Abstract
We report on a six years old boy with several features of Greig cephalopolysyndactyly syndrome (GCPS) including craniofacial dysmorphism, hypertelorism, heart defect, preaxial hexadactyly of toes, partial agenesis of corpus callosum, and severe developmental delay. Greig cephalopolysyndactyly (GCPS) can be caused by GLI3 deletions. In patients with large deletions which include additional genes, it is termed Greig cephalopolysyndactyly-contiguous gene syndrome (GCPS-CGS). It is generally believed that the deletion size correlates with disease severity. Nearly all cases appear to be a result of GLI3 de novo deletions. Chromosome analysis of our patient revealed a large deletion in chromosome 7(p13–p14). Unlike most previously described cases, we found that this deletion resulted from a paternal balanced insertional translocation of 7p13–14 into the long arm of chromosome 5.
Introduction
Greig cephalopolysyndactyly (GCPS; OMIM # 175700) has been found to result from mutations, duplications or deletions of GLI3 (CitationVortkamp et al 1991; CitationJohnston et al 2005). When the deletion is large and includes other genes, it is termed Greig cephalopolysyndactyly-contiguous gene syndrome (GCPS-CGS). In patients with GCPS-CGS, deletion size seems to correlate with the level of disability, ie, smaller deletions are associated with a lesser degree of developmental impairment both in motor milestones and language. Characteristic clinical features in GCPS-CGS include craniofacial malformations such as macrocephaly, frontal bossing, hypertelorism, broad base of the nose as well as post- or preaxial polydactyly of the hands, broad hallux, and syndactyly of the hands and feet. However, it should be noted that the clinical manifestations are variable.
Severe cases of Greig syndrome show significant clinical overlap with acrocallosal syndrome (ACLS). The overlapping features between GCPS and ACLS are likely to result from a deletion of similar genes, or from genes that function in a common pathway close to the GLI3 locus (CitationJohnston et al 2003). For distinction of ACLS from GCPS-CGS, clinical findings of ACLS such as mental retardation, agenesis of the corpus callosum, cerebellar hypoplasia as well as intracranial cysts are widely used (CitationKoenig et al 2002). With the exception of complete agenesis of the corpus callosum, however, these criteria may be unreliable.
Here we report a new case of an interstitial 7p13–14 deletion in a boy with GCPS-CGS. The father presented with the same deleted chromosome 7 along with an insertion of the 7p13–14 segment into the long arm of chromosome 5.
Case report
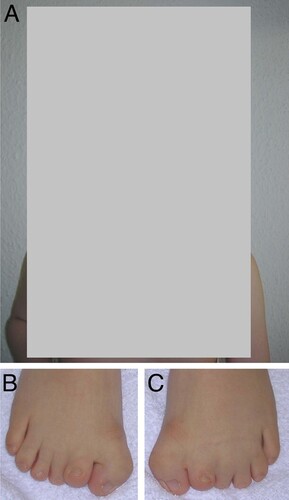
The proband, now six years old, was the first child born to healthy, nonconsanguinous parents. Sonography during pregnancy revealed growth retardation, cerebral malformation, cloverleaf skull, heart defect, omphalocele, and a single umbilical artery. He was delivered at 37 weeks of gestation by cesarean section because of gestosis. The birth weight was 2,200 g (3rd centile), length was 45 cm (<3rd centile), and the head circumference (HD) was 33 cm (3rd centile). At birth, laryngeal hypoplasia and cricoid cartilage stenosis were observed requiring tracheostomy. The feet displayed a bilateral duplication of the big toes as well as cutaneous syndactyly between the 2nd and 4th toe. Other abnormalities noted at birth included exomphalos, ventricular septal defect and penile hypospadias. At the age of six years, facial dysmorphism was evident (). Microcephaly (HD: 50.5 cm) with bitemporal bossing, unusual facial appearance with hypertelorism, broad nasal tip, flat nasal bridge, anteverted nares, long philtrum, high-arched palate and low-set, posteriorly rotated ears were noted. His length was 109 cm (<3rd centile) and his weight was 20 kg (25th centile). Well healed umbilical and inguinal hernia scars were noted. Cryptorchism had surgically been corrected on the left side. On the right side an orchidectomy had been performed. In addition, we found brachydactyly, broad thumbs, widely spaced nipples and scoliosis. Ocular abnormalities included nystagmus and myopia. Development was delayed. He sat unsupported at 3¾ years, and at the age of 6 years he was able to walk only with assistance. Moreover, learning disabilities with poor speech articulation, and conduction deafness were apparent. On the Münchener Developmental Scale at a chronological age of 5 years and 7 months his mental age was 15 months with delays in all subscales. A MRI scan of the brain showed partial agenesis of the corpus callosum. The family history is negative for any hand or foot anomalies as well as for mental retardation.
Figure 1 A) Facial view of the patient with GCPS-CGS at the age of 6 years. Note, broad nasal root and skull abnormality. B) and C) right and left foot with polydactyly and syndactyly.
Cytogenetic and molecular analysis
Chromosome preparations were made from PHA-stimulated peripheral blood lymphocytes and analyzed by standard GTG-banding procedures at a banding level of 550 (ISCN). FISH studies were performed using YAC clones selected from the MCN Reference Center at the Max Planck Institute for Molecular Genetics in Berlin (Supplemental ). DNA probes were labelled directly by NICK translation with SpectrumOrange-dUTP or SpectrumGreen-dUTP (ABBOTT, Illinois, USA). Labelled YAC clones were dissolved in hybridisation mixture (50% formamide, 2 × SSC, 10% dextran sulfate). After hybridization, metaphases were counterstained with DAPI and viewed with selective filters. Images were captured with the Cytovision digital imaging system (Applied Imaging, Sunderland, UK).
Table 1 Genotype analysis in the GCPS-CGS interval
Microsatellite markers from chromosome 7p were retrieved from the National Center for Biotechnology Information database (see www.ncbi.nlm.nih.gov). Cy5-labeled primers for polymerase chain reaction (PCR) were commercially synthesized (MWG-Biotech, Ebersberg, Germany) and standard PCR analysis was performed. PCR products were resolved on an ALFexpress sequencer (Amersham Biosciences: see www.amershambiosciences. com) and analyzed by the Fragment Manager Program of DNAsis software (MiraiBio, Inc., Alameda, CA, USA).
Results
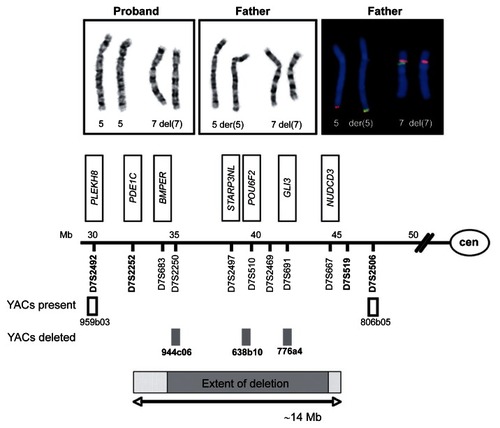
Conventional cytogenetic analysis revealed an aberrant male karyotype in the proband. For further elucidation, conventional chromosome analysis of the parents as well as FISH-analyses with YAC probes from 7p were carried out. The proband was shown to have a deletion in chromosome 7, comprising bands 7p13 and 7p14. The father presented with an identical 7p deletion, but in addition a derivative chromosome 5 was found with the 7p13–14 segment inserted into the long arm of chromosome 5. Interpretation of the proband’s karyotype was 46,XY,der(7)ins(5; 7) (q35; p13p14)pat. The results of the STR-analysis are given in and . The distal breakpoint of the deletion was determined between markers D7S2252 and D7S2250, localized at 7p14.3. The proximal breakpoint of the deletion is situated between D7S667 and D7S519, localised at 7p13. From the mapping coordinates retrieved from the NCBI STS map we estimated the deletion to have an extent of approximately 9.2 to 14.4 Mbp (7p13–14.3). It comprises more than 50 genes including the recently identified commonly deleted genes INHBA, GLI3, PGAM, and GCK. Whether the BMPER gene is also deleted in our patient could not be elucidated because markers D7S683 and D7S460 were not informative in this family.
Figure 2 A and B G-banded chromosomes 5 (left pair) and 7 (right pair) of the proband A) and his father B). In the proband, both chromosomes 5 show an inconspicious banding pattern, whereas one chromosome 7 (right homologue) shows an interstitial deletion of the short arm. In the father, a derivative chromosome 5 (right homologue) with an insertional translocation to the long arm was detected in addition to a deleted chromosome 7 (right homologue). C) examples of FISH experiments on chromosomes 5 (left pair) and 7 (right pair) of the proband’s father. The insertional translocation is highlighted with YAC 944c06 from 7p14.2. The subtelomeric 5q probe in red is distal to YAC 944c06 in green on the derivative chromosome 5 (right homologue). On the derivative chromosome 7, YAC 944c06 is missing, but YAC 959b03 (in red) is present at 7p14.3. D) schematic representation of breakpoints of chromosomal region 7p13 to 7p14. Microsatellite markers and YACs used for FISH characterization of the deletion are depicted along with a selection of genes located within the deleted region (boxed). Microsatellite markers present are depicted in bold. The minimal extent of the deletion is set in grey.
Discussion
There is considerable phenotypic overlap between ACLS and severe forms of GCPS. Both syndromes have similar patterns of polydactyly of hands and feet as well as macrocephaly. Features observed in our patient which usually distinguish ACLS from GCPS are agenesis of the corpus callosum, severe mental retardation, heart defects as well as umbilical and inguinal hernias. However, corpus callosum agenesis and severe mental retardation were also occasionally reported in GCPS, emphasizing the phenotypic and genotypic heterogeneity of both syndromes.
CitationJohnston and colleagues (2003) suggested that for some GCPS patients a phenotype difficult to distinguish from ACLS can be caused by deletions in 7p13, and in some cases involves the deletion of genes flanking GLI3. The identification of all disease-related genes within the deletions is not only invaluable for research on gene function, but is also essential for proper care, prevention and information for the patient’s family.
Craniosynostosis is an occasional finding in GCPS-CGS. Our patient has pronounced skull abnormalities, however, craniosynostosis is uncertain. In addition, macrocephaly is a frequent finding in GCPS patients with smaller deletions (CitationJohnston et al 2005). It should be noted that our patient despite the presence of skull abnormalities had a microcephaly. Previously, microcephaly has been reported in a single boy with a telomere-telomere fusion of the short arms of chromosome 7 and 22 that was accompanied by a large interstitial deletion in 7p (7p11.2–p15.1) (CitationZneimer et al 2000). Like in this patient, the deletion interval detected in our patient extends distally beyond most GCPS-CGS deletions. This may suggest haploinsuffi ciency of a gene or genes located in 7p14.2 to result in microcephaly.
In GCPS-CGS patients, no common breakpoints or deletion hot spots have been found (CitationJohnston et al 2007). CitationSchwarzbraun and colleagues (2006) identified deletion sizes from 4.5 to 15 Mb. Interestingly, all deletions examined were of paternal origin. Cases of GCPS reported so far were caused by de novo interstitial 7p deletions (CitationSchwarzbraun et al 2006). Our patient, however, inherited the deleted chromosome 7 from the father, who is the carrier of a balanced insertional translocation within chromosome 5q. The insertional breakpoint of chromosome 7p material into chromosome 5 is located distal to the marker WI-5924/WI-7554 (YAC 962f06). Thus, the father has a 50% risk for unbalanced offspring, as not only deletions but also duplications of 7p material cause clinical phenotypes (CitationCai et al 1999).
A commonly deleted segment of about 2.8 Mb including the genes INHBA, GLI3, PGAM2, and GCK apparently contributes to the GCPS-CGS phenotype in the majority of GCPS patients. Deletion of the POU6F2 gene may further predispose patients to Wilms tumor, since germline mutations in POU6F2 located in 7p14.1 have recently been identified in Wilms tumor patients (CitationPerotti et al 2004). The impact of haploinsufficiency of most other genes contained in the deletion interval of our patient, is largely unknown. For the development of a more precise genotype-phenotype correlation in GCPS-CGS, it is highly desirable to identify all disease-related genes within the deletion intervals.
Disclosure
The authors report no conflicts of interest in this work.
References
- CaiTYuPTagleDA1999Duplication of 7p21.2 – >pter due to maternal 7p; 21q translocation: Implications for critical segment assignment in the 7p duplication syndromeAm J Med Genet863051110494083
- JohnstonJJOlivos-GlanderITurnerJ2003Clinical and molecular delineation of the greig cephalopolysyndactyly contiguous gene deletion syndrome and its distinction from acrocallosal syndromeAm J Med Genet A12323642
- JohnstonJJOlivos-GlanderIKilloranC2005Molecular and clinical analyses of greig cephalopolysyndactyly and pallister-hall syndromes: Robust phenotype prediction from the type and position of gli3 mutationsAm J Hum Genet766092215739154
- JohnstonJJWalkerRLDavisS2007Zoom-in comparative genomic hybridisation arrays for the characterisation of variable breakpoint contiguous gene syndromesJ Med Genet44e5917098889
- KoenigRBachAWoelkiU2002Spectrum of the acrocallosal syndromeAm J Med Genet10871111857542
- PerottiDDe VecchiGTestiMA2004Germline mutations of the pou6f2 gene in wilms tumors with loss of heterozygosity on chromosome 7p14Hum Mutat24400715459955
- SchwarzbraunTWindpassingerCOfnerL2006Genomic analysis of five chromosome 7p deletion patients with greig cephalopolysyndactyly syndrome (gcps)Eur J Med Genet493384516829355
- VortkampAGesslerMGrzeschikKH1991Gli3 zinc-finger gene interrupted by translocations in greig syndrome familiesNature352539401650914
- ZneimerSMCotterPDStewartSD2000Telomere-telomere (end to end) fusion of chromosomes 7 and 22 with an interstitial deletion of chromosome 7p11.2 – >p15.1: Phenotypic consequences and possible mechanismsClin Genet581293311005146