
Abstract
Ankylosing spondylitis (AS) is a complex disease involving multiple risk factors, both genetic and environmental. AS patients are predominantly young men, and the disease is characterized by inflammation and ankylosis, mainly at the cartilage–bone interface and enthesis. HLA-B27 has been known to be the major AS-susceptibility gene for more than 40 years. Despite advances made in the past few years, progress in the search for non-human leukocyte antigen susceptibility genes has been hampered by the heterogeneity of the disease. Compared to other complex diseases, such as inflammatory bowel disease (IBD), fewer susceptibility loci have been identified in AS. Furthermore, non-major histocompatibility-complex susceptibility loci discovered, such as ERAP1 and IL23R, are likely contributors to joint inflammation. Identification and confirmation of functional variants remains a significant challenge of investigations involving genome-wide association studies (GWAS). It remains unclear why none of the AS-susceptibility genes identified in GWAS appear to be directly involved in the ankylosing process. Numerous reviews have recently been published on the genetics of AS. Therefore, aside from a brief summary of what AS GWAS has successfully achieved thus far, this review will focus on directions that could address unanswered questions raised by GWAS.
Introduction
Ankylosing spondylitis
Ankylosing spondylitis (AS) is a subset of spondyloarthritis (SpA), which is characterized by inflammation of the sacroiliac joints, peripheral inflammatory arthropathy, and the absence of rheumatoid factor. Other subsets of SpA include reactive arthritis, psoriatic arthritis, colitic arthropathies (inflammatory bowel disease [IBD]-related SpA), and undifferentiated SpA. With a prevalence of 0.1%–1.4%, AS is an under-recognized form of chronic arthritis.Citation1 It can lead to significant spinal disease and peripheral arthritis, which can manifest as chronic back pain and a progressive spinal ankylosis. The disease strikes predominantly men between the ages of 20 and 40 years, in their peak productive years, leading to significant loss of work productivity and decreased quality of life.
Diagnosis, progression, and current management of AS
AS is usually diagnosed according to the modified New York criteria,Citation2 which include a combination of such clinical features as limited motion of the lumbar spine, persistent lower-back pain, limited chest expansion, and radiographic evidence of sacroiliitis. The hallmark of AS is neo-ossification at the site of joint inflammation.Citation3 Although joint inflammation can be detected early in the disease process, eg, in the first year of symptoms, using magnetic resonance-imaging technology, this is not a definitive diagnosis test for AS. The subsequent spinal structural changes, as visualized on radiographs, appear relatively late. This explains in part why it can take 5–10 years to confirm a diagnosis of AS after the initial onset of symptoms.Citation4,Citation5
Nonsteroidal anti-inflammatory drugs (NSAIDs) are the first-line drug treatment for AS patients with symptomatic disease.Citation6 Continuous use of NSAIDs appears to slow radiographic progression.Citation7,Citation8 If NSAID treatment fails, biologics, particularly tumor necrosis factor (TNF)-α inhibitors, are used in patients with active disease. The TNF blockers adalimumab, etanercept, infliximab, golimumab, and certolizumab have all been proven to be highly effective in controlling inflammation and in improving the quality of life of most AS patients. Whether these TNF blockers could halt progression of structural changes remains a controversial issue. Some studies showed no structural impact,Citation9 but a recent study indicated that TNF inhibitors impact structural damage, such as new syndesmophyte appearance, growth of existing syndesmophytes, and development of ankylosis in AS.Citation10 However, a substantial proportion of AS patients (∼25%) do not respond to TNF-inhibitor therapy, and some initial responders developed secondary unresponsiveness.
Joint ankylosis
Current concepts regard new bone formation at the enthesis as a pathological response to injury, and that joint inflammation precedes ossification. Early stages of ankylosis involve squaring of the vertebral bodies and formation of syndesmophytes. Total spinal ankylosis and kyphosis are found in the most severe cases. Radiographic changes of the cervical and lumbar spine in AS patients are scored using the modified Stokes AS Spine Score (mSASSS).Citation11 Scoring of sequential radiographs from the same patient over time determines the change in mSASSS per year. Systematic evaluation of ankylosis progression in AS patients is subjective and time-consuming.Citation12,Citation13
There remains some uncertainty whether joint inflammation and ankylosis in AS are linked events or independent processes.Citation14–Citation16 For clinicians, this critical issue affects the management strategy for SpA: early intervention with anti-TNF therapy would prevent ankylosis development only if inflammation and ankylosis are sequential and linked processes.
Genetics and AS
Based on family and twin studies, it has been established for a long time that AS has a strong genetic component. The sibling recurrence risk of AS is 9.2% (λs is 82) compared to 0.1% in the general population.Citation17 The heritability of AS is estimated to be >95%. The strong association with human leukocyte antigen (HLA)-B27 was discovered in the early 1970s. Population studies have indicated that 2% of HLA-B27-positive individuals develop AS,Citation18 implying that other factors – genetic, environmental, or stochastic – contribute importantly to disease development. The most direct evidence that genetic polymorphisms other than HLA-B27 contribute to AS is the difference in concordance rates reported for HLA-B27-positive monozygotic twins (17 of 27 [63%]) and HLA-B27-positive dizygotic twins (four of 15 [27%]).Citation19 These same statistics indicate that the predisposition to AS is not entirely genetically determined. Nongenetic factors might correspond to an environmental effect, such as a specific microbial infection, as implicated in reactive arthritis, or it might correspond to a stochastic event in development, such as the emergence of specific immune cells.
Recent genome-wide association study findings
A number of updated reviews on AS genetics,Citation20–Citation24 including genome-wide association study (GWAS) results, have recently been published, and thus we only briefly summarize the key findings here. Based on the most recent report on AS susceptibility loci detected by ImmunoChip (Illumina, San Diego, CA, USA) genotyping of the largest cohort (more than 10,000 individuals for both cases and controls examined),Citation25 there are at least 25 AS non-major histocompatibility complex (MHC) immune-related risk loci (summarized in ), eleven of which have been identified previously.Citation26 A major achievement of these studies relates to the identification of important biological pathways likely responsible for AS pathogenesis.
Table 1 Summary of ankylosing spondylitis-susceptibility genes identified by genome-wide association studies
Role of the interleukin-23-related pathway
AS-associated genetic variants relating to this pathway include interleukin (IL)-23R, IL-12β, Tyk2, IL27, and IL-6R. Susceptibility to loci involved in this pathway is shared among many inflammatory diseases, including IBD and psoriasis. It remains to be elucidated how influence on one common pathway leads to the development of different inflammatory diseases. A recent elegant study in Cell illustrates the power of using a combination of genetic, clinical, and functional analyses in both mice and humans to unravel how a noncoding single-nucleotide polymorphism (SNP) can influence the disease process.Citation27 Intriguingly, this FOXO3 variant (rs12212067), which regulates cytokine production in monocytes, was not associated with susceptibility to the rheumatoid arthritis or Crohn’s disease.
It is established that IL-23 drives the differentiation of CD4-positive Th17 cells, which produce IL-17. IL-17 in turn can facilitate the production of other factors (such as IL-6, IL-8, TNF, chemokines, matrix metalloproteinases, and receptor activator of nuclear factor κB ligand) from a wide range of cell types.Citation28 A French SpA study illustrated that variants at loci in the IL-23/Th17 pathway influence expression levels of genes involved in the differentiation of Th17/Th1 cells, and it is likely that the pathological outcome is dictated by combinatorial assortments of multiple variants.Citation29 A comprehensive discussion on how IL-23/IL-17 pathways impact on AS pathogenesis is beyond the scope of this review. An excellent review on this aspect has just been published.Citation28
Role of aminopeptidases
In addition to ERAP1 and ERAP2, two other aminopeptidases (LNPEP and NPEPPS) are associated with AS,Citation23 reiterating the importance of antigen presentation in AS pathogenesis. Both protective and susceptible ERAP1 variants associated with AS have been identified. The relative attributable risk of ERAP1 to AS is about 25%, whereas that of HLA-B27 is about 50%. These two genes combined provide the two most powerful disease risk factors to AS. Intriguingly, the association of ERAP1 is restricted to HLA-B27-positive AS patients.Citation25 One recent functional study showed that ERAP1 variants affect HLA-B27 antigen presentation and stability in vivo.Citation30 Protective variants lead to less ERAP1 activity, and less efficient trimming of HLA-B27 ligands. Another study supported the notion that AS-associated ERAP1 variants alter the composition and length of HLA-B27 ligands.Citation31 A more in-depth review on the role of ERAP1 in AS pathogenesis will be discussed in later sections.
ERAP2 is unique to humans, and does not exist in mice. However, a high-frequency variant, when present in homozygosity (about 25% of the population), results in the absence of ERAP2 protein in these individuals. In ERAP2-deficient human B cells, surface MHC-I expression is reduced.Citation32 It remains unclear whether the absence of ERAP2 might alter/modulate antigen presentation in these individuals, especially patients with such diseases as AS and Crohn’s disease in which disease-associated ERAP2 variants exist. Intriguingly, one ERAP2 variant (rs2549782) confers natural resistance to human immunodeficiency virus-1 infection.Citation33 Results from the most recent GWAS indicated that ERAP2 variants are associated with AS in HLA-B27-negative cases.Citation25
Despite substantial sequence homology, similar overall domain organization and structures between ERAP1 and ERAP2, the N-terminal peptide specificities between these two aminopeptidases are quite different, as explained by their crystal structures.Citation34 We showed that an ERAP1 ERAP2 haplotype (rs27044[G] rs30187[T] rs2549782[T]) is associated with familial AS.Citation35 A recent study using sequencing haplotypes in 20 individuals showed that this haplotype occurs naturally.Citation36 Amino acid variants coded by ERAP2 rs2549782 (N392K) alter both the specificity and activity of ERAP2. Amino acid variants coded by ERAP1 rs27044 (Q730E) and rs30187 (K528R) affect peptide-trimming activity. To date, there has been only one study that assessed the effects of naturally occurring ERAP haplotypes.Citation36 Importantly, results from this study showed that ERAP SNPs, when assessed in combination (as a haplotype), showed different effects compared to those assessed singly. Though there are human cells deficient in ERAP2, currently no ERAP1-deficient human cells are available for accurate assessments of the effects of natural ERAP haplotypes, and this poses a limitation on these types of studies. It is expected that different ERAP haplotypes would impact on natural killer cell and cytotoxic T-lymphocyte functions.
Additional AS risk loci contribute to variations in T-cell lineages (EOMES, IL7R, RUNX3, and ZM1Z1 for CD8+ T cells, and BACH2 and SH2B3 for CD4+ T cells). Variants in G-protein-coupled receptors (GPR35, GPR37, GPR65, and GPR25) were also identified, but their involvement with AS pathogenesis is less clear. It has been estimated that all these non-MHC risk loci only account for 4.3% of heritability in AS, while HLA-B27 contribute 20.1%, implying that a majority of risk loci (about 75%) remain undefined.
Another insight emerged from GWAS relates to common risk loci shared among some inflammatory diseases. Most notably is the largest number of loci shared between AS and IBDCitation37,Citation38 (12 and 11 AS loci shared with Crohn’s disease and ulcerative colitis [UC] respectively). At least 163 risk loci have been identified in IBD and about 28 of them were shared between Crohn’s disease and ulcerative colitis. The significant number of risk loci shared between AS and IBD supports the recent concept that gut involvement contributes to disease pathogenesis in a large subset (up to 60%) of AS patients. This issue is further addressed in a later section of this review.
Limitations of GWAS
In general, despite the wealth of new information obtained from GWAS, some unexpected challenges also emerged. Following are examples: 1) The number of risk loci uncovered was not only high (in terms of thousands in some complex traits) but also increased proportionally with the cohort size analyzed.Citation39 There are some indications that variants from different steps of the same biological pathway could contribute similarly to the eventual clinical outcome; 2) The effect size of individual risk loci are usually very modest (odds ratio ∼1.05–1.4), and GWAS can only detect common variants with a minor allele frequency of >0.05. For minor causal variants, deep sequencing of the regions of interest is required;Citation40,Citation41 3) Identification of causal variants with direct or indirect functional relevance to disease risk has proved to be difficult. It is challenging to prove that functional consequences of variants (usually assessed singly) contribute to disease pathogenesis. For complex traits, different combinations of risk factors can lead to similar clinical outcomes, leading to disease heterogeneity. In IBD, of 163 risk loci identified, only one (nucleotide-binding oligomerization domain-containing protein 2) has been shown to correlate with clinical outcomes.Citation42 More importantly, the lack of large cohorts with detailed clinical parameters and well-characterized disease outcome renders meaningful analyses of genotypes obtained in GWAS a major challenge; 4) Known AS susceptibility locus (such as the IL1 gene cluster) can be missed. In the earlier AS genetic studies, positive and negative results were obtained with respect to the association of variants within the IL1 gene cluster in AS.Citation43–Citation47 This locus was not detected in recent AS GWAS. However, a recent French study showed that IL1A is associated with AS susceptibility or sacroiliitis in AS.Citation48 Likely reasons contributing to the discrepancies among studies include disease heterogeneity, study design, and power limitations. The precise role of IL-1 in AS pathogenesis remains unclear, though it could contribute singly or in combination with other pathways with susceptibility variants (such as IL-23). Despite these inconsistent results from different studies, the latest International Genetics of Ankylosing Spondylitis Consortium GWASCitation25 concluded that AS is associated with the IL1R1–IL1R2 locus located on chromosome 2q11. There were two signals, one in each of the IL1R genes.
Are there risk loci relating to neo-ossification/ankylosis in AS patients?
In the AS GWAS result published in 2010,Citation26 ANTXR2 (CMG2) was identified as one of the risk loci. Unfortunately, no SNP in this locus was included in the most recent AS GWAS, and thus it is unclear whether this association is replicable. ANTXR2 is not associated with AS in the Han Chinese,Citation49 and the minor allele frequency was too low to analyze this in Koreans. Anthrax toxin-receptor 2 could potentially affect new bone formation, as it is a membrane-bound molecule that can interact with low-density lipoprotein receptor-related protein (LRP)-6.Citation50 LRP6 is an important surface receptor in the Wnt/β–catenin pathway, and thus can affect osteoblastic activity. More work in discerning whether ANTXR2 plays a role in AS pathogenesis is warranted.
A recent GWAS performed in Han Chinese with ASCitation49 detected two risk loci likely with relevance to bone formation (HAPLN1–EDIL3 at 5q14.3 and ANO6 at 12q12.1). HAPLN1 has been shown to be involved with osteophyte formationCitation51 in Japanese women with spinal osteoarthritis. EDIL3 has an inhibitory effect on Wnt/β-catenin signaling.Citation52 ANO6 plays a role in osteoclastogenesis.Citation53 The most recent GWAS on East Asians using the ImmunoChip failed to replicate association of these two loci.Citation25 One explanation is the low frequencies of the variants. This scenario is supported by the absence of association in IL23R variants for Han Chinese AS, but recent sequencing of this locus revealed a few rare variants with potential functional relevance.Citation54 However, it is also possible that the absence of ANTRX2, HAPLN1–EDIL3, and ANO6 association in Han Chinese AS GWAS might be due to other ethnic differences.
There are a few studies implicating the role of IL-23 in excess bone ossification in AS. In a French population, an IL-23R variant was associated with radiographic sacroiliitis in AS.Citation55 A more indirect finding relates to elevated levels of IL-23 detected in bone marrow cells from AS spinal facet joints obtained in corrective surgery.Citation56 A mouse study also illustrated that IL-23 could drive enthesitis via entheseal resident T cells (positive for IL-23R, retinoic acid receptor-related orphan receptor-γt, and CD3) in an IL-22-dependent manner.Citation57
Considering the strength of association of ERAP1 with AS, we questioned whether genes involved in the antigen-processing pathway could also be a marker of severity in addition to susceptibility. For this study,Citation58 a total of 241 Caucasian patients with AS from spondylitis clinics in Toronto and Edmonton were included. Those patients who had at least two full sets of radiographs for mSASSS scoring at a minimum interval of 1.5 years were included in the analysis for genetic predictors of progression. Genotyping was done for a panel of 13 coding region SNPs in the ERAP1, LMP2, LMP7, TAP1, and TAP2 genes. In the univariate analysis for predictors of baseline radiographic severity, the duration of disease was the strongest, followed by male sex and LMP2 and ERAP1 variants. In multivariate analysis, the only genetic predictor that remained strongly associated with severity was LMP2. This is very interesting, as LMP2 has previously been reported to be associated with AS and uveitis in AS patients. Due to the association with uveitis, LMP2 may in fact be a marker of a more aggressive form of AS that could lead to more structural damage. Moreover, the proteasome helps to break down β-catenin, and abnormalities in LMP2 could lead to excess Wnt/β-catenin signaling and osteoblastic activity.
A candidate-gene approach to unravel ankylosis-related risk loci in AS; the role of ANKH and TNAP
Inorganic pyrophosphate (PPi) plays an important role in regulating mineralization and bone formation.Citation59 The sources of PPi for the cartilage matrix include intracellular-to-extracellular transport of PPi through the cell membrane in association with a membrane transport system that involves the ANKH (human homolog of progressive ankylosis) proteinCitation60 and generation of PPi at the cell surface by tissue-nonspecific alkaline phosphatases (TNAPs) or ectonucleotidases.Citation61,Citation62
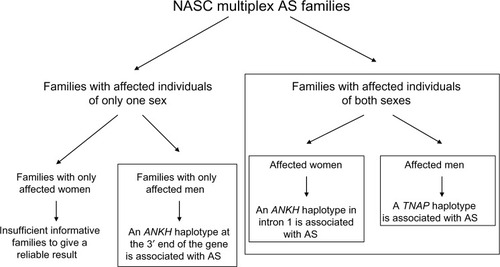
Using a candidate-gene approach, our linkage- and family-based association analysesCitation63 demonstrated that North American Caucasian AS patients have excess sharing of the ANKH gene region, and that AS is significantly associated with a specific ANKH haplotype.Citation63 Significantly, we further showed that there are sex differences in the ANKH variants associated with AS.Citation64 Intriguingly, there is heterogeneity even in multiplex AS families. There were three types of families in our cohort of multiplex AS families: 1) families with affected individuals of both sexes, 2) families with only men affected, and 3) families with only women affected. In the first type of families with affected individuals of both sexes, two ANKH SNPs (rs28006[C] rs25957[C]) were associated with AS only in affected women. This haplotype was transmitted to affected women 79% of the time (15 of 19), but to affected men only 27% of the time (three of eleven). This effect is substantial, as the odds ratio for increased risk approaches 3.0 (0.79/0.27=2.92). In another haplotype (rs26307[C] rs27356[C]), the frequency of transmission was 70% to affected men (21 of 30) and 43% to affected women (13 of 30). In the subset of families with only men affected, 94% of the time (16 of 17), this haplotype was transmitted to affected men. There were too few informative families with only women affected with this haplotype, and thus we do not have a reliable assessment of the frequency at which this haplotype was transmitted to affected women in this subset for comparison.
Using Family-Based Association Test (Harvard School of Public Health, Boston, MA, USA) analysis in the subset of AS families with affected individuals of both sexes, we also found that a TNAP variant (Tyr263His) is significantly associated with AS.Citation65 Furthermore, this TNAP variant is significantly associated with AS in affected men (dominant model, P=0.005). More impressively, the frequency of transmission of this allele was 92.3% (12 of 13) to affected men and only 64.7% (eleven of 17) to affected women. It appears that in this subset of AS families with affected individuals of both sexes, an ANKH polymorphism in the intron 1, close to exon 2 region, predisposes the affected women to AS, while a TNAP variant (possibly Tyr263His) predisposes the affected men to AS. The TNAP Tyr263His is a functional variant,Citation66 and is associated with bone mineral density in elderly women.Citation67 Osteoporosis is a common occurrence in AS. Investigation on whether this functional TNAP variant (Tyr263His) is associated with osteoporosis in AS patients is warranted.
In our cohort of North American Caucasian multiplex families, both ANKH and TNAP were associated with AS (summarized in ). Subsequently, there have been reports of ANKH but not TNAP association in JapaneseCitation68 and Han ChineseCitation69 cohorts. In an earlier UK study, no ANKH association was detected.Citation70 A recent presentation at the annual meeting of the American College of Rheumatology reported that ANKH variants were associated with disease severity in Korean AS.Citation71
Figure 1 Summary of family-based association analyses using multiplex ankylosing spondylitis (AS) families from the North American Spondylitis Consortium (NASC).
Gene–gene interactions: the role of ERAP1 and HLA-B27 in AS
The discovery of the association of ERAP with AS opened new opportunities for investigating AS. Gain or loss of ERAP1 function could be used in experimental settings to further decipher the mechanisms by which ERAP1 contributes to the overall pathogenesis of AS. The mechanisms by which ERAP1 and HLA-B27 contribute to AS are under study. The main function of ERAP1 is to trim peptides within the ER into the right length before binding to MHC-I molecules. Abnormal function of ERAP1 can lead to generation of anomalous peptides of incorrect length and sequence, which subsequent to MHC-I binding (eg, HLA-B27) can lead to peptide–MHC-I complex misfolding. These misfolded proteins could either accumulate within the ER, thus contributing to the ER stress, or be transported to the cell surface as a free heavy chain. Both of these mechanisms have been implicated as contributing to AS pathogenesis, but the degree of their contribution to the overall pathogenesis of AS remains speculative. Loss of ERAP1 function leads to decreased surface expression of MHC-I molecules. Reduced surface expression has been suggested to contribute to the overall pathogenesis of AS. Recent data from our laboratory analyzing immune response to influenza virus have shown a change in the overall flu-peptide repertoire in mice deficient in ERAP function (ERAP1−/−).Citation72 Our results confirm the findings of other investigators: ERAP1−/− mice had changes in the presentation of viral epitopes of different types, indicating the importance of ERAP in viral peptide generation and presentation. While these studies implicate the presence of an arthritogenic peptide in AS, the identity of this epitope is unknown. Since loss of ERAP1 function seems to be protective in AS, ERAP1 may be involved in the generation of arthritogenic peptides. However, to date, there has been no direct evidence either confirming or refuting this hypothesis.
Gene–gene interactions: the role of ERAP1 in AS patients following influenza infection
ERAP1 plays an integral role in the immune response following an infection. It can either enhance or diminish the immune response by changing the peptide repertoire presented by antigen-presenting cells.Citation73–Citation75 ERAP1, for instance, has been shown to affect the repertoire of peptides generated following influenza infection, one of the most commonly encountered human viruses. However, there are no reports investigating the host response to flu infection in AS patients. Although there are suggestions that an arthritogenic peptide or peptides plays a central role in AS,Citation76,Citation77 so far no such peptides have been discovered. Prior flu infection may influence the presentation of such arthritogenic peptides. In addition, AS is a multigenic disease, and as such, studies of AS patients with prior flu infection would be complex to analyze.Citation77 Given these limitations, there is a high need for an AS animal model to investigate the role of these aforementioned factors in vivo. An animal model could also be useful to address gene–gene interaction in AS, which is highlighted by the fact that ERAP1 is associated only with B27-positive AS and not with B27-negative AS.
To overcome this limitation, we have used human HLA transgenic (HLA Tg) mice, which lack both ERAP1 and endogenous MHC-I molecule expression and which express HLA-B27 (ie, Tg HLA-B27/ERAP−/−) to investigate the interaction of ERAP1 and HLA-B27 in AS. These two genes are the strongest genetic associations with AS to date,Citation78 yet B27/ERAP−/− mice manifest no articular abnormalities (unpublished results). It should be noted in this regard that loss-of-function variants of ERAP1 are protective in AS. On the other hand, since the flu peptides presented by B27 are well known, this animal model can provide important insights into the role of ERAP in generating B27-specific antigenic peptides. Initial studies of flu infection of Tg HLA-B27/ERAP−/− mice suggest that ERAP1 may play a central role in the phenomenon of immunodominance after flu infection.Citation79 This animal model could prove valuable in deciphering the interaction of ERAP1 and HLA-B27 in an in vivo context.
Animal models with axial ankylosis
Though some AS patients have peripheral arthritis, AS is mainly an axial disease. An informative animal model could unravel the underlying mechanisms that lead to the development of axial inflammation and eventual ankylosis. Because AS is a multifactorial complex disease, it is unlikely that a perfect animal model for AS exists. Since HLA-B27 showed the strongest association with AS, transgenic rats highly expressing HLA-B27 and human β2-microglobulin could be a reasonable model. These transgenic rats have spontaneous peripheral and axial inflammation, as well as gut disease.Citation80 With elevated β2-microglobulin expression, ankylosis developed with concurrent suppression of gut disease and an unfolded protein response.Citation81
Two mouse models overexpressing TNFα (hTNFtgCitation82 and TNF AU-rich elements [ΔARE]Citation83) develop systemic inflammation, gut disease, and sacroiliitis, but no ankylosis. A recent studyCitation84 reported that mechanotransduction can lead to enthesitis and neo-ossification at entheseal sites in tail-suspended TNFΔARE mice. Enthesitis induction involves a number of pathways, including signaling via IL-23R-positive entheseal resident cells.
The proteoglycan-induced spondylitis (PGISp) model mimics many features of human AS, including axial inflammation and ankylosis.Citation85,Citation86 The spines of the PGISp mice showed decreased levels of Wnt-signaling antagonists (such as Dickkopf-related protein 1 and sclerostin [SOST]).Citation87 There is evidence suggesting that enhanced Wnt/β-catenin signaling contributes to ankylosis in AS patients. Dickkopf-related protein 1, an antagonist of Wnt/β-catenin signaling, has been reported to be either dysfunctionalCitation88 or present at lower-than-normal levels in AS patients.Citation89 Serum levels of SOST are also lower in AS patients than healthy individuals.Citation90 Our recent work on ank/ank (progressive ankylosis) mice that have peripheral and spinal ankylosis showed enhanced Wnt/β-catenin signaling in the joints of these mutant mice.Citation91
Old male DBA/1 mice develop peripheral arthritis and it has been shown that noggin (NOG) can rescue the enthesopathy.Citation92 In contrast, we showed that NOG treatment of ank/ank mice led to more severe ankylosis, with concurrent generation of high levels of immunoglobulin (Ig)-G immune complexes (ICs) in which the autoantigens are either NOG (a bone morphogenetic protein-signaling antagonist) or SOST (a Wnt/β-catenin signaling antagonist).Citation93 These results from the mutant ank/ank mice led to our novel finding of similar NOG/SOST IgG ICs in humans, and AS patients have significantly elevated serum levels of these ICs.Citation93 An intriguing possibility is that ICs involving autoantibodies against NOG and SOST at their interacting sites may mimic the inhibitory interaction that naturally occurs between these two proteins. By this mechanism, these autoantibodies would be predicted to play a physiological role in bone homeostasis in normal individuals. However, overabundance of these autoantibodies would lead to reduced levels of functional NOG and SOST, resulting in enhanced bone morphogenetic protein and β-catenin signaling, neo-ossification and eventual spinal ankylosis.
Lessons from GWAS: how might they help in disease management?
Biologics involving inhibition of TNFα have proven to have dramatic efficacy in about 70% of AS patients, but they do not cure the disease. In many patients, challenges remain regarding how best to achieve and maintain remission. As mentioned earlier, treatment targeting IL-1 using anakinra was not very promising.Citation94 IL6R is one of the AS-susceptibility genes detected in GWAS, yet tocilizumab (anti-human IL-6-receptor monoclonal antibody)Citation95 and sarilumab (a fully human monoclonal antibody against IL-6Rα)Citation96 appear to have no efficacy in AS patients. Genetic studies implicated the importance of the IL-23 signaling pathway. A recent prospective clinical trial showed promising efficacy and safety in the use of ustekinumabCitation97 (anti-IL-12/23p40) to treat AS patients. Promising results were shown in IL-17 blockade.Citation98 More studies are warranted to explore novel therapies for AS patients especially for TNF-inhibitor nonresponders.
Future directions
As mentioned earlier, AS is a very heterogeneous complex disease. Different combinations of risk loci, together with other nongenetic factors, would lead to a similar clinical outcome, ie, AS disease. Studies using patient cohorts with bias toward certain subsets would result in controversial and inconsistent results. Genetic analysis of AS subsets would lead to the identification of subset-specific risk loci. The association of ERAP1 being restricted to HLA-B27-positive AS patients represents a good example.
It has been known for decades that AS patients have extra-articular manifestations. A recent systematic meta-analysisCitation99 confirmed that about 25.8% of AS patients have acute anterior uveitis (AAU); 9.3% and 6.8% have concomitant psoriasis and IBD, respectively. Intriguingly, the large overlap of AS patients with AAU is associated with disease duration, implying that the disease process in AS renders the patient susceptible to AAU. In contrast, it was reported that occurrence of IBD or psoriasis in AS was not associated with disease duration. In some such AS patients, IBD and psoriasis might be present prior to the diagnosis of AS. The implication of this is that certain AS subsets might have different etiologic pathways underlying the disease. For example, histopathological findings demonstrated that 50%–60% of AS patients have evidence of gut inflammation,Citation100 although this is clinically evident in only 5%–10% of AS patients.Citation101 It may be that AS patients with subclinical versus clinical gut inflammation represent distinct subsets of the disease. It is possible that AS patients whose gut and joint inflammation might be triggered by specific microbial exposure and thus leading to distinctive, pathogenic immune responses. In support of this, we recently found that higher-than-normal levels of NOG and SOST IgG immune complexes were detected in sera of AS patients, as well as in patients with both AS and IBD.Citation93 A recent GWAS has already revealed common risk loci between AS and IBD (such as ERAP1 and IL23R).Citation25 It is anticipated that performing GWAS using samples from AS patients with subclinical versus clinical gut inflammation would unravel novel risk loci specific for this AS subset.
In summary, recent GWAS findings provide invaluable clues on pathways key to the development of AS. The logical lines of investigation to follow should focus on the biology of the risk factors, especially in terms of how these risk factors would influence disease pathogenesis and clinical outcome.
Acknowledgments
This work was funded by the Arthritis Society of Canada, the Canadian Institutes of Health Research (CIHR), and the CIHR Institute of Musculoskeletal Health and Arthritis (IMHA).
Disclosure
The authors report no conflicts of interest in this work.
References
- PalBAnkylosing spondylitis, a seronegative spondyloarthritisPractitioner19872317857933321017
- Van der LindenSValkenburgHCatsAEvaluation of diagnostic criteria for ankylosing spondylitis. A proposal for modification of the New York criteriaArthritis Rheum1984273613686231933
- DakwarEReddyJValeFLUribeJSA review of the pathogenesis of ankylosing spondylitisNeurosurg Focus200824E218290740
- FeldtkellerEKhanMAvan der HeijdeDvan der LindenSBraunJAge at disease onset and diagnosis delay in HLA-B27 negative vs positive patients with ankylosing spondylitisRheumatol Int200323616612634937
- RudwaleitMvan der HeijdeDKhanMABraunJSieperJHow to diagnose axial spondyloarthritis earlyAnn Rheum Dis20046353554315082484
- SongIHPoddubnyyDARudwaleitMSieperJBenefits and risks of ankylosing spondylitis treatment with nonsteroidal anti-inflammatory drugsArthritis Rheum20085892993818383378
- WandersAvan der HeijdeDLandewéRNonsteroidal antiinflammatory drugs reduce radiographic progression in patients with ankylosing spondylitis: a randomized clinical trialArthritis Rheum2005521756176515934081
- PoddubnyyDRudwaleitMHaibelHEffect of nonsteroidal anti-inflammatory drugs on radiographic spinal progression in patients with axial spondyloarthritis: results from the German Spondyloarthritis Inception CohortAnn Rheum Dis2012711616162222459541
- Van der HeijdeDLandewéREinsteinSRadiographic progression of ankylosing spondylitis after up to two years of treatment with etanerceptArthritis Rheum2008581324133118438853
- HaroonNInmanRDLearchTJThe impact of tumor necrosis factor α inhibitors on radiographic progression in ankylosing spondylitisArthritis Rheum2013652645265423818109
- AvernsHLOxtobyJTaylorHGJonesPWDziedzicKDawesPTRadiological outcome in ankylosing spondylitis: use of the Stoke Ankylosing Spondylitis Spine Score (SASSS)Br J Rheumatol1996353733768624642
- WandersAJLandewéRBSpoorenbergAWhat is the most appropriate radiologic scoring method for ankylosing spondylitis? A comparison of the available methods based on the outcome measures in rheumatology clinical trials filterArthritis Rheum2004502622263215334477
- CreemersMCFranssenMJvan’t HofMAGribnauFWvan de PutteLBvan RielPLAssessment of outcome in ankylosing spondylitis: an extended radiographic scoring systemAnn Rheum Dis20056412712915051621
- BaraliakosXListingJRudwaleitMThe relationship between inflammation and new bone formation in patients with ankylosing spondylitisArthritis Res Ther200810R10418761747
- LoriesRJDougadosMInflammation and ankylosis: still an enigmatic relationship in spondyloarthritisAnn Rheum Dis20127131731822315213
- MaksymowychWPElewautDSchettGMotion for debate: the development of ankylosis in ankylosing spondylitis is largely dependent on inflammationArthritis Rheum2012641713171922354725
- BrownMALavalSHBrophySCalinARecurrence risk modeling of the genetic susceptibility to ankylosing spondylitisAnn Rheum Dis20005988388611053066
- BraunJBollowMRemlingerGPrevalence of spondylarthropathies in HLA-B27 positive and negative blood donorsArthritis Rheum19984158679433870
- BrownMAKennedyLGMacGregorAJSusceptibility to ankylosing spondylitis in twins: the role of genes, HLA and the environmentArthritis Rheum199740182318289336417
- Diaz-PeñaRLópez-VázquezALópez-LarreaCOld and new HLA associations with ankylosing spondylitisTissue Antigens20128020521322881057
- ReveilleJDGenetics of spondyloarthritis – beyond the MHCNat Rev Rheumatol2012829630422487796
- RobinsonPCBrownMAThe genetics of ankylosing spondylitis and axial spondyloarthritisRheum Dis Clin North Am20123853955323083754
- RobinsonPCBrownMAGenetics of ankylosing spondylitisMol Immunol20145721123916070
- JinGXDuanJZGuoWLLiLCuiSQWangHAssociation between IL-1RN gene polymorphisms and susceptibility to ankylosing spondylitis: a large Human Genome Epidemiology review and meta-analysisGenet Mol Res2013121720173023765978
- International Genetics of Ankylosing Spondylitis Consortium (IGAS)CortesAHadlerJIdentification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related lociNat Genet20134573073823749187
- Australo-Anglo-American Spondyloarthritis Consortium (TASC)ReveilleJDSimsAMGenome-wide association study of ankylosing spondylitis identifies non-MHC susceptibility lociNat Genet20104212312720062062
- LeeJCEspéliMAndersonCAHuman SNP links differential outcomes in inflammatory and infectious disease to a FOXO3-regulated pathwayCell2013155576924035192
- SmithJAColbertRAThe interleukin-23/interleukin-17 axis in spondyloarthritis pathogenesis: Th17 and beyondArthritis Rheum201466231241
- CoffreMRournierMRybczynskaMCombinatorial control of Th17 and Th1 cell functions by genetic variations in genes associated with the interleukin-23 signaling pathway in spondyloarthritisArthritis Rheum2013651510152123508476
- García-MedelNSanz-BravoAVan NguyenDFunctional interaction of the ankylosing spondylitis-associated endoplasmic reticulum aminopeptidase I polymorphism and HLA-B27 in vivoMol Cell Proteomics2012111416142922918227
- ChenLFischerRPengYCritical role of endoplasmic reticulum aminopeptidase I in determining the length and sequence of peptides bound and presented by HLA-B27Arthritis Rheum201466284294
- AndrésAMDennisMYKretzschmarWWBalancing selection maintains a form of ERAP2 that undergoes nonsense-mediated decay and affects antigen presentationPLoS Genet20106e100115720976248
- CaglianiRRivaSBiasinMGenetic diversity at endoplasmic reticulum aminopeptidases is maintained by balancing selection and is associated with natural resistance to HIV-1 infectionHum Mol Genet2010194705471420843824
- BirtleyJRSaridakisEStratikosEMavridisIMThe crystal structure of human endoplasmic reticulum aminopeptidase 2 reveals the atomic basis for distinct roles in antigen processingBiochemistry20125128629522106953
- TsuiFWHaroonNReveilleJAssociation of an ERAP1 ERAP2 haplotype with familial ankylosing spondylitisAnn Rheum Dis20106973373619433412
- ReevesEEdwardsCJElliottTJamesENaturally occurring ERAP1 haplotypes encode functionally distinct alleles with fine substrate specificityJ Immunol2013191354323733883
- LeesCW1BarrettJCParkesMSatsangiJNew IBD genetics: common pathways with other diseasesGut2011601739175321300624
- JostinsLRipkeSWeersmaRKHost-microbe interactions have shaped the genetic architecture of inflammatory bowel diseaseNature201249111912423128233
- VisscherPMBrownMAMcCarthyMIYangJFive years of GWAS discoveryAm J Hum Genet20129072422243964
- LinDYTangZZA general framework for detecting disease associations with rare variants in sequencing studiesAm J Hum Genet20118935436721885029
- BeaudoinMGoyettePBoucherGDeep resequencing of GWAS loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitisPLoS Genet20139e100372324068945
- WoltersFLRusselMGSijbrandijJDisease outcome of inflammatory bowel disease patients: general outline of a Europe-wide population-based 10-year clinical follow-up studyScand J Gastroenterol Suppl2006243465416782622
- DjouadiKNedeiecBTamouzaRInterleukin 1 gene cluster polymorphisms in multiplex families with spondylarthropathiesCytokine2001139810311145849
- MaksymowychWPRahmanPReeveJPGladmanDDPeddleLInmanRDAssociation of the IL1 gene cluster with susceptibility to ankylosing spondylitis: an analysis of three Canadian populationsArthritis Rheum20065497498516508980
- TimmsAECraneAMSimsAMThe interleukin 1 gene cluster contains a major susceptibility locus for ankylosing spondylitisAm J Hum Genet20047558759515309690
- Van der PaardtMCrusiusJBGarcía-GonzálezMAInterleukin-1 beta and interleukin-1 receptor antagonist gene polymorphisms in ankylosing spondylitisRheumatology (Oxford)2001401359136411752505
- SimsAMTimmsAEBruges-ArmasJProspective meta-analysis of interleukin 1 gene complex polymorphisms confirms associations with ankylosing spondylitisAnn Rheum Dis2008671305130918063673
- MonnetDKadiAIzacBAssociation between the IL-1 family gene cluster and spondyloarthritisAnn Rheum Dis20127188589022312160
- LinZBeiJXShenMA genome-wide association study in Han Chinese identifies new susceptibility loci for ankylosing spondylitisNat Genet201144737722138694
- DeuquetJLauschESuperti-FurgaAvan der GootFGThe dark sides of capillary morphogenesis gene 2EMBO J20123131322215446
- UranoTNarusawaKShirakiMSingle-nucleotide polymorphism in the hyaluronan and proteoglycan link protein 1 (HAPLN1) gene is associated with spinal osteophyte formation and disc degeneration in Japanese womenEur Spine J20112057257720953637
- TakaiAInomataHArakawaAYakuraRMatsuo-TakasakiMSasaiYAnterior neural development requires Del1, a matrix-associated protein that attenuates canonical Wnt signaling via the Ror2 pathwayDevelopment20101373293330220823067
- SuzukiJUmedaMSimsPJNagataSCalcium-dependent phospholipid scrambling by TMEM16FNature201046883483821107324
- DavidsonSIJiangLCortesABrief report: High-throughput sequencing of IL23R reveals a low-frequency, nonsynonymous single-nucleotide polymorphism that is associated with ankylosing spondylitis in a Han Chinese populationArthritis Rheum2013651747175223606107
- KadiACosstantinoFIzacBBrief report: The IL23R nonsynonymous polymorphism rs11209026 is associated with radiographic sacroiliitis in spondyloarthritisArthritis Rheum2013652655266023818276
- AppelHMaierRBleilJIn situ analysis of interleukin-23- and interleukin-12-positive cells in the spine of patients with ankylosing spondylitisArthritis Rheum2013651522152923508523
- SherlockJPJoyce-ShaikhBTurnerSPIL-23 induces spondyloarthropathy by acting on ROR-γ+CD3+CD4–CD8– entheseal resident T cellsNat Med2012181069107622772566
- HaroonNMaksymowychWPRahmanPTsuiFWO’SheaFDInmanRDRadiographic severity of ankylosing spondylitis is associated with polymorphism of the large multifunctional peptidase 2 gene in the Spondyloarthritis Research Consortium of Canada cohortArthritis Rheum2012641119112622034108
- MeyerJLCan biological calcification occur in the presence of pyrophosphate? Arch Biochem Biophys1984231186326671
- HoAMJohnsonMDKingsleyDMRole of the mouse ank gene in control of tissue calcification and arthritisScience200028926527010894769
- GodingJWGrobbenBSlegersHPhysiological and pathophysiological functions of the ecto-nucleotide pyrophosphatase/phosphodiesterase familyBiochim Biophys Acta2003163811912757929
- TerkeltaubRAInorganic pyrophosphate generation and disposition in pathophysiologyAm J Physiol Cell Physiol2001281C1C1111401820
- TsuiFWTsuiHWChengEYNovel genetic markers in the 5′-flanking region of ANKH are associated with ankylosing spondylitisArthritis Rheum20034879179712632434
- TsuiHWInmanRDPatersonADReveilleJDTsuiFWANKH variants associated with ankylosing spondylitis: gender differencesArthritis Res Ther20057R513R52515899038
- TsuiHWInmanRDReveilleJDTsuiFWAssociation of a TNAP haplotype with ankylosing spondylitisArthritis Rheum20075623424317195227
- Goseki-SoneMSogabeNFukushi-IrieMFunctional analysis of the single nucleotide polymorphism (787T>C) in the tissue-nonspecific alkaline phosphatase gene associated with BMDJ Bone Miner Res20052077378215824850
- SogabeNOdaKNakamuraHMolecular effects of the tissue-nonspecific alkaline phosphatase gene polymorphism (787T>C) associated with bone mineral densityBiomed Res20082921321918724009
- FuruichiTMaedaKChouCRAssociation of the MSX2 gene polymorphisms with ankylosing spondylitis in JapaneseJ Hum Genet20085341942418299954
- LiuZCuiYZhouXZhangXHanJAssociation of mineralization-related genes TNAP and ANKH polymorphisms with ankylosing spondylitis in the Chinese Han populationBiosci Trends20137899223612078
- TimmsAEZhangYBradburyLWordsworthBPBrownMAInvestigation of the role of ANKH in ankylosing spondylitisArthritis Rheum2003482898290214558096
- KimTHYoungBJBangSYGenetic variants in OPG and ANKH are associated with severe ankylosis in patients with AS in Korean cohortArthritis Rheum201365SupplS10 Abstract 1901
- AkramAInmanRDInfluenza infection of MHC-1 transgenic mice reveals that ERAP is necessary and sufficient for generation of the B27-specific immunodominant epitopesArthritis Rheum201365SupplS531
- AkramAInmanRDCo-expression of HLA-B7 and HLA-B27 alleles is associated with B7-restricted immunodominant responses following influenza infectionEur J Immunol2013433254326724113999
- BlanchardNShastriNCoping with loss of perfection in the MHC class I peptide repertoireCurr Opin Immunol200820828818243675
- KanasekiTShastriNEndoplasmic reticulum aminopeptidases associated with antigen processing regulates quality of processed peptides presented by MHC class I moleculesJ Immunol20081816275628218941218
- HaroonNEndoplasmic reticulum aminopeptidase 1 and interleukin-23 receptor in ankylosing spondylitisCurr Rheumatol Rep20101438338922782541
- HaroonMInmanRDEndoplasmic reticulum aminopeptidases: biology and pathogenic potentialNat Rev Rheumatol2010646146720531381
- EvansDMSpencerCCPointonJJInteraction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibilityNat Genet20114376176721743469
- AkramAInmanRDImmunodominance: a pivotal principle in host response to viral infectionsClin Immunol20121439911522391152
- HammerREMaikaSDRichardsonJATangJPTaurogJDSpontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human β2m: an animal model of HLA-B27-associated human disordersCell199063109911122257626
- TranTMDorrisMISaturntiraNAdditional human beta-microglobulin curbs HLA-B27 misfolding and promotes arthritis and spondylitis without colitis in male HLA-B27-transgenic ratsArthritis Rheum2006541317132716575857
- RedlichKGörtzBHayerSOverexpression of tumor necrosis factor causes bilateral sacroiliitisArthritis Rheum2004501001100515022345
- KontoyiannisDPasparakisMPizarroTTCominelliFKolliasGImpaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologiesImmunity19991038739810204494
- JacquesPLambrechtSVerheugenEProof of concept: enthesitis and new bone formation in spondyloarthritis are driven by mechanical strain and stromal cellsAnn Rheum Dis20147343744523921997
- GlantTTMikeczKArzoumanianAPooleARProteoglycan-induced arthritis in BALB/c mice. Clinical features and histopathologyArthritis Rheum1987302012123827960
- BárdosTSzabóZCzipriMA longitudinal study on an autoimmune murine model of ankylosing spondylitisAnn Rheum Dis20056498198715640265
- HaynesKRPettitARDuanRExcessive bone formation in a mouse model of ankylosing spondylitis is associated with decreases in Wnt pathway inhibitorsArthritis Res Ther201214R25323171658
- DaoussisDLiossissSCSolomouEEEvidence that Dkk-1 is dysfunctional in ankylosing spondylitisArthritis Rheum20106215015820039407
- KwonSRLimMJSuhCHDickkopf-1 level is lower in patients with ankylosing spondylitis than in healthy people and is not influenced by anti-tumor necrosis factor therapyRheumatol Int2012322523252721833531
- AppelHRuiz-HeilandGListingJAltered skeletal expression of sclerostin and its link to radiographic progression in ankylosing spondylitisArthritis Rheum2009603257326219877044
- Las HerasFPritzkerKPHSoAAberrant chondrocyte hypertrophy and activation of β-catenin signaling precede joint ankylosis in ank/ank miceJ Rheumatol20123958359322298904
- LoriesRJDereseILuytenFPModulation of bone morphogenetic protein signaling inhibits the onset and progression of ankylosing spondylitisJ Clin Invest20051151571157915902307
- TsuiFWTsuiHWLasHeras FPritzkerKPInmanRDSerum levels of novel noggin and sclerostin-immune complexes are elevated in ankylosing spondylitisAnn Rheum Dis Epub7262013
- HaibelHRudwaleitMListingJSieperJOpen label trial of anakinra in active ankylosing spondylitis over 24 weeksAnn Rheum Dis20056429629815208175
- DudlerJAubry-RozierVTocilizumab in axial spondyloarthropathies: about 18 casesAnn Rheum Dis201170Suppl 312821062853
- SieperJBraunJKayJSarilumab for the treatment of ankylosing spondylitis: results of a Phase II, randomized, double-blind, placebo-controlled study (ALIGN)Ann Rheum Dis Epub2182014
- PoddubnyyDHermannKGCallhoffJListingJSieperJUstekinumab for the treatment of patients with active ankylosing spondylitis: results of a 28-week, prospective, open-label, proof-of-concept study (TOPAS)Ann Rheum Dis Epub January 3, 2014
- BaetenDBaraliakosXBraunJAnti-interleukin-17A monoclonal antibody seculinumab in treatment of ankylosing spondylitis: a randomized, double-blind, placebo-controlled trialLancet20133821705171324035250
- StolwijkCvan TubergenACastillo-OrtizJDBoonenAPrevalence of extra-articular manifestations in patients with ankylosing spondylitis: a systematic review and meta-analysisAnn Rheum Dis Epub922013
- Van PraetLvan den BoschFEJacquesPMicroscopic gut inflammation in axial spondyloarthritis: a multiparametric predictive modelAnn Rheum Dis20137241441723139267
- BremanderAPeterssonIFBergmanSEnglundMPopulation-based estimates of common comorbidities and cardiovascular disease in ankylosing spondylitisArthritis Care Res (Hoboken)20116355055621452267