Abstract
Phelan–McDermid syndrome is caused by the loss of terminal regions of different sizes at 22q13. There is a wide range of severity of symptoms in patients with a 22q13 deletion, but these patients usually show neonatal hypotonia, global developmental delay, and dysmorphic traits. We carried out a clinical and molecular characterization of a patient with neonatal hypotonia and dysmorphic features. Array-based comparative genomic hybridization showed an 8.24 Mb terminal deletion associated with a 0.20 Mb duplication. Characterization of patients with Phelan–McDermid syndrome both clinically and at the molecular level allows genotype-phenotype correlations that provide clues to help elucidate the clinical implications.
Introduction
The 22q13.3 deletion syndrome, also known as Phelan–McDermid syndrome (PMS, OMIM # 606232), is characterized by global developmental delay, absent or impaired speech, neonatal hypotonia, autistic traits, and mild dysmorphic features.Citation1,Citation2 This syndrome results from loss of segments of varying sizes involving the terminal region of long arm of chromosome 22, due to a simple deletion (75% of PMS cases), ring chromosomes, or unbalanced translocations.Citation3–Citation5 Affected individuals have deletions ranging in size from 95 Kb to 9.22 Mb, and no common breakpoint has been observed.Citation6,Citation7
Due to lack of clinical recognition, the cryptic nature of this deletion in a significant fraction of cases, and the difficulty of performing an appropriate diagnostic test, PMS is considered to be underdiagnosed and its true prevalence is unknown.Citation4 A subtelomeric fluorescence in situ hybridization analysis of more than 11,000 patients with developmental disabilities suggested that deletion 22q13 was second to deletion 1p36 as the most frequent subtelomeric rearrangement, and was identified in 0.2% of those evaluated.Citation8 The number of malformation syndromes attributed to these microdeletions is increasing as more are being identified through molecular diagnostic techniques. Until recently, the diagnosis was based on cytogenetic banding and fluorescence in situ hybridization. Oligo-array comparative genomic hybridization (CGH) is able to detect smaller deletions and accurately measure deletion size and breakpoints.
Most of the published reports consider that the haploinsufficiency of SHANK3, which encodes a synapse structural protein and is located approximately 130 kb from the telomere, was responsible for the major neurobehavioral symptoms of this syndrome.Citation9,Citation10 A patient with developmental delay, speech delay and minor dysmorphic facial features (ptosis, epicanthal folds and cupper ears) carrying a de novo interstitial deletion disrupting only SHANK3 gene, supported that happloinsufficiency of SHANK3 alone, and no other genes telomeric to it, was responsible for PMS.Citation10
Recent genotype–phenotype studies have shown more severe phenotypes associated with larger deletions.Citation5,Citation6,Citation11 These results suggested that other genomic regions proximal to SHANK3 are responsible for speech and developmental delay, and some dysmorphic features, when deleted.
Clinical report
The patient is a Caucasian male, the first child of healthy parents. His mother was 29 and his father 30 years old when he was born. The father, grandfather, and great-grandfather presented toe syndactyly. First fetal movements were felt by the mother at 21 weeks. The pregnancy was complicated by maternal preeclampsia. He was born at gestational age 38 weeks by cesarean. Fetal monitoring before labor registered few movements. Birth weight was 2770 g, length 48 cm, and cranial perimeter 35.5 cm.
In the neonatal period, echocardiography showed a persistent ductus arteriosus and patent foramen ovale. Axial hypotonia was present. A cerebral scan was performed with a normal result. Dysmorphic features have been described in and can be seen in . He showed facial features that included large ears, full cheeks and pointed chin, large extremities, and a short webbed neck.
Figure 1 Dysmorphic features of patient described in this study. (A) Short webbed neck. (B) Large extremities, left talus valgus deformity, large penis and testes.
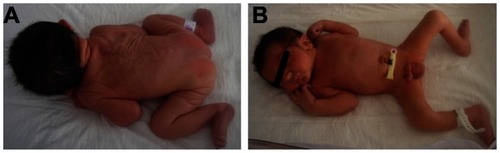
Table 1 Dysmorphic features of the patient described in this study
At 23 months old he had a bilateral hearing deficit and recurrent respiratory infections. Sacral ultrasound demonstrated renal crystal depositions.
Cytogenetics
The deletion was cytogenetically visible by conventional G-banding karyotype (). The parents were tested, displaying normal karyotypes, so the deletion was considered as de novo. Molecular testing for fragile X was negative.
Figure 2 Characterization of the deletion: cytogenetic and molecular studies. (A) Conventional G-banding chromosome 22. The deleted chromosome is on the right. (B) CGH-array. Reduced dosage for probes is shown to the left of the control two-copy line and increased dosage is shown to the right.
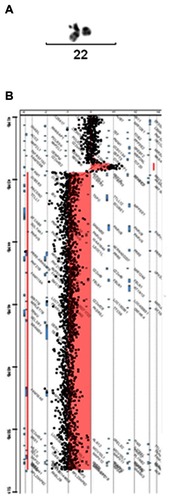
High resolution CGH-array (400K; Agilent Technologies, Santa Clara, CA) was carried out to determine the size and extent of the deletion (). This was determined to be a terminal deletion of 8.24 Mb associated with a duplication of 0.20 Mb adjacent to the deleted region, 46,XY,arr cgh 22q13.2(42694206-42889651)x3, 22q13.22q13.3(42944814-51186390)x1 (location according NCBI Build 37.3). The deleted region has 137 genes in the Genes on Sequences NCBI map, of which 61 are in the OMIM database. The duplicated region contains two genes, one of which (NFAM1) is in the OMIM database.
Discussion
The clinical features of PMS are highly variable. Patients’ deletion sizes are also highly variable. Recent genotype–phenotype studies using CGH-array have found correlations between deletion size and the severity of selected phenotypes.Citation6,Citation7 Different features have been associated with larger deletions including large hands,Citation7 and dysmorphic features related to ears, toenails, and philtrum, as well as male genital anomalies.Citation6,Citation12 Our patient has a phenotype similar to those reported with similar deletion sizes. This genotype–phenotype correlation suggested that there are clinically important genes located proximal to SHANK3 contributing to PMS phenotype. In agreement with this, Wilson et alCitation11 reported two cases with intact SHANK3 that showed development delay and dysmorphic features.
The interval deleted in our patient contains 137 genes. Some of these genes (besides SHANK3) are responsible for clinically significant disorders: UPK3A (renal adysplasia), FBLN1 (synpolydactyly associated with metacarpal and metatarsal synostoses), ATXN10 (spinocerebellar ataxia 10), TRMU (liver failure, transient infantile, deafness, aminoglycoside-induced).
Our patient carries an 8.24 Mb deletion associated with a 0.20 Mb duplication. Only two similar cases with deletion-duplications in 22q13 have been previously reported, although these were not the same as our patient’s. Lindquist et alCitation13 described a patient with a 7.9 Mb duplication and a 4.2 Mb deletion of 22q13 who had generalized developmental delay, delayed speech, hypotonia, dysmorphic features (including epicanthal folds, flat midface, and full cheeks), apneic spells with seizures, persistent ductus arteriosus and Pierre Robin sequence (never reported before in PMS). Koolen et alCitation5 reported a case with a 3.9 Mb subtelomeric deletion associated with a 2.0 Mb duplication. This patient had signs of retinitis pigmentosa (never reported before in PMS). There are a growing number of similar cases that have been reported at different chromosomal ends: 1p, 2q, 4p, 5p, and 8p.Citation14–Citation18 This type of chromosome rearrangement may be more common than previously thought, and it can be detected by high-resolution CGH-array.
A phenotype present in our patient had never been observed in 22q13 deletion: short webbed neck. This feature had been previously observed in 22q13 duplication syndrome.Citation19 This feature is only present when 22q13.2 is duplicated. Patients with duplications 22q13.3-qter do not show short neck. So, this phenotype is possibly attributable to the duplication.
In summary, this study underscores the utility of array CGH for the characterization of the size and nature of subtelomeric rearrangements and to predict the severity of phenotypes. Associations between phenotypes and deleted/duplicated regions provide valuable information about clinically important genes, micro-RNAs or regulatory elements, and will allow investigation of their role in the phenotypes of this syndrome.
Acknowledgments
The authors wish to thank Dr José Miguel García-Sagredo and Dr Jorge J Prieto for their critical comments on the manuscript and Miss Jéssica Calvo for technical support. This work was supported by CITOLAB and Cátedra de Biomedicina Reproductiva Vistahermosa.
Disclosure
The authors report no conflicts of interest in this work.
References
- PhelanMCRogersRCSaulRA22q13 deletion syndromeAm J Med Genet20011012919911391650
- HavensJMVisootsakJPhelanMCGrahamJMJr22q13 deletion syndrome: an update and review for the primary pediatricianClin pediatr (Phila)2004431435314968892
- LucianiJJde MasPDepetrisDTelomeric 22q13 deletions resulting from rings, simple deletions, and translocations: cytogenetic, molecular, and clinical analyses of 32 new observationsJ Med Genet200340969069612960216
- PhelanMCDeletion 22q13.3 syndromeOrphanet J Rare Dis200831418505557
- KoolenDAReardonWRosserEMMolecular characterisation of patients with subtelomeric 22q abnormalities using chromosome specific array-based comparative genomic hybridisationEur J Hum Genet20051391019102415986041
- SarasuaSMDwivediABoccutoLAssociation between deletion size and important phenotypes expands the genomic region of interest in Phelan-McDermid syndrome (22q13 deletion syndrome)J Med Genet2011481176176621984749
- DharSUdel GaudioDGermanJR22q13.3 deletion syndrome: clinical and molecular analysis using array CGHAm J Med Genet A2010152A357358120186804
- RavnanJBTepperbergJHPapenhausenPSubtelomere FISH analysis of 11688 cases: an evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilitiesJ Med Genet200643647848916199540
- WilsonHLWongACShawSRMolecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptomsJ Med Genet200340857558412920066
- DelahayeAToutainAAbouraAChromosome 22q13.3 deletion syndrome with a de novo interstitial 22q13.3 cryptic deletion disrupting SHANK3Eur J Med Genet200952532833219454329
- WilsonHLCrollaJAWalkerDInterstitial 22q13 deletions: genes other than SHANK3 have major effects on cognitive and language developmentEur J Hum Genet200816111301131018523453
- JeffriesARCurranSElmslieFMolecular and phenotypic characterization of ring chromosome 22Am J Med Genet A2005137213914716059935
- LindquistSGKirchhoffMLundsteenCFurther delineation of the 22q13 deletion syndromeClin Dysmorphol2005142556015770125
- CotterPDKaffeSLiLGershinIFHirschhornKLoss of subtelomeric sequence associated with a terminal inversion duplication of the short arm of chromosome 4Am J Med Genet20011021768011471177
- FloridiaGPiantanidaMMinelliAThe same molecular mechanism at the maternal meiosis I produces mono- and dicentric 8p duplicationsAm J Hum Genet19965847857968644743
- BonagliaMCGiordaRPoggiGInverted duplications are recurrent rearrangements always associated with a distal deletion: description of a new case involving 2qEur J Hum Genet20008859760310951522
- BallifBCYuWShawCAKashorkCDShafferLGMonosomy 1p36 breakpoint junctions suggest pre-meiotic breakage-fusion-bridge cycles are involved in generating terminal deletionsHum Mol Genet200312172153216512915474
- IzzoAGenesioRRongaV40 Mb duplication in chromosome band 5p13.1p15.33 with 800 kb terminal deletion in a foetus with mild phenotypic featuresEur J Med Genet201255214014422269966
- FeenstraIKoolenDAVan der PasJCryptic duplication of the distal segment of 22q due to a translocation (21;22): three case reports and a review of the literatureEur J Med Genet200649538439516503209