsulinism and Beckwith–Wiedemann syndromeArch Dis Child Fetal Neonatal Ed200184F67F6911124932
- WilsonMPetersGBennettsBThe clinical phenotype of mosaicism for genome-wide paternal uniparental disomy: two new reportsAm J Med Genet2008146A13714818033734
- GosdenRTraslerJLuciferoDFaddyMRare congenital disorders, imprinted genes, and assisted reproductive technologyLancet20033611975197712801753
- HallidayJOkeKBrehenySAlgarEBeckwith–Wiedemann syndrome and IVF: a case control studyAm J Hum Genet200475526.e815284956
- StrawnEYJrBickDSwansonAIs it the patient or the IVF? Beckwith–Wiedemann syndrome in both spontaneous and assisted reproductive conceptionsFertil Steril201094754.e1754.e220338562
- CalvelloMTabanoSColapietroPQuantitative DNA methylation analysis improves epigenotype-phenotype correlations in Beckwith–Wiedemann syndromeEpigenetics201381053106023917791
- WiedemannHRTumors: an hemihypertrophy associated with Wiedemann-Beckwith syndromeEur J Pediatr1983141129
- TanTYAmorDJTumour surveillance in Beckwith–Wiedemann syndrome and hemihyperplasia: a critical review of the evidence and suggested guidelines for local practiceJ Paediatr Child Health20064248649016925531
- DeBaunMRTuckerMARisk of cancer during the first four years of life in children from the Beckwith–Wiedemann Syndrome RegistryJ Pediatr19981323984009544889
- ClericuzioCLMartinRADiagnostic criteria and tumor screening for individuals with isolated hemiphyperplasiaGenet Med20091122022219367194
- BrioudeFLacosteANetchineIBeckwith–Wiedemann syndrome: growth pattern and tumor risk according to molecular mechanism, and guidelines for tumor surveillanceHorm Res Paediatr20138045746524335096
- ShumanCBeckwithJBSmithACWeksbergRBeckwith–Wiedemann syndrome. December 14, 2010PagonRAAdamMPBirdTDGeneReviews®Seattle, WA, USAUniversity of Washington, Seattle1993–2014 Available from: http://www.ncbi.nlm.nih.gov/books/NBK1394/Accessed July 30, 2014
- RomanelliVBelinchónABenito-SanzSCDKN1C (p57(Kip2)) analysis in Beckwith–Wiedemann syndrome (BWS) patients: genotype-phenotype correlations, novel mutations, and polymorphismsAm J Med Genet A2010152A1390139720503313
- LamWWHatadaIOhishiSAnalysis of germline CDKN1C (p57KIP2) mutations in familial and sporadic Beckwith–Wiedemann syndrome (BWS) provides a novel genotype-phenotype correlationJ Med Genet19993651852310424811
- WeksbergRShumanCSmithACBeckwith–Wiedemann syndromeAm J Med Genet Part C2005137C122316010676
- WelshHIStockleyTLParkinsonNArdingerHHCDKN1C mutations and genital anomaliesAm J Med Genet A2012158A26522140035
- BliekJMaasSMRuijterJMIncreased tumour risk for BWS patients correlates with aberrant H19 and not KCNQ1OT1 methylation: occurrence of KCNQ1OT1 hypomethylation in familial cases of BWSHum Mol Genet20011046747611181570
- WeksbergRNishikawaJCaluseriuOTumor development in the Beckwith Wiedemann syndrome is associated with a variety of constitutional molecular 11p15 alterations including imprinting defects of KCNQ1OT1Hum Mol Genet2001102989300011751681
- ChitayatDFriedmanJMDimmickJENeuroblastoma in a child with Wiedemann Beckwith syndromeAm J Med Genet Part C201035433436
- DelgadoALozanoMJRuffaGBeckwith–Wiedemann syndrome. Presentation of 5 casesMinerva Pediatr1975272452611226194
- ElliottMBaylyRColeTTempleIKMaherERClinical features and natural history of Beckwith–Wiedemann syndrome: Presentation of 74 new casesClin Genet1994461681747820926
- KimYShibutaniTHirotaYMahbubSFMatsuuraHAnesthetic considerations of two sisters with Beckwith–Wiedemann syndromeAnesth Prog199643242810323122
- LarocheCTestelinSDevauchelleBCleft palate and Beck-with–Wiedemann syndromeCleft Palate Craniofac J20054221221715748114
- RomanelliVBelinchonACampos-BarrosACDKN1C mutations in HELLP/preeclamptic mothers of Beckwith–Wiedemann syndrome (BWS) patientsPlacenta20093055155419386358
- VilainELe MerrerMLecointreCIMAGe, a new clinical association of intrauterine growth retardation, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital anomaliesJ Clin Endocrinol Metab1999844335434010599684
- PedreiraCCSavarirayanRZacharinMRIMAGe syndrome: a complex disorder affecting growth, adrenal and gonadal function, and skeletal developmentJ Pediatr200414427427714760276
- AmanoNNaoakiHIshiiTRadiological evolution in IMAGe association: a case reportAm J Med Genet A2008146A2130213318627061
- BalasubramanianMSpriggAJohnsonDSIMAGe syndrome: case report with a previously unreported feature and review of published literatureAm J Med Genet A2010152A3138314221108398
- KatoFHamajimaTHasegawaTIMAGe syndrome: clinical and genetic implications based on investigations in three Japanese patientsClin Endocrinol (Oxf)20148070671324313804
- SoejimaHHigashimotoKEpigenetic and genetic alterations of the imprinting disorder Beckwith–Wiedemann syndrome and related disordersJ Hum Genet20135840240923719190
- DiasRPMaherERAn imprinted IMAGe: insights into growth regulation through genomic analysis of a rare diseaseGenome Med201246022839767
- HamajimaNJohmuraYSuzukiSNakanishiMSaitohSIncreased protein stability of CDKN1C causes a gain-of-function phenotype in patients with IMAGe syndromePLoS One20138e7513724098681
- HatadaIOhashiHFukushimaYAn imprinted gene p57KIP2 is mutated in Beckwith–Wiedemann syndromeNat Genet1996141711738841187
- BennettJSchrier VerganoSDeardorffMAIMAGe syndromePagonRAAdamMPBirdTDGeneReviews®Seattle, WA, USAUniversity of Washington, Seattle19932014 Available from: http://www.ncbi.nlm.nih.gov/books/NBK190103/Accessed July 30, 2014
- BorrielloACaldarelliIBencivengaDp57(Kip2) and cancer: time for a critical appraisalMol Cancer Res201191269128421816904