
Abstract
Bipolar disorder is a common, complex genetic disorder, but the mode of transmission remains to be discovered. Many researchers assume that common genomic variants carry some risk for manifesting the disease. The research community has celebrated the first genome-wide significant associations between common single nucleotide polymorphisms (SNPs) and bipolar disorder. Currently, attempts are under way to translate these findings into clinical practice, genetic counseling, and predictive testing. However, some experts remain cautious. After all, common variants explain only a very small percentage of the genetic risk, and functional consequences of the discovered SNPs are inconclusive. Furthermore, the associated SNPs are not disease specific, and the majority of individuals with a “risk” allele are healthy. On the other hand, population-based genome-wide studies in psychiatric disorders have rediscovered rare structural variants and mutations in genes, which were previously known to cause genetic syndromes and monogenic Mendelian disorders. In many Mendelian syndromes, psychiatric symptoms are prevalent. Although these conditions do not fit the classic description of any specific psychiatric disorder, they often show nonspecific psychiatric symptoms that cross diagnostic boundaries, including intellectual disability, behavioral abnormalities, mood disorders, anxiety disorders, attention deficit, impulse control deficit, and psychosis. Although testing for chromosomal disorders and monogenic Mendelian disorders is well established, testing for common variants is still controversial. The standard concept of genetic testing includes at least three broad criteria that need to be fulfilled before new genetic tests should be introduced: analytical validity, clinical validity, and clinical utility. These criteria are currently not fulfilled for common genomic variants in psychiatric disorders. Further work is clearly needed before genetic testing for common variants in psychiatric disorders should be established.
Introduction
Bipolar disorder is a severe and common mental disorder. It is present in approximately 5.7 million American adults, or 2.6 percent of the US population aged 18 years and older in any given year.Citation1 At the core of the disease are dramatic and unpredictable mood swings between mania and depression. The diagnosis is usually made based on a combination of clinical indicators from a list of diagnostic criteria.Citation2 Bipolar disorder has a characteristic disease course, but the individual symptoms of bipolar disorder are not specific, and they may vary considerably from person to person and over the disease course. In some individuals, symptoms of depression prevail; in others, the clinical presentation is dominated by elevated or irritable mood with excessive energy, hyperactivity, and even aggressiveness.Citation3 About half of the individuals diagnosed with bipolar disorder also suffer from distorted experiences of reality, known as hallucinations and delusions.Citation4 Because the symptoms of bipolar disorder are shared with many psychiatric disorders, the diagnostic boundaries are not clearly defined. Not uncommonly, patients change diagnoses over the course of a lifetime. The clinical presentation is highly variable; hence, bipolar disorder has also been conceptualized as a group of related mood disorders, referred to as bipolar spectrum disorders. In addition, anxiety disorders, abuse of illegal substances, alcohol dependence, and attention-deficit/hyperactivity disorder often co-occur with bipolar disorder.Citation5–Citation8 This phenomenon is not well understood. Although some experts believe that these conditions share common genetic risk factors with bipolar disorder, others have been more cautious. The disease onset of bipolar disorder is during adolescence and early adulthood, but the diagnosis is often delayed by many years.Citation9 A contributing factor is the complex clinical picture, with sometimes very subtle symptoms, at disease onset. This is particularly tragic, as about half of the individuals with bipolar disorder attempt suicide at least once in their lifetime, and many complete the attempt.Citation10,Citation11 Despite severe symptoms, treatment can be successful if the correct diagnosis is made and treatment is initiated early.Citation12 Consequently, enormous efforts have been made to identify genetic risk factors or biomarkers that would identify individuals at risk and could facilitate early diagnosis and treatment.
Bipolar as a common complex disorder
Bipolar disorder is a complex and multifactorial disorder. The heritability of bipolar disorder based on concordance rates for bipolar disorder in twin studies has been estimated to be between 60% and 80%.Citation13 Slightly lower estimates of genetic risk have been suggested based on family studies and large population cohorts.Citation14 Even though this evidence for genetic risk factors is convincing, most clinicians would agree that a positive family history of bipolar disorder is actually not very common in everyday clinical practice. In fact, it is quite rare to find families in which bipolar disorder affects multiple members over several generations, as would be expected for a monogenic Mendelian disorder. Many patients are isolated cases. In addition to genetic risk factors, nongenetic risk factors might contribute to the manifestation of bipolar disorder, as well, such as alcohol and drug dependence or physical and sexual abuse.Citation15 It has been well established that environmental and social risk factors play a significant role in schizophrenia.Citation16–Citation19 A similar picture might evolve in bipolar disorder, as well.Citation20
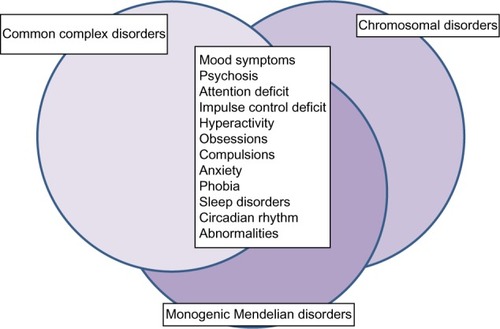
In addition to inherited genomic variants, recent evidence supports a significant role of de novo protein-damaging mutations in psychiatric disorders.Citation21 Experts in the field agree that susceptibility to bipolar disorder is most likely influenced by many genetic risk factors with small to moderate effect. Individual-specific and family-specific environmental factors might play a role, as well. The results of genome-wide association studies have supported this disease model. In very large population-based studies of thousands of individuals, a handful of replicated association signals have emerged at the level of genome-wide statistical significance.Citation22 In general, these variants have had very small effect sizes. Due to space constraints, I shall focus only on single nucleotide polymorphisms (SNPs) in the genes CACNA1C, ODZ4, and NCAN, which have emerged as promising candidate genes for bipolar disorder in genome-wide association studies. I admit that this selection could be disputed, but it is beyond the scope of this review to mention all candidate genes for bipolar disorder that have emerged so far. As no gene could be considered as an undeniable risk factor, the interested reader is referred to other recent reviews on this subject.Citation23 On the other hand, rare genetic syndromes and monogenic Mendelian disorders could present with psychiatric symptoms that closely resemble bipolar disorder or schizophrenia. These genetic disorders are often not properly diagnosed (). Therefore, it is important to alert clinicians to genetic syndromes that could resemble primary psychiatric disorders. In many cases, the genetic cause is already known, and genetic testing could assist in the differential diagnosis.
Figure 1 Symptoms of mood disorders are shared among common complex disorders, rare chromosomal disorders, and monogenic Mendelian disorders. Although many common complex disorders and rare Mendelian disorders share psychiatric symptoms, they do not always share the same genetic risk factors. Common complex disorders, such as bipolar disorder, are likely influenced by many genetic variants with small effect, in addition to environmental risk factors. Rare chromosomal disorders are characterized by large chromosomal deletions and duplications, which could potentially affect hundreds of genes. Rare monogenic Mendelian disorders are caused by characteristic mutations in a single gene. These differences in genetic risk factors have important consequences for risk prediction, genetic testing, and counseling.
Common variants as risk factors for bipolar disorder
CACNA1C (calcium channel, voltage-dependent, L type, alpha-1C subunit)
The SNP rs1006737 in the gene CACNA1C is the most replicated and most studied common genomic variant associated with bipolar disorder to date.Citation24–Citation26 The SNP is located in an intronic region and it occurs with significant allele frequency differences in all ethnic populations. The A allele, which is thought to be a risk factor for bipolar disorder, is present in 31% of European populations and in only 6% of Asian populations, but in almost 56% of individuals of African descent. As a result of these differences in allele frequencies, this SNP is vulnerable to confounding effects of ethnic admixture in genome-wide association studies. In fact, the association between the A allele of rs1006737 and bipolar disorder, first reported in a Caucasian population, could not be replicated with genome-wide significance in individuals of African descent or in some studies in Europe and Asia.Citation27–Citation29 Although the A allele seems to increase the risk of bipolar disorder in some population subgroups, most individuals who carry the “minor” allele are healthy. Therefore, the question remains how rs1006737 could influence disease processes in bipolar disorder. As the SNP is not located in the coding region of the gene CACNA1C, researchers have hypothesized that the variant could influence gene expression. In postmortem brain studies, scientists found evidence of reduced CACNA1C gene expression in individuals with the A allele, particularly in the cerebellum but not in other brain regions.Citation30 However, this finding could not be replicated by other researchers, and some groups have even found increased gene expression.Citation31 Further research is clearly needed to explain these discrepancies before definite conclusions can be reached.
Investigators have studied functional impairment in individuals with bipolar disorder who carried the A allele, particularly in the domains of executive function and emotional face processing.Citation32,Citation33 Although some researchers have found no effect, others reported significant reduction in cognitive function, but only in individuals who carried two A alleles, compared with patients with only one or no A allele.Citation34 Several groups could not replicate these results or even reported effects in the opposite direction.Citation35,Citation36 The results in patients with bipolar disorder have been inconclusive. Despite these discrepancies, studies generally agree that significant effects have not been observed in healthy individuals. Several factors could have contributed to the conflicting results, including small effect sizes and small sample sizes, in addition to differences in methodology, analysis, and interpretation across studies. Additional complexity has been added to the debate after researchers discovered a significant association signal with rs1006737 in a study in which patients with major affective disorder, bipolar disorder, autism spectrum disorder, attention-deficit/hyperactivity disorder, and schizophrenia had been combined. This finding indicated a lack of clinical specificity.Citation37 Recently, studies have claimed additional intronic SNPs in and around CACNA1C as being associated with psychiatric disorders, but replication and functional studies are still lacking.Citation38,Citation39
Psychiatric genetics is rooted in the assumption that common genomic variants tag functional variants in or around the gene closest to the common variant. Therefore, it is often implied that the closest gene might have some functional role in the disease processes of the associated disorder. CACNA1C is a large gene with over 11,541 known variants. Most of these variants are located in the introns and regions downstream of the gene. Missense mutations in exons are very rare. CACNA1C codes for the alpha-1 subunit of a voltage-dependent calcium channel. This subunit forms a transmembrane channel through which calcium ions enter the cell. Calcium channels are important neuronal regulators of muscle contraction in the heart, but they are also involved in skeletal muscle contraction. In the brain, CACNA1C could be involved in axon guidance and synaptic transmission. Mutations in the coding region of CACNA1C are the cause of Timothy syndrome (Mendelian Inheritance in Man [MIM] #601005), a rare Mendelian disorder, also known as long QT syndrome with syndactyly.Citation40–Citation42
Timothy syndrome is a multiorgan disease with lethal arrhythmias and congenital heart disease, developmental abnormalities of fingers and toes, immune deficiency, intermittent hypoglycemia, cognitive impairment, and behavioral abnormalities resembling autism. The syndrome is caused by one of two known heterozygote mutations (p.Gly402Ser and p.Gly406Arg) in exon 8 of the gene. This functional region encodes the transmembrane calcium channel. The mutated codon is a key regulator of calcium transport across the cell membrane. If one of these mutations is present, increased calcium influx into the cell occurs, resulting in increased excitability of the cell. Because of these severe consequences, children with Timothy syndrome often die at the age of 2 or 3 years. Survival into adulthood is very rare. However, a milder form of the disorder, Brugada syndrome type 3 (MIM #611875), has been described.Citation43 Brugada syndrome is characterized by cardiac arrhythmia with characteristic electrocardiogram changes. Sudden death could occur due to ventricular fibrillation, even though most individuals with this syndrome survive into adulthood. The disorder is caused by heterozygote mutations in different functional regions of CACNA1C (Gly490Arg in exon 10 and Ala39Val in exon 2). Current knowledge suggests that mutations in the coding region of the gene CACNA1C do not lead to bipolar disorder but rather to a monogenic Mendelian disorder with cognitive disability and autistic features.
In mice, homozygous mutations that completely eliminate the CACNA1C protein are lethal. Mutations that impaired the protein function resulted in reduced insulin secretion, glucose intolerance, poor motor coordination, increased anxiety, and hypoactivity in some studies.Citation44 In more than 69 papers, researchers have explored the relationship between rs1006737 and bipolar disorder, as well as other psychiatric disorders.Citation45 However, further work is clearly needed before the SNP rs1006737 could be established as an undisputed genetic risk factor for bipolar disorder.
ODZ4 (teneurin transmembrane protein 4)
The common variant rs12576775 in the intron of gene ODZ4 has been associated with bipolar disorder. However, significant association has also been established with autism spectrum disorders, attention-deficit/hyperactivity disorder, major depressive disorder, and schizophrenia in a combined analysis of these psychiatric disorders.Citation22,Citation37,Citation38,Citation46 The variant has a minor allele frequency of about 10% across all ethnic populations with the exception of European populations, in which the minor allele occurs in about 20% of individuals. Researchers have studied the effect of rs12576775 in individuals with bipolar disorder and also in healthy individuals using functional and structural brain imaging; however, the results of these studies have been inconclusive.Citation47–Citation50
ODZ4 is a large transmembrane protein and its structure resembles signal transduction molecules.Citation51 During brain development, ODZ4 appears to play a central role in the regulation of neuronal and synaptic connectivity.Citation52 At later stages of brain maturation, ODZ4 has been shown to orchestrate the development and differentiation of oligodendrocytes and the myelination of neuronal axons.Citation53 The gene has 14,410 known variants, but mutations in the coding region of the gene are not known to cause a Mendelian disorder. Knowledge about ODZ4 is still very limited, and more functional studies are clearly needed before clinical applications could be considered.
NCAN (neurocan)
NCAN is a large gene located on chromosome 19p13.11. The SNP rs1064395, which is found in an intronic region of the gene, has been associated with bipolar disorder in one study,Citation54 but so far not all studies have replicated this finding.Citation55 Even though association between variants in NCAN and schizophrenia has been tested, no genome-wide significant results have been found.Citation56 Rs1064395 affects only one of five alternative transcripts of the gene. Significant differences in allele frequencies of the associated allele have been observed across all ethnic populations. Although the overall frequency of the A allele (or disease-associated allele) is about 23%, it is the major allele in 51% of African populations. The disease-associated allele is present in only 12% of individuals in Asia and in 15% of individuals in Europe. Studies in postmortem brains of individuals with bipolar disorder or schizophrenia found increased cortical folding in some brain regions of patients with the A allele compared with healthy controls; however, no effect of the A allele was detected in the comparison of healthy individuals with the A allele and healthy individuals with the alternative allele.Citation57
The gene NCAN codes for a large secreted protein that is found predominantly in the extracellular space, the lumen of the Golgi apparatus, and the lysosomal cavities. Public databases list 741 known variants in the gene, but coding variants have not been associated with any Mendelian disorder. The protein is involved in the modulation of cell adhesion, cell migration, and axon guidance. However, knockout mice had normal brain structure and function. Mild deficits in synaptic plasticity in neurons of the hippocampus have been observed,Citation58 but only the complete knockout of four related proteoglycans resulted in severe structural and functional abnormalities.Citation59 These findings indicate that alternative mechanisms might exist to compensate for the loss of function of the NCAN protein.
Associations with genomic variants in NCAN do not appear to be disease specific. SNPs close to the gene have been associated with abnormalities in lipid metabolism at the genome-wide level of statistical significance in various ethnic populations;Citation60–Citation64 however, negative results have also been reported.Citation65
The question remains how to interpret statistically significant association signals with common genomic variants and how to translate the results into clinical practice. It remains to be seen to what degree statistically significant association signals indicate genetic risk factors and how they could explain disease processes leading to bipolar disorder. Based on current knowledge, any genetic testing for bipolar disorder involving common genetic variants lacks scientific support and is clearly premature.
Chromosomal disorders with symptoms of bipolar disorder
Most clinicians would agree that bipolar disorder is a complex and multifactorial disorder with a relatively low recurrence risk. Psychiatric symptoms, however, are also common in certain rare monogenic Mendelian disorders and chromosomal disorders, also known as duplication and deletion syndromes. Even though these diseases do not meet the classic description of bipolar disorder or schizophrenia, they could present with acute symptoms of mania, severe depression, or psychosis; therefore, the differential diagnosis could be challenging. Even though these conditions are individually very rare, together they present a nonignorable fraction of cases with psychiatric symptoms. The Online Mendelian Inheritance in Man (OMIM) database lists about 88 entries with mood symptoms, and about 64 of these conditions are chromosomal disorders.Citation66 Psychosis in Mendelian disorders is even more common than extreme mood symptoms. In fact, symptoms of hallucinations and delusions are described in about 138 entries in OMIM, and 93 of these are chromosomal disorders. Some of these disorders are rare dominant Mendelian conditions or structural abnormalities shared with a parent. Under these circumstances, the recurrence risk could be as high as 50%. Therefore, it is essential to consider these disorders in the differential diagnosis of bipolar disorder or schizophrenia so that the correct diagnosis can be established and the families counseled about the increased recurrence risk. Although it is beyond the scope of this review to cover this topic comprehensively, I will present four examples of chromosomal disorders and one example of a rare Mendelian disorder in which mood symptoms and psychosis are part of the clinical presentation ().
Table 1 Phenotype comparison of four rare chromosomal disorders and one monogenic Mendelian disorder reveals strong similarities and overlapping psychiatric symptomsTable Footnotea
Prader–Willi syndrome (MIM #176270) (chromosome 15q11 deletion syndrome)
Prader–Willi syndrome is a classic example of a chromosomal disorder with prominent mood symptoms and psychosis. This syndrome, also known as chromosome 15q11 deletion syndrome, is characterized by obesity, small hands and feet, characteristic facial features, and mild to moderate intellectual disability. Children with Prader–Willi syndrome develop severe mood disorder with frequent mood swings, irritability, and aggressive outbursts, as well as obsessive–compulsive behavior.Citation67 Attention deficit, autism, and language delay are also commonly observed.Citation68,Citation69 During early adulthood, hallucinations and delusions occur in almost 30% of cases while mood symptoms persist. Seizure disorders are not uncommon in Prader–Willi syndrome.Citation70 The syndrome is caused by an imbalance between maternal and paternal genetic material on chromosome 15q11. These imbalances could result from a small deletion on the paternal chromosome, which could vary considerably in size. The smallest deletion described in the literature removed a differentially methylated 5′ regulatory exon of the gene SNRPN (small nuclear ribonucleoprotein polypeptide N), which changed the methylation pattern and, consequently, the gene expression levels of hundreds of genes.Citation71 Less commonly, a duplication of maternal material is found. Even though most cases develop spontaneously, familial transmission has been observed, and transmitting mothers are asymptomatic.Citation72 The syndrome occurs with a prevalence of about one in 22,000 worldwide. The chromosomal changes in this syndrome are disease specific and highly penetrant. If the clinical presentation suggests Prader–Willi syndrome, genetic testing and counseling are recommended.
Chromosome 15q13.3 deletion syndrome (MIM #612001)
Adjacent to the Prader–Willi region, a large two megabase deletion on chromosome 15q13.3 has been described. Patients with this chromosomal abnormality have mild to moderate intellectual disability and mild dysmorphic features of the hands and face.Citation73,Citation74 Frequently, carriers of the deletion develop seizure disorders and autistic traits.Citation75–Citation77 The clinical presentation of individuals with the 15q13.3 deletion syndrome is highly variable, ranging from asymptomatic to severe intellectual impairment.Citation78 Behavioral abnormalities are common and include aggressiveness, impulse control problems, attention deficits, and hyperactivity.Citation79,Citation80 Individuals are sometimes misdiagnosed with bipolar disorder or obsessive–compulsive disorder. Anxiety disorders and phobias have also been described. Psychotic symptoms could lead to the diagnosis of schizophrenia in some cases, even though the syndromic gestalt of the disorder does not fit classical descriptions of schizophrenia.Citation81,Citation82 Increasing evidence suggests that reduced function of the gene CHRNA7 is responsible for the neuropsychiatric deficits in this deletion syndrome.Citation83,Citation84 Carriers of deletions involving the gene CHRNA7 have severe mental disability, seizure disorders, and low muscle tone.Citation85 On brain imaging studies, mild hypogenesis of the corpus callosum has been detected. The 15q13.3 microdeletion is a contiguous gene deletion inherited in an autosomal dominant manner. Approximately 25% of 15q13.3 microdeletions are de novo; approximately 75% are inherited.Citation86 Offspring of an individual with the 15q13.3 microdeletion have a 50% chance of inheriting the deletion. Although prenatal testing is technically feasible, it is not possible to reliably predict the phenotype based on the laboratory finding of a 15q13.3 microdeletion, because of reduced penetrance. Mutations on the nondeleted chromosome could have disease-modifying effects.
Chromosome 10q26 deletion syndrome (MIM #609625)
The chromosome 10q26 deletion syndrome is characterized by mild to moderate intellectual disability, short stature, small head, and characteristic facial features. Cardiac, renal, and genital abnormalities have been described in some individuals.Citation87 Rapid mood swings are common: eg, very affectionate behavior could unpredictably turn into aggressive and provocative actions.Citation88 Hyperactivity and attention deficits are also common.Citation89,Citation90 Poor speech and language development, as well as autistic traits, have been described. Cognitive impairment is highly variable and can range from mild learning disabilities to severe mental handicap and absent speech. Familial transmission has been described,Citation91 and in these instances the risk could be as high as 50%.
Velo–cardio–facial syndrome (MIM #602054) (chromosome 22q11 deletion syndrome)
Velo–cardio–facial syndrome is caused by a three megabase deletion on chromosome 22q11 spanning about 40 genes. The deletion has a population prevalence of about one in 2,000. The syndrome is a multiorgan, complex disorder. The spectrum of symptoms is wide-ranging, from near normal to severe impairment and even life-threatening manifestations.Citation92 Familial transmission and intrafamilial variability have been reported.Citation93 Psychiatric symptoms are common. Children and adolescents with velo–cardio–facial syndrome are at increased risk for depression, anxiety, and attention-deficit/hyperactivity disorders.Citation94,Citation95 Obsessive–compulsive behavior and autistic features are also not uncommon. About 30% of individuals with the deletion develop behavioral symptoms that could resemble bipolar disorder, or even hallucinations and delusions.Citation96,Citation97 Some individuals have been diagnosed with schizophrenia because of intellectual decline after the onset of psychosis.Citation98,Citation99
Monogenic disorders with symptoms of bipolar disorder
Point mutations and other genomic changes in a single gene could change the structure and function of the encoded protein. Sometimes, the loss of a single gene can result in severe complex disorders, also known as monogenic Mendelian disorders. Familial transmission of these disorders is very rare in severely affected individuals with early onset, but it is not uncommon in milder cases or disorders with onset in adulthood.
Smith–Magenis syndrome (MIM #182290)
Smith–Magenis syndrome is a genetic syndrome characterized by mild to moderate intellectual disability, self-injurious behaviors, obesity, skeletal abnormalities, and characteristic facial features.Citation100 Sleep disturbances are common due to abnormal circadian rhythms. The syndrome could be caused by a microdeletion on chromosome 17p11.2, but most of the symptoms have been traced back to the loss of function of a single gene, RAI1 (retinoic acid induced 1). Small deletions, frameshift mutations, premature stop codons, and missense mutations in RAI1 could result in all the major features of the syndrome.Citation101,Citation102 Smith–Magenis syndrome affects approximately one in 25,000 individuals.Citation103 Genetic testing for this disorder is well established, and genetic counseling is recommended.
Summary
In common complex disorders, genome-wide association studies have identified common variants that might indicate a small increase in genetic risk. Based on these results, the possibility of genetic testing has been discussed among clinicians, researchers, and patients alike.Citation104 Although genetic testing is well established for chromosomal disorders or monogenic Mendelian disorder, the issue is more ambiguous in common complex multigenic conditions, due to the nature of the identified genomic variants. In the current debate, it might be advisable to learn from established principles and practices. The National Institutes of Health/Department of Energy Task Force on Genetic Testing has established three criteria that should be fulfilled before a new genetic test can be introduced, particularly in the context of genetic screening.Citation105 These criteria are analytical validity, clinical validity, and clinical utility. Analytical validity refers to the accuracy and precision of a genetic test performed in a clinical laboratory. The second criterion, clinical validity, refers to a range of clinical performance measures, including clinical sensitivity, clinical specificity, and positive predictive value. The third requirement, clinical utility, refers to the usefulness of a test in clinical settings to improve the health of individuals.
Chromosomal disorders and monogenic Mendelian disorders with mood symptoms and psychosis are caused by rare structural genomic variants and protein-damaging mutations in single genes. Rare genetic mutations in these conditions are functional, disease specific, and highly penetrant. Even though these conditions are individually rare, they could still pose a nonignorable risk because the recurrence rate could be as high as 50% if the genetic risk factor is also present in one of the parents. As transmitting parents might be asymptomatic, genetic testing is recommended for every individual in whom mood symptoms are combined with neurological abnormalities or cognitive symptoms suggesting a Mendelian disorder or genetic syndrome.
Although genetic tests for Mendelian disorders and chromosomal disorders are well established, specific, and sensitive with high predictive value, the issue is more controversial for common complex disorders. After all, most individuals who carry the common “risk” allele are healthy and are not expected to develop the disease. In addition, the associated variants are usually not disease specific but have been associated with a wide range of disorders. Therefore, common variants are not suitable to predict the presence or absence of a clinical disease or the likelihood of developing a disease. In summary, the clinical utility of genetic tests for common complex disorders has not been clearly established, and the risk–benefit ratio remains unclear. Therefore, the risk of misinterpretation and misuse of genetic tests in common complex psychiatric disorders is high, especially if the tests are directly marketed to consumers. The potential for stigmatization, discrimination, anxiety, and burden on family relationships is real and should not be underestimated. As the field of psychiatric genetics advances, community interests, particularly regarding those who carry a “risk” allele – in some cases more than 50% of a specific ethnic population – should be assessed and risk–benefit ratios discussed. Clearly, more work is needed before genetic testing for common complex disorders can be established.
Acknowledgments
This work was supported by a grant from the National Institute of Mental Health R01MH085744-03 and a fellowship from the Brain and Behavior Research Foundation (NARSAD Young Investigator Grant). I am grateful to Jake Carpenter for editorial assistance.
Disclosure
The author reports no conflicts of interest in this work.
References
- National Institute of Mental HealthBipolar disorder among adults Available from: http://www.nimh.nih.gov/statistics/1bipolar_adult.shtmlAccessed January 10, 2014
- American Psychiatric Association [homepage on the internet] Available from: http://www.psychiatry.org/homeAccessed January 10, 2014
- MerikangasKRCuiLHeatonLIndependence of familial transmission of mania and depression: results of the NIMH family study of affective spectrum disordersMol Psychiatry Epub10152013
- VandeleurCLMerikangasKRStrippoliMPCastelaoEPreisigMSpecificity of psychosis, mania and major depression in a contemporary family studyMol Psychiatry Epub10152013
- CorryJGreenMRobertsGAnxiety, stress and perfectionism in bipolar disorderJ Affect Disord201315131016102424064398
- van Emmerik-van OortmerssenKvan de GlindGKoeterMWPsychiatric comorbidity in treatment seeking substance use disorder patients with and without ADHD; results of the IASP studyAddiction Epub1042013
- Di FlorioACraddockNvan den BreeMAlcohol misuse in bipolar disorder. A systematic review and meta-analysis of comorbidity ratesEur Psychiatry Epub9252013
- Di NicolaMSalaLRomoLAdult attention-deficit/hyperactivity disorder in major depressed and bipolar subjects: role of personality traits and clinical implicationsEur Arch Psychiatry Clin Neurosci Epub9282013
- LeboyerMKupferDJBipolar disorder: new perspectives in health care and preventionJ Clin Psychiatry201071121689169521190640
- FagioliniAKupferDJRucciPScottJANovickDMFrankESuicide attempts and ideation in patients with bipolar I disorderJ Clin Psychiatry200465450951415119913
- HauserMGallingBCorrellCUSuicidal ideation and suicide attempts in children and adolescents with bipolar disorder: a systematic review of prevalence and incidence rates, correlates, and targeted interventionsBipolar Disord201315550752323829436
- PostRMLeverichGSKupkaRWEarly-onset bipolar disorder and treatment delay are risk factors for poor outcome in adulthoodJ Clin Psychiatry201071786487220667291
- SmollerJWFinnCTFamily, twin, and adoption studies of bipolar disorderAm J Med Genet C Semin Med Genet2003123C1485814601036
- WrayNRGottesmanIIUsing summary data from the Danish national registers to estimate heritabilities for schizophrenia, bipolar disorder, and major depressive disorderFront Genet2012311822783273
- AasMEtainBBellivierFAdditive effects of childhood abuse and cannabis abuse on clinical expressions of bipolar disordersPsychol Med Epub9132013
- BrownASThe environment and susceptibility to schizophreniaProg Neurobiol2011931235820955757
- MeliGOttlBPaladiniACataldiLPrenatal and perinatal risk factors of schizophreniaJ Matern Fetal Neonatal Med201225122559256322646662
- CanettaSEBrownASPrenatal infection, maternal immune activation, and risk for schizophreniaTransl Neurosci20123432032723956839
- KhandakerGMZimbronJLewisGJonesPBPrenatal maternal infection, neurodevelopment and adult schizophrenia: a systematic review of population-based studiesPsychol Med201343223925722717193
- HaukvikUKMcNeilTLangeEHPre- and perinatal hypoxia associated with hippocampus/amygdala volume in bipolar disorderPsychol Med20132711123803260
- KennyEMCormicanPFurlongSExcess of rare novel loss-of-function variants in synaptic genes in schizophrenia and autism spectrum disordersMol Psychiatry Epub10152013
- CraddockNSklarPGenetics of bipolar disorderLancet201338198781654166223663951
- SzczepankiewiczAEvidence for single nucleotide polymorphisms and their association with bipolar disorderNeuropsychiatr Dis Treat201391573158224143106
- SklarPSmollerJWFanJWhole-genome association study of bipolar disorderMol Psychiatry200813655856918317468
- FerreiraMAO‘DonovanMCMengYAWellcome Trust Case Control Consortium. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorderNat Genet20084091056105818711365
- MoskvinaVCraddockNHolmansPGene-wide analyses of genome-wide association data sets: evidence for multiple common risk alleles for schizophrenia and bipolar disorder and for overlap in genetic riskMol Psychiatry200914325226019065143
- KloiberSCzamaraDKarbalaiNANK3 and CACNA1C: missing genetic link for bipolar disorder and major depressive disorder in two German case-control samplesJ Psychiatr Res201246897397922647524
- ZhangJCaiJZhangXDoes the bipolar disorder-associated CACNA1C gene confer susceptibility to schizophrenia in Han Chinese?J Mol Neurosci201351247447723900723
- ZhangXZhangCWuZAssociation of genetic variation in CACNA1C with bipolar disorder in Han ChineseJ Affect Disord2013150226126523680436
- GershonESGrennanKBusnelloJA rare mutation of CACNA1C in a patient with bipolar disorder, and decreased gene expression associated with a bipolar-associated common SNP of CACNA1C in brainMol Psychiatry Epub8272013
- BigosKLMattayVSCallicottJHGenetic variation in CACNA1C affects brain circuitries related to mental illnessArch Gen Psychiatry201067993994520819988
- WangFMcIntoshAMHeYGelernterJBlumbergHPThe association of genetic variation in CACNA1C with structure and function of a frontotemporal systemBipolar Disord2011137–869670022085483
- Soeiro-de-SouzaMGOtaduyMCDiasCZBioDSMachado-VieiraRMorenoRAThe impact of the CACNA1C risk allele on limbic structures and facial emotions recognition in bipolar disorder subjects and healthy controlsJ Affect Disord201214119410122464935
- Soeiro-de-SouzaMGBioDSDiasVVVietaEMachado-VieiraRMorenoRAThe CACNA1C risk allele selectively impacts on executive function in bipolar type I disorderActa Psychiatr Scand2013128536236923406546
- ZhangQShenQXuZThe effects of CACNA1C gene polymorphism on spatial working memory in both healthy controls and patients with schizophrenia or bipolar disorderNeuropsychopharmacology201237367768422012475
- PaulusFMBedenbenderJKrachSAssociation of rs1006737 in CACNA1C with alterations in prefrontal activation and fronto-hippocampal connectivityHum Brain Mapp Epub2132013
- Cross-Disorder Group of the Psychiatric Genomics ConsortiumSmollerJWCraddockNIdentification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysisLancet201338198751371137923453885
- GreenEKHamshereMFortyLReplication of bipolar disorder susceptibility alleles and identification of two novel genome-wide significant associations in a new bipolar disorder case-control sampleMol Psychiatry201318121302130723070075
- HamshereMLWaltersJTSmithRGenome-wide significant associations in schizophrenia to ITIH3/4, CACNA1C and SDCCAG8, and extensive replication of associations reported by the Schizophrenia PGCMol Psychiatry201318670871222614287
- SplawskiITimothyKWSharpeLMCa(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autismCell20041191193115454078
- HsiaoPYTienHCLoCPJuangJMWangYHSungRJGene mutations in cardiac arrhythmias: a review of recent evidence in ion channelopathiesAppl Clin Genet2013611323837003
- BoczekNJBestJMTesterDJExome sequencing and systems biology converge to identify novel mutations in the L-type calcium channel, CACNA1C, linked to autosomal dominant long QT syndromeCirc Cardiovasc Genet20136327928923677916
- BrugadaJBrugadaPFurther characterization of the syndrome of right bundle branch block, ST segment elevation, and sudden cardiac deathJ Cardiovasc Electrophysiol1997833253319083883
- DaoDTMahonPBCaiXMood disorder susceptibility gene CACNA1C modifies mood-related behaviors in mice and interacts with sex to influence behavior in mice and diagnosis in humansBiol Psychiatry201068980181020723887
- PubMed [homepage on the internet] Available from: http://www.ncbi.nlm.nih.gov/pubmed/Accessed January 10, 2014
- Psychiatric GWAS Consortium Bipolar Disorder Working GroupLarge-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4Nat Genet2011431097798321926972
- TesliMEgelandRSønderbyIENo evidence for association between bipolar disorder risk gene variants and brain structural phenotypesJ Affect Disord2013151129129723820096
- HeinrichALourdusamyATzschoppeJThe risk variant in ODZ4 for bipolar disorder impacts on amygdala activation during reward processingBipolar Disord Epub4242013
- TesliMSkatunKCOusdalOTCACNA1C risk variant and amygdala activity in bipolar disorder, schizophrenia and healthy controlsPLoS One201382e5697023437284
- KrugAWittSHBackesHA genome-wide supported variant in CACNA1C influences hippocampal activation during episodic memory encoding and retrievalEur Arch Psychiatry Clin Neurosci Epub7172013
- Ben-ZurTFeigeEMotroBWidesRThe mammalian Odz gene family: homologs of a Drosophila pair-rule gene with expression implying distinct yet overlapping developmental rolesDev Biol2000217110712010625539
- ZhouXHBrandauOFengKThe murine Ten-m/Odz genes show distinct but overlapping expression patterns during development and in adult brainGene Expr Patterns20033439740512915301
- SuzukiNFukushiMKosakiKTeneurin-4 is a novel regulator of oligodendrocyte differentiation and myelination of small-diameter axons in the CNSJ Neurosci20123234115861159922915103
- CichonSMühleisenTWDegenhardtFAGenome-wide association study identifies genetic variation in neurocan as a susceptibility factor for bipolar disorderAm J Hum Genet201188337238121353194
- OručLKapur-PojskićLRamićJPojskićNBajrovićKAssessment of relatedness between neurocan gene as bipolar disorder susceptibility locus and schizophreniaBosn J Basic Med Sci201212424524823198940
- MühleisenTWMattheisenMStrohmaierJAssociation between schizophrenia and common variation in neurocan (NCAN), a genetic risk factor for bipolar disorderSchizophr Res20121381697322497794
- SchultzCCMühleisenTWNenadicICommon variation in NCAN, a risk factor for bipolar disorder and schizophrenia, influences local cortical folding in schizophreniaPsychol Med Epub6242013
- ZhouXHBrakebuschCMatthiesHNeurocan is dispensable for brain developmentMol Cell Biol200121175970597811486035
- RauchUZhouXHRoosGExtracellular matrix alterations in brains lacking four of its componentsBiochem Biophys Res Commun2005328260861715694392
- LuYFeskensEJBoerJMExploring genetic determinants of plasma total cholesterol levels and their predictive value in a longitudinal studyAtherosclerosis2010213120020520832063
- YanTTYinRXLiQSex-specific association of rs16996148 SNP in the NCAN/CILP2/PBX4 and serum lipid levels in the Mulao and Han populationsLipids Health Dis20111024822208664
- ZhouLDingHZhangXGenetic variants at newly identified lipid loci are associated with coronary heart disease in a Chinese Han populationPLoS One2011611e2748122110658
- SpeliotesEKYerges-ArmstrongLMWuJGenome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traitsPLoS Genet201173e100132421423719
- GordenAYangRYerges-ArmstrongLMGenetic variation at NCAN locus is associated with inflammation and fibrosis in nonalcoholic fatty liver disease in morbid obesityHum Hered2013751344323594525
- NakayamaKBayasgalanTYamanakaKLarge scale replication analysis of loci associated with lipid concentrations in a Japanese populationJ Med Genet200946637037419487539
- Online Mendelian Inheritance in Man (OMIM) [homepage on the internet] Available from: http://www.ncbi.nlm.nih.gov/omimAccessed January 10, 2014
- BoerHHollandAWhittingtonJButlerJWebbTClarkeDPsychotic illness in people with Prader Willi syndrome due to chromosome 15 maternal uniparental disomyLancet2002359930113513611809260
- BurnsideRDPasionRMikhailFMMicrodeletion/microduplication of proximal 15q11.2 between BP1 and BP2: a susceptibility region for neurological dysfunction including developmental and language delayHum Genet2011130451752821359847
- SkokauskasNSweenyEMeehanJGallagherLMental health problems in children with Prader-Willi syndromeJ Can Acad Child Adolesc Psychiatry201221319420322876265
- FanZGreenwoodRFisherAPendyalSPowellCMCharacteristics and frequency of seizure disorder in 56 patients with Prader-Willi syndromeAm J Med Genet A2009149A71581158419533781
- SutcliffeJSNakaoMChristianSDeletions of a differentially methylated CpG island at the SNRPN gene define a putative imprinting control regionNat Genet19948152587987392
- HallBDSmithDWPrader-Willi syndrome. A resumé of 32 cases including an instance of affected first cousins, one of whom is of normal stature and intelligenceJ Pediatr19728122862935042487
- AutioSPihkoHTengströmCClinical features in a de novo interstitial deletion 15q13 to q15Clin Genet19883452932983228997
- NinomiyaSYokoyamaYKawakamiMUneTMaruyamaHMorishimaTUnique maternal deletion of 15q in a patient with some symptoms of Prader-Willi syndromePediatr Int200547554154516190961
- SharpAJMeffordHCLiKA recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizuresNat Genet200840332232818278044
- HelbigIMeffordHCSharpAJ15q13.3 microdeletions increase risk of idiopathic generalized epilepsyNat Genet200941216016219136953
- de KovelCGTrucksHHelbigIRecurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsiesBrain2010133Pt 1233219843651
- van BonBWMeffordHCMentenBFurther delineation of the 15q13 microdeletion and duplication syndromes: a clinical spectrum varying from non-pathogenic to a severe outcomeJ Med Genet200946851152319372089
- MillerDTShenYWeissLAMicrodeletion/duplication at 15q13.2q13.3 among individuals with features of autism and other neuropsychiatric disordersJ Med Genet200946424224818805830
- PagnamentaATWingKSadighi AkhaEA 15q13.3 microdeletion segregating with autismEur J Hum Genet200917568769219050728
- Ben-ShacharSLanpherBGermanJRMicrodeletion 15q13.3: a locus with incomplete penetrance for autism, mental retardation, and psychiatric disordersJ Med Genet200946638238819289393
- SahooTTheisenARosenfeldJACopy number variants of schizophrenia susceptibility loci are associated with a spectrum of speech and developmental delays and behavior problemsGenet Med2011131086888021792059
- ShinawiMSchaafCPBhattSSA small recurrent deletion within 15q13.3 is associated with a range of neurodevelopmental phenotypesNat Genet200941121269127119898479
- Hoppman-ChaneyNWainKSegerPRSuperneauDWHodgeJCIdentification of single gene deletions at 15q13.3: further evidence that CHRNA7 causes the 15q13.3 microdeletion syndrome phenotypeClin Genet201383434535122775350
- EndrisVHackmannKNeuhannTMHomozygous loss of CHRNA7 on chromosome 15q13.3 causes severe encephalopathy with seizures and hypotoniaAm J Med Genet A2010152A112908291120979196
- GeneReviews™ [internet]PagonRoberta AAdamMargaret PBirdThomas DDolanCynthia RFongChin-ToSmithRichard JHStephensand KarenSeattle (WA)University of Washington, Seattle1993–2014 Available from http://www.ncbi.nlm.nih.gov/books/Accessed October 18, 2013
- MehtaLDuckettDPYoungIDBehaviour disorder in monosomy 10qterJ Med Genet19872431851863573005
- CourtensWWuytsWRoomsLPeraSBWautersJA subterminal deletion of the long arm of chromosome 10: a clinical report and reviewAm J Med Genet A2006140440240916419133
- MillerNDNanceMAWohlerESMolecular (SNP) analyses of overlapping hemizygous deletions of 10q25.3 to 10qter in four patients: evidence for HMX2 and HMX3 as candidate genes in hearing and vestibular functionAm J Med Genet A2009149A466968019253379
- YatsenkoSAKruerMCBaderPIIdentification of critical regions for clinical features of distal 10q deletion syndromeClin Genet2009761546219558528
- IrvingMHansonHTurnpennyPDeletion of the distal long arm of chromosome 10; is there a characteristic phenotype? A report of 15 de novo and familial casesAm J Med Genet A2003123A215316314598339
- BassettASChowEWHustedJClinical features of 78 adults with 22q11 deletion syndromeAm J Med Genet A2005138430731316208694
- McLeanSDSaalHMSpinnerNBEmanuelBSDriscollDAVelo-cardio-facial syndrome. Intrafamilial variability of the phenotypeAm J Dis Child199314711121212168237917
- JolinEMWellerRAWellerEBOccurrence of affective disorders compared to other psychiatric disorders in children and adolescents with 22q11.2 deletion syndromeJ Affect Disord2012136322222821215459
- AnejaAFremontWPAntshelKMManic symptoms and behavioral dysregulation in youth with velocardiofacial syndrome (22q11.2 deletion syndrome)J Child Adolesc Psychopharmacol200717110511417343558
- ShprintzenRJGoldbergRBYoungDWolfordLThe velo-cardio-facial syndrome: a clinical and genetic analysisPediatrics19816721671727243439
- ShprintzenRJVelo-cardio-facial syndrome: 30 Years of studyDev Disabil Res Rev200814131018636631
- EversLJVermaakMPEngelenJJCurfsLMThe velocardiofacial syndrome in older age: dementia and autistic featuresGenet Couns200617333334017100202
- ButcherNJChowEWCostainGKarasDHoABassettASFunctional outcomes of adults with 22q11.2 deletion syndromeGenet Med2012141083684322744446
- De LeersnyderHSmith-Magenis syndromeHandb Clin Neurol2013111295296623622179
- GirirajanSElsasLJ2ndDevriendtKElseaSHRAI1 variations in Smith-Magenis syndrome patients without 17p11.2 deletionsJ Med Genet2005421182082815788730
- ElseaSHWilliamsSRSmith-Magenis syndrome: haploinsufficiency of RAI1 results in altered gene regulation in neurological and metabolic pathwaysExpert Rev Mol Med201113e1421545756
- JuyalRCFigueraLEHaugeXMolecular analyses of 17p11.2 deletion in 62 Smith-Magenis syndrome patientsAm J Med Genet1996589981007
- GershonESAlliey-RodriguezNNew ethical issues for genetic counseling in common mental disordersAm J Psychiatry2013170996897623897273
- WilfondBSThomsonEJModels of public health genetic policy developmentKhouryMJBurkeWThomsonEJGenetics and Public Health in the 21th CenturyOxford, UKOxford University Press20006181