Abstract
The purpose of this review is to assess the current literature on deafness nonsyndromic autosomal dominant 2 (DFNA2) hearing loss and the mutations linked to this disorder. Hearing impairment, particularly nonsyndromic hearing loss, affects multiple families across the world. After the identification of the DFNA2 locus on chromosome 1p34, multiple pathogenic mutations in two genes (GJB3 and KCNQ4) have been reported. The overwhelming majority of pathogenic mutations linked to this form of nonsyndromic hearing loss have been identified in the KCNQ4 gene encoding a voltage-gated potassium channel. It is believed that KCNQ4 channels are present in outer hair cells and possibly inner hair cells and the central auditory pathway. This form of hearing loss is both phenotypically and genetically heterogeneous and there are still DFNA2 pedigrees that have not been associated with changes in either GJB3 or KCNQ4, suggesting that a possible third gene exists at this locus. Further studies of the DFNA2 locus will lead to a better understanding of progressive hearing loss and provide a better means of early detection and treatment.
Introduction
Hearing loss is one of the most common sensory deficits seen in the population. Approximately one in 1000 people in the US is born deaf or will develop profound deafness in early childhood.Citation1 It is estimated that more than 50% of cases of childhood deafness are genetic, while the rest are a result of environmental factors.Citation1 Certain forms of progressive hearing loss, such as presbycusis, are likely a result of the combination of genetic factors and environmental insults.
Hearing impairment can occur in isolation (nonsyndromic) or in combination with other defects (syndromic). Approximately 70% of inherited hearing disorders are estimated to be nonsyndromic in nature.Citation1 Nonsyndromic hearing loss has a variety of inheritance patterns, including X-linked deafness (DFN), autosomal dominant (DFNA), and autosomal recessive (DFNB) patterns. Generally, recessive traits present with severe hearing loss at an early age, while autosomal dominant traits present as slowly progressive hearing loss later in life.
A systematic literature review of DFNA2 hearing loss was conducted utilizing the US National Library of Medicine Medline database. Over 50 relevant articles relating to DFNA2-associated hearing loss were identified and critically evaluated using evidence-based techniques. All articles with significant contributions to this topic were included in the review, beginning with the first articles in the late 1990s by Xia and Kubisch.Citation3,Citation4
To date, over 50 loci and nearly 30 genes have been identified for autosomal dominant nonsyndromic hearing loss, while there are over 100 gene loci associated with all subtypes of nonsyndromic deafness. DFNA2 hearing loss affects numerous families from diverse backgrounds, including those of European, American, and Asian origins, thus demonstrating its genetic heterogeneity.Citation2 Two genes have been linked to the DFNA2 locus and some have postulated the existence of a third causative gene.Citation3–Citation5 However, pathogenic mutations in the KCNQ4 gene appear to represent all of DFNA2 associated-hearing impairment, outside of two Chinese families associated with GJB3 mutations.Citation3 Genes associated with most forms of autosomal dominant hearing loss have been linked to only a single or few families; however, due to the global impact of DFNA2 mutations, it has been the subject of much interest in recent years.Citation1 Kubisch and Xia reported the first mutations in families with DFNA2 hearing loss, and since that time, a wide variety of pathogenic mutations in KCNQ4 have been described in many families.Citation3,Citation4 It is anticipated that a better understanding of the mechanism of DFNA2 loss could serve as a potential model for the detection and treatment of progressive hearing loss.
Clinical presentation
DFNA2-associated hearing loss has a presentation that is typically later in onset and progressive over time, as opposed to early onset and severe loss in recessive forms. High frequency hearing loss may be detected in early childhood, and typically progresses to reach moderate to mid frequency levels of 40–60 decibels (dB) at about 50 years of age, and can exceed 80 dB at 1 kHz in later years, causing significant problems with speech discrimination in affected patients.Citation6,Citation7 Most individuals suffer from severe to profound hearing loss by the age of 70 years, and most are fitted with hearing aids by early adulthood.Citation6 Studies of other affected families have shown an annual threshold increase of about 1 dB per year, with the higher frequencies (1–8 kHz) exhibiting significantly more rapid progression than the lower frequencies (0.25–0.5 kHz).Citation8 Other studies have demonstrated similar rates of progression, but no significant evidence of vestibular dysfunction.Citation9 One study found a small number of DFNA2 patients (three cases) with vestibular hyperreactivity who were prone to motion sickness; however, the remaining individuals were free of balance issues.Citation7 Temporal bone imaging is unremarkable, and physical examination is usually normal. Family history reveals a dominant pattern of inheritance of hearing impairment. The age at initial presentation and progression of hearing loss has shown variability between DFNA2 families, reinforcing the phenotypic heterogeneity of the disease.
Identification of DFNA2 locus
Identification of the locus associated with DFNA2 hearing loss was accomplished through the study of a large Indonesian family with over 100 members affected by nonsyndromic progressive autosomal dominant hearing loss. Members with the impairment noted decreased hearing in the second or third decades of life, and audiometry revealed that all affected members had a loss of at least 30 dB by 30 years of age, with profound loss within 10 years of onset.Citation10
Genetic linkage analysis was performed in this Indonesian family as well as in an American and Dutch family with similar audiometric characteristics. First attempts to link the Indonesian family with previously described loci for hereditary deafness on chromosome 5q and 13q were unsuccessful, so microsatellite markers were used to search the entire genome. Linkage was ultimately detected on the short arm of chromosome 1, also known as 1p34. Association with this locus was also confirmed in the American family, but the Dutch family could not be linked to this site, reaffirming that autosomal dominant deafness is not only clinically but also genetically heterogeneous.Citation10
Connexins and hearing loss
Mutations in GJB2 encoding connexin 26, the most common cause of nonsyndromic hearing loss, have previously been shown to cause autosomal recessive (DFNB1) and dominant (DFNA3) forms of hearing impairment.Citation11 This mutation, as well as mutant connexin 30, are both found at the DFNA3 locus.Citation12,Citation13 Xia et al proposed that other members of the connexin family could be involved in DFNA2 hearing loss, so the gene (GJB3) encoding gap junction protein beta-3 (connexin 31) was cloned.Citation3 This gene was mapped to chromosome 1p34.
GJB3 is comprised of two exons, only the second of which is a coding exon. In order to determine if there was any association of connexin 31 with DFNA2 hearing loss, the coding regions of 42 families with hereditary diseases linked to the short arm of chromosome 1 were sequenced. These diseases included sensorineural hearing loss, erythrokeratodermia, Charcot-Marie-Tooth disease, and ptosis. In two small Chinese families with sensorineural hearing impairment, a missense and a nonsense mutation were identified in the coding region of GJB3, while the individuals with the other mentioned disease states did not exhibit any mutations ().Citation3 This was the first identification of a connexin gene linked to the DFNA2 locus. Nucleotide changes of GJB2 were identified in these families, but these were felt to be polymorphisms unrelated to the hearing loss in this group.Citation3
Table 1 GJB3 mutations
Some controversy exists over the role of GJB3 mutations in DFNA2 hearing loss. Some have stated that the evidence presented supporting the role of GJB3 in nonsyndromic hearing loss is not convincing.Citation14 In the Chinese families evaluated, one female carrier of the mutation exhibited normal hearing by audiogram, suggesting incomplete penetrance, which typically is not seen with autosomal dominant forms of hearing loss.Citation3,Citation14 Further studies of a five-generation family with a progressive, high frequency autosomal dominant hearing loss revealed a one-sequence alteration in the first noncoding exon of GJB3. However, this mutation was identified in both affected and unaffected family members, again suggesting that GJB3 may not be directly involved in this type of progressive hearing impairment.Citation5
Additionally, in 1998, when the two Chinese families with GJB3 mutations were being studied, mutations in the voltage-gated potassium channel (KCNQ4), now linked with DFNA2 hearing loss, was yet to be identified. Therefore, it is possible that these families had an existing mutation in the KCNQ4 voltage-gated potassium channel that remained undetected.Citation14 To date, no other DFNA2 families have demonstrated the GJB3 mutation. Attention has now shifted towards the elucidation of mutations in voltage-gated ion channels. This has proven to be an extremely prosperous endeavor because various mutations in potassium channels have been linked to hearing loss in families from around the world ().
Table 2 Known KCNQ4 mutations
Role of potassium channels in hearing loss
Ion channels have a central role in regulation of the ionic composition of intracellular and extracellular fluid, which are key for hair cell activation in the inner ear. Mutations in these channels have long been suspected to play a part in hearing loss, particularly due to the importance of ion regulation in perilymph and endolymph in the inner ear. Signal transduction in the cochlea depends on maintenance of a high potassium concentration and a positive potential in the endolymph. This is mediated through ion transport across the stria vascularis into the hair cells and efflux of K+, likely through voltage-gated ion channels.Citation15 Inner hair cells provide the brain with electrical input while outer hair cells amplify acoustic vibrations.Citation15
Mutations leading to human disease including cardiac arrhythmias, epilepsy, and congenital deafness have been found in approximately ten potassium channel genes. Four of these known genes encode K+ channels from the KCNQ family.Citation15 Since previous relationships between KCNQ1 mutation and deafness in the Jervell and Lange-Nielsen syndrome were elucidated, KCNQ4 has been thought to be a candidate gene for individuals with slowly progressive hearing loss.Citation16
KCNQ4 structure and function
In recent years, increased attention has been directed towards identification of mutations and pathogenic mechanisms of the KCNQ4 gene and its role in hearing loss. This is a complex gene with 14 exons encoding a protein of 695 amino acids. It has a long C-terminus and six transmembrane domains with the fourth domain (S4) serving as the voltage sensor segment. The fifth and sixth domains constitute the pore region with the peptides in between the domains composing the P-loop. This single pore region, which forms the selectivity filter of the pore, comprises only 3% of the KCNQ4 protein, yet is where the majority of mutations linked to nonprogressive autosomal dominant hearing loss have been identified.Citation15,Citation17,Citation18
Studies of murine cochlear RNA demonstrate KCNQ4 transcripts in the brain and in the outer hair cells of the cochlea.Citation4,Citation19 Inner and outer hair cell apical membranes are in contact with endolymph that fills the scala media which has a high potassium concentration and low sodium concentration. Apical potassium channels lead to an influx of K+ while basolateral channels likely mediate efflux of K+. The location of the KCNQ4 potassium channel on the basal membrane of the outer hair cell prohibits function as a mechanosensitive channel or motor protein.Citation19 It is suspected that KCNQ4 contributes to basolateral K+ conductance of the ion and removal of intracellular K+ taken up from the endolymph.Citation4 Further studies have suggested that mutation of KCNQ4 may cause chronic K+ overload, causing slow degeneration of outer hair cells, therefore explaining the progressive nature of DFNA2 hearing loss.Citation15
Identification of KCNQ4 mutations
Since the first identification of a KCNQ4 mutation linked to DFNA2 hearing impairment in 1999, there have been 18 additional families having similar hearing loss with mutations of KCNQ4 ( and ). Although this gene has 14 exons, the mutations seem to cluster around exons 4, 5, 6, and 7, with missense mutations being the most common.
Figure 1 Structure of KCNQ4.
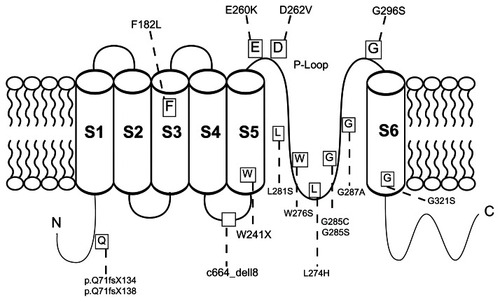
Initial studies of the KCNQ4 gene identified a missense mutation (G285S) in exon 6 in a heterozygous state in three members of a French family with profound hearing loss.Citation4 Potassium channel pores have a highly conserved glycine in the GYG K+ channel signature sequence that is found in different classes of channels in all species. This missense mutation, along with another mutation, G285C, identified in an American family, were found to affect the first glycine in this sequence which disrupts the channel selectivity filter and channel function.Citation4,Citation20 These mutations prevent the ability of the channel to distinguish between K+ and Na+ ions.Citation16 Mutant channels did not have any detectable currents when expressed in Xenopus oocytes, and when injected into a wild-type KCNQ4 model to mimic a heterozygous state, there was a significant reduction of channel currents by about 90%. This led to the proposal that mutant KCNQ4 exerts a dominant negative effect on the wild-type gene.Citation4,Citation15
Further analysis of DFNA2 families revealed that the majority of the missense mutations clustered around the pore region. The G296S mutation, located in the pore region, identified in a small Spanish family, also demonstrated a strong dominant negative effect on wild-type channels.Citation21 Another missense mutation, G287A, identified by Arnett et al in an American family was also found to disrupt the GYG sequence just as in the initial reported KCNQ4 mutation.Citation22
The only missense mutation that has been identified in multiple families of diverse backgrounds is W276S, which has led to the proposal that this represents a mutational hot spot.Citation20,Citation23–Citation25 With this mutation, Trp276 is altered. This amino acid is located in the pore loop of KCNQ4 channels adjacent to another highly conserved tryptophan residue which has been proposed to function in maintaining the appropriate diameter of the pore.Citation26
Further mutations at the pore region include L281S and L274S identified in an American and Dutch family, respectively.Citation18,Citation27 It is proposed that replacing these hydrophobic leucine residues with hydrophilic amino acids disrupts the structure of the KCNQ4 channel.Citation28 Another unique mutation in a Dutch family was a G321S mutation at the S6 transmembrane domain, which also contributes to pore structure.Citation20 To date, only one mutation in a Taiwanese family has been linked to the S3 transmembrane domain. This represents a missense mutation that is not linked to pore formation.Citation29
Thus far, only three mutations resulting in truncated peptides have been elucidated.Citation20,Citation30,Citation31 The first FS71 mutation was identified in a Belgian family. Interestingly, this mutation was not in the pore region, but in the N-terminal cytoplasmic region.Citation20 The second, found in a Japanese family, was a one-base deletion in exon 1, causing a frameshift mutation with a resultant truncated peptide before the first transmembrane domain.Citation30 The third stop codon mutation, identified in an American family on exon 5, is predicted to result in a truncated peptide that lacks most of the S5 transmembrane domain, the S6 domain, and the C-terminal region.Citation31
It has been suggested that the type, rather than the location, of the mutation has the greatest effect on patient phenotype. This was demonstrated in a Dutch family with a S6 transmembrane domain missense mutation that exhibited a phenotype similar to that of patients with pore region missense mutations.Citation18,Citation20 Individuals with the mutation identified by Kamada et al, ie, a nonsense mutation in the N-terminal cytoplasmic region, had late-onset and pure high frequency hearing loss.Citation30 Coucke et al found a similar phenotype in patients with a nonsense mutation in FS71 in the S6 domain, suggesting that deletions in general are associated with later-onset hearing loss that is mild when compared with missense mutations.Citation20 However, the stop codon identified by Hildebrand et al did not fit this phenotype, because the affected individual had a more severe form of hearing loss requiring cochlear implantation during childhood.Citation31
Analysis of a Korean family identified an 18 nucleotide deletion (c.664_del18) in exon 4 of the KCNQ4 gene which is suspected to result in a six amino acid deletion in the intramembrane loop between the S4 and S5 transmembrane domains.Citation17 Human embryonic kidney cells were cotransfected with the wild-type KCNQ4 and the exon 4 deletion, W276S mutation, and G285C mutations. Cells cotransfected with previously identified pore-loop missense mutations,W276S and G285C, as well as the exon 4 deletion exhibited normal protein synthesis and membrane expression of the ion channel, but there was no appreciable ion flow through the membrane. This suggests an inhibition of KCNQ4 channel function through a dominant negative effect and illustrates the importance of the S4–S5 intramembrane loop.Citation17
In deletion mutations, haploinsufficiency has been proposed as the pathogenic mechanism of action.Citation30 Haploinsufficiency may result from a null mutation, but it is possible that there is still KCNQ4 function from the intact allele. This may provide sufficient KCNQ4 channels to cause only mild hearing impairment, therefore suggesting a genotype-phenotype correlation.Citation30 Studies in mice with total KCNQ4 knockout showed a more rapidly progressive hearing loss in comparison with those heterozygous for the dominant negative mutation. Because K+ channels form tetramers, it can be assumed that 1/16 of potassium channels in a functional state can lead to a significant delay in hair cell degeneration. Those heterozygous for the knockout mutation did not show any hearing abnormalities, suggesting that only 50% of currents are required for normal hearing.Citation32
Hearing loss of 50–60 dB in these mice would correlate with a selective loss of outer hair cell function.Citation32 However, DFNA2 pedigrees with hearing loss > 60 dB has led others to suggest that outer hair cell dysfunction alone cannot account for the degree of hearing loss seen in these patients.Citation33
KCNQ4 not just in outer hair cells
Different mechanisms of KCNQ4-mediated hearing loss have been proposed, including the previously mentioned K+ recirculation in outer hair cells.Citation4,Citation15 Interestingly, it has also been noted that there is higher expression of KCNQ4 potassium channels in the basal turn of the cochlea, which is responsible for high frequency hearing and would thus explain the high frequency hearing loss seen early in patients with DFNA2.Citation4
Others have proposed defective signal transmission in the central auditory pathway as a potential cause of progressive hearing loss.Citation19,Citation33 In addition to the basolateral membranes of outer hair cells, immunocytochemistry has revealed the presence of KCNQ4 in type 1 vestibular hair cells, as well as several nuclei of the central auditory pathway in the brainstem.Citation19 The role of KCNQ4 in vestibular hair cells is unclear because DFNA2 patients do not typically exhibit vestibular symptoms. This was confirmed with further experiments where mice had a constitutive knockout of KCNQ4.Citation32
An additional proposed mechanism of KCNQ4 hearing loss centers on dysfunctional basal inner hair cells and spiral ganglion neurons.Citation33 The belief that defective outer hair cells cannot account for the progressive nature of hearing impairment in DFNA2 pedigrees led to the study of the distribution of KCNQ4 protein and related it to expression of Kcnq4 splice variants.Citation34 Four splice variants were identified, with expression of the third variant (Kcnq4_v3) restricted primarily to the cochlea and with the highest expression in the basal spiral ganglion and inner hair cells.Citation34 It is proposed that this pattern of expression could explain the high frequency hearing loss seen in DFNA2 families.Citation34 Furthermore, the expression patterns in the murine organ of Corti and spiral ganglion cells have led to the hypothesis that progressive hearing loss could be a result of increasing mutation load toward the apex of the inner hair cells and neurons of the spiral ganglion, resulting in accumulation of defective proteins and, ultimately, a negative effect on cochlear function.Citation34
Possible third gene responsible for DFNA2 hearing loss
To date, two genes have been linked to the DFNA2 locus, ie, GJB3, encoding connexin 31, and KCNQ4, encoding a voltage-gated potassium channel.Citation3,Citation4 Controversy does exist over the association between changes found in GJB3 and DFNA2-associated deafness, as described above. Others have proposed that a third gene may exist at this locus which has yet to be identified. An Indonesian family that was mapped to the DFNA2 locus did not have an identifiable mutation in connexin 31 or the KCNQ4 voltage-gated potassium channel, suggesting that there is potentially another causative gene.Citation10,Citation18
The UCSF-99 family, a large five-generation family with a pattern of hearing loss characteristic of DFNA2 was linked to chromosome 1p34, the site of the DFNA2 locus. While sequence changes were seen in this family for both GJB3 and KCNQ4, these mutations were seen in both affected and unaffected family members, suggesting that neither gene was truly responsible for the hearing impairment.Citation5 This particular family does differ from other DFNA2 pedigrees in that the onset of hearing loss is later and progresses at a slower rate, providing further evidence that the DFNA2 locus is genetically heterogeneous.
Proposed candidates for this unidentified gene include POU3F1 and COL9A2 which are located within the mapped interval described by Goldstein et al.Citation5 Two members of the POU transcription factor family, which are expressed in embryonal stem cells and the brain, have been implicated in hearing loss. POU3F4 has been associated with DFN3 (deafness with fixation of the stapes) which is the most common X-linked form of hearing impairment, while POU4F3 has been linked with DFNA15 hearing loss, another form of nonsyndromic hearing loss.Citation35,Citation36 This makes POU3F1 an attractive candidate gene for further analysis.
Collagen proteins have also been linked with hearing impairment, such as the association of COL11A2 with DFNA13 hearing loss which primarily affects the mid frequencies. A connection has also been identified between type IV collagen mutations and Alport syndrome.Citation5COL9A2 will also likely see further attention in the coming years as a possible causal gene in nonsyndromic progressive hearing loss.
Currently, detection of DFNA2 hearing loss is based on characteristic clinical findings, including audiometric characteristics as well as a family history of dominant later-onset progressive hearing loss.
The only definitive means of diagnosing DFNA2-associated hearing impairment is molecular genetic testing. Through elucidation of the pathogenic mechanisms of DFNA2-associated hearing impairment, early diagnosis and eventually gene therapy for this condition will hopefully become available. In addition, this could potentially serve as a model for further study of more common forms of progressive hearing loss.
Conclusion
DFNA2-associated hearing loss is both clinically and genetically heterogeneous. To date, two genes, KCNQ4 and GJB3, have been linked to the DFNA2 locus on the short arm of chromosome 1 with most, if not all, pathogenic mutations identified in KCNQ4. Due to the inability to link certain DFNA2 families to mutations of GJB3 or KNCQ4, it has been proposed that a third causative gene exists. The majority of focus has been directed towards mutations of the KCNQ4 voltage-gated ion channel, and a variety of missense and nonsense mutations have been identified in families from diverse backgrounds. With the large population that is affected by hearing loss, it is presumed that a better understanding of the molecular mechanisms of DFNA2 hearing impairment will lead to improved methods of diagnosis and, potentially, treatment of this condition, as well as other forms of progressive hearing loss.
Acknowledgment
We would like to thank Kevin A Heraldo for assistance with graphics.
Disclosure
The authors report no conflicts of interest in this work.
References
- MortonNEGenetic epidemiology of hearing impairmentAnn N Y Acad Sci199163016311952587
- Van CampGSmithRJHHereditary hearing loss Available from: http://hereditaryhearingloss.orgAccessed April 11, 2012.
- XiaJHLiuCYTangBSMutations in the gene encoding gap junction protein beta-3 associated with autosomal dominant hearing impairmentNat Genet1998203703739843210
- KubischCSchroederBCFriedrichTKCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutations in dominant deafnessCell19999643744610025409
- GoldsteinJALalwaniAKFurther evidence for a third deafness gene within the DFNA2 locusAm J Med Genet200210830430911920835
- DeLeenheerEMEnsinkRJKunstHPDFNA2/KCNQ4 and its manifestationsAdv Otorhinolaryngol200261414612408061
- De LeenheerEMHuygenPLCoukePJAdmiraalRJVan CampGCremersCWLongitudinal and cross-sectional phenotype analysis in a new, large Dutch DFNA2/KCOQ4 familyAnn Otol Rhinol Laryngol200211126727411915881
- KunstHMarresHHuygenPNonsyndromic autosomal dominant progressive sensorineural hearing loss: audiologic analysis of a pedigree linked to DFNA2Laryngoscope199810874809432071
- MarresHvan EwijkMHuygenPInherited nonsyndromic hearing loss. An audiovestibular study in a large family with autosomal dominant progressive hearing loss related to DFNA2Arch Otolaryngol Head Neck Surg19971235735779193215
- CouckePJVan CampGDjoyodiharjoBLinkage of autosomal dominant hearing loss to the short arm of chromosome 1 in two familiesN Engl J Med19943314254318035838
- DenoyelleFLina-GranadeGPlauchuHConnexin 26 gene linked to a dominant deafnessNature19983933193209620796
- KelsellDPDunlopJStevensHPConnexin 26 mutations in hereditary nonsyndromic sensorineural deafnessNature199738780839139825
- GrifaAWagnerCAD’AmbrosioLMutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locusNat Genet199923161810471490
- SmithRJHildebrandMDFNA2 nonsyndromic hearing lossGene Reviews at Gene Tests: Medical Genetics Information Resources (database online) Available from: http://www.genetests.org. Updated February 17, 2011Accessed April 11, 2012
- JentschTJNeuronal KCNQ potassium channels: physiology and role in diseaseNat Rev Neurosci20001213011252765
- NeyroudNTessonFDenjoyIA novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndromeNat Genet1997151861899020846
- BaekJIParkHJParkKPathogenic effects of a novel mutation (c.664_681del) in KCNQ4 channels associated with auditory pathologyBiochim Biophys Acta2011181253654320832469
- Van HauwePCoukePJEnsinkRJHuygenPCremersCWVan CampGMutations in the KCNQ4 K+ channel gene, responsible for autosomal dominant hearing loss, cluster in the channel pore regionAm J Med Genet20009318418710925378
- KharkovetsTHardelinJPSafieddineSKCNQ, a K+ channel mutated in a form of dominant deafness, is expressed in the inner ear and the central auditory pathwayProc Natl Acad Sci USA20009784333433810760300
- CouckePJVan HauwePKelleyPMMutations in the KCNQ4 gene are responsible for autosomal dominant deafness in four DFNA2 familiesHum Mol Genet199981321132810369879
- MenciaAGonzalez-NietoDModamio-HoybjorSA novel mutation KCNQ4 pore-region mutation (p.G296S) causes deafness by impairing cell-surface channel expressionHum Genet2008123415318030493
- ArnettJEmerySBKimTBAutosomal dominant progressive sensorineural hEaring loss due to a novel mutation in the KCNQ4 geneArch Otolaryngol Head Neck Surg2011137545921242547
- TopsakalVPenningsRJte BrinkeHPhenotype determination guides swift genotyping of a DFNA2/KCNQ4 family with a hot spot mutation (W276S)Otol Neurotol200526525815699719
- Van CampGCoukePJAkitaJA mutational hot spot in the KCNQ4 gene responsible for autosomal dominant hearing impairmentHum Mutat200220151912112653
- AkitaJAbeSShinkawaHKimerlingWJUsamiSClinical and genetic features of nonsyndromic autosomal dominant sensorineural hearing loss: KCNQ4 is a gene responsible in JapaneseJ Hum Genet20014635536111450843
- DoyleDAMorais CabralJPfuetznerRAThe structure of the potassium channel: molecular basis of K+ conduction and selectivityScience199828069779525859
- TalebizadehZKelleyPMAskewJWBeiselKWSmithSDNovel mutation in the KCNQ4 gene in a large kindred with dominant progressive hearing lossHum Mutat19991449350110571947
- NieLMutations in KCNQ4 channels associated with nonsyndromic progressive sensorineural hearing lossCurr Opin Otlolaryngol Head Neck Surg200816441444
- SuCCYangJJShiehJCSuMCLiSYIdentification of novel mutations in the KCNQ4 gene of patients with nonsyndromic deafness from TaiwanAudiol Neurolotol2007122026
- KamadaFKureSKudoTA novel KCNQ4 one-base deletion in a large pedigree with hearing loss: implication for the genotype-phenotype correlationJ Hum Genet20065145546016596322
- HildebrandMSTackDMcMordieSJAudioprofile-directed screening identified novel mutations in KCNQ4 causing hearing loss at the DFNA2 locusGenet Med20081079780418941426
- KharkovetsTDedekKMaierHMice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafnessEMBO J20062564265216437162
- BeiselKWNelsonNCDelimontDCFritzschBLongitudinal gradients of KCNQ4 expression in spiral ganglion and cochlear hair cells correlate with progressive hearing loss in DFNA2Brain Res Mol Brain Res20008213714911042367
- BeiselKWRocha-SanchezSMMorrisKADifferential expression of KCNQ4 in inner hair cells and sensory neurons is the basis of progressive high-frequency hearing lossJ Neurosci2005259285929316207888
- De KokYJJvan der MaaerlSMBitner-GlindziczMAssociation between X-linked mixed deafness and mutations in the POU domain gene POU3F4Science19952676856887839145
- VahavaOMorellRLynchEDMutations in transcription factor POUF43 associated with inherited progressive hearing loss in humansScience1998279195019549506947