Abstract
Background
Dubowitz syndrome is a rare, autosomal recessive disorder characterized by intrauterine and postnatal growth retardation, severe microcephaly, psychomotor retardation, hyperactivity, eczema, and characteristic dysmorphic facial features. Although many cases have been reported, the cause of this disease is still unknown.
Case
We present here the case of a Lebanese girl with Dubowitz syndrome in whom an unpleasant urine odor was persistently reported since birth.
Conclusion
Although Dubowitz syndrome has been largely described in the medical literature, this is the first time that a peculiar urine odor was reported. This case report adds a new and unusual feature to the numerous findings related to this rare polymorphous syndrome.
Introduction
Dubowitz syndrome is a condition characterized by multiple congenital anomalies.Citation1 It is described as a rare autosomal recessive disorder with a unique set of clinical features, including microcephaly, short stature, broad nasal bridge, sloping forehead and micrognathia, facial asymmetry, blepharophimosis, sparse hair and eyebrows, low-set ears, growth and mental retardation, and susceptibility to tumor formation. Children with this syndrome have similar behavioral characteristics. Hyperactivity, minimal attention, aggressiveness, shyness, dislike of crowds, food refusal, and bedwetting have all been reported. In addition, Dubowitz syndrome is known to be associated with other medical problems that increase the risk for a shorter life span. Although Dubowitz syndrome has been well described in the literature since it was first identified in 1965, the exact cause of this disorder is still unknown and diagnosis is based mainly on clinical features.Citation2–Citation5
Here, we report the case of a girl having typical features of Dubowitz syndrome (facial dysmorphism, eczema, microcephaly, and developmental and growth delay), with an unexplained odorous urine that has never been reported to be associated with the syndrome.
Case presentation
A 5-year-old girl was presented to our institution because of failure to thrive. She was the fifth child of unrelated parents originating from the same village in the south of Lebanon. She was born vaginally at term to a 33-year-old mother; her birth weight was 2,200 grams. The course of pregnancy was normal and the mother denied any medication use, smoking, or alcohol intake. The girl’s perinatal history was significant for neonatal jaundice requiring phototherapy. The family history was unremarkable except for mental retardation in a maternal uncle and a congenital heart defect in an older brother.
In early infancy, the girl had feeding difficulties and failed to thrive. She also had a history of recurrent respiratory infections and eczematous skin changes. In addition, since birth she had had a musty urine odor that had never been reported in her family. Urine analyses were repeatedly normal.
At consultation, developmental evaluation revealed a hyperactive child with significant speech delay. Her vocabulary consisted of 10–15 words. She used few two-word sentences. Otherwise, the gross and fine motor abilities were adequate for her age. She made good eye contact and could understand and carry out simple verbal instructions, but she had a short attention span and impulsive behavior.
Upon examination, her height was reported at 84 cm, her weight was 10 kg, and her head circumference was 44 cm, all below the third percentile according to US Centers for Disease Control growth charts.Citation6
She had fair, dry skin and sparse, fine blond hair. Her face was elongated with a high sloping forehead. She had sparse lateral eyebrows, long eyelashes, wide-set eyes (telecanthus) with hypertelorism, low-set ears, a broad nasal bridge with a prominent tip, micrognathia, a thin upper lip, a high-arched palate, dental caries, a sacral dimple, and eczematous skin changes on the extremities, mainly on the flexural areas ( and ).
Figure 1 High forehead with flat supraorbital ridges and widely spaced eyes.
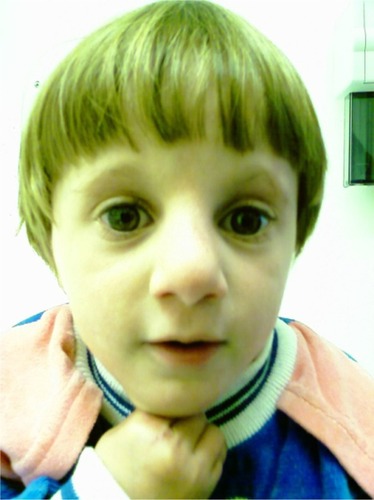
Figure 2 Prominent nasal bridge and sparse eyebrows.

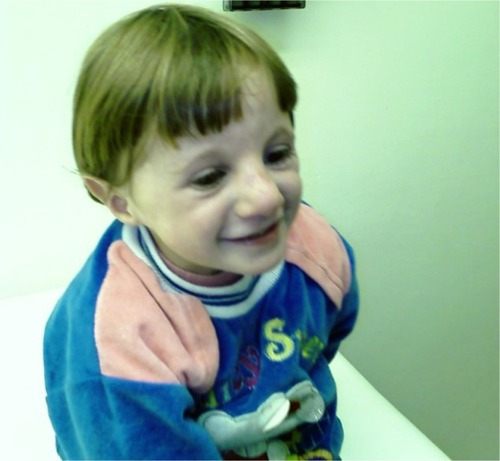
When she smiled, her facial skin wrinkled and she looked like a prematurely aged girl (). The remainder of the physical examination was normal.
Figure 3 Prematurely aged appearance of the proband when she smiles.
Laboratory workup yielded normal results for the following tests: complete blood count, serum blood urea nitrogen, creatinine, sodium, potassium, chloride, total serum protein, liver function, lipid profile, thyroid hormone, urine organic acids, plasma acylcarnitine profile and free carnitine level, plasma amino acids, serum lactate and pyruvate levels, plasma zinc, plasma immunoglobulins including normal immunoglobulin E levels, urinalysis, urine cultures, stool studies, insulin-like growth factor 1, antiendomyseal antibodies, sweat chloride, and 46, XX karyotype.
Other tests included MRI examination of the brain, X-rays of the spine, a voiding cystourethrogram, and an upper gastrointestinal series; all were normal. An echocardiography revealed a trace mitral regurgitation. Bone age was 2-years delayed compared with the chronological age.
Discussion
This case report presents a patient with Dubowitz syndrome. This is the first case reported from Lebanon. Although direct consanguinity has been reported in a number of cases,Citation7–Citation9 Dubowitz syndrome is underreported in the Arab population, which is known to have a high consanguinity rate.Citation7
Dubowitz syndrome is a very rare childhood disorder. The direct cause of the disorder is still unknown; however, most of the published data refer to possible genetic etiology.Citation2 Indeed, a recent study has reported the NSUN2 gene as the first potential causal gene with a relationship to the Dubowitz syndrome spectrum phenotype.Citation10
So far, diagnosis of Dubowitz syndrome is reached primarily through observational methods, mainly based on examination of facial appearance, growth records, and medical history.Citation2,Citation3 Although most reported cases in the literature share the classic characteristics that define this syndrome,Citation2,Citation3,Citation5 it is still difficult to diagnose the disorder as many of the features may not be prominent or concordant in different cases. In addition, Dubowitz syndrome shares symptoms of mental and growth retardation, chromosomal instability, and skin lesions with other disorders such as Bloom syndrome, Fanconi anemia, and fetal alcohol syndrome,Citation2–Citation4 suggesting that Dubowitz syndrome can easily be confused with these other disorders and thus easily underdiagnosed or misreported.
The main feature that distinguishes Dubowitz syndrome from Bloom syndrome and Fanconi anemia is facial appearance.Citation11 On the other hand, fetal alcohol syndrome may present with comparable facial features, as well as microcephaly, hyperactivity, growth and mental retardation – all of which can be present in Dubowitz syndrome.Citation5,Citation9 However, absence of prenatal alcohol exposure in this case definitely rules out fetal alcohol syndrome.Citation2–Citation4,Citation11
Our patient had the classic clinical findings of Dubowitz syndrome; however, a new finding stands out in our case: the presence of an unpleasant urine odor. To our knowledge, this case report is the first to describe this condition in a patient with Dubowitz syndrome. Odorous urine can be the result of dehydration, urinary infection, or various metabolic etiologies.Citation12 An odor of burnt sugar or maple syrup is characteristic of maple syrup urine disease, a sweaty-feet odor can be found in isovaleric acidemia, a cat-like odor is associated with multiple carboxylase deficiency and 3-methylcrotonylglycinuria, and a cabbage-like odor is found in tyrosinemia and methionine malabsorption.Citation13
In our patient, infectious and metabolic causes have been ruled out. Previous case reports have suggested that Dubowitz syndrome is a result of a certain cholesterol metabolism disorder that is reflected in a low lipoprotein profile.Citation14,Citation15 However, the lipid profile of our patient was normal. One main differential diagnosis resulting from eczematous dermatitis and a musty or mousey odor of urine in children is phenylketonuria,Citation13 and this condition was ruled out as well. Odorous urine may be reportedly the result of certain medications. However, our patient did not report intake of any drugs.Citation16
Although Dubowitz syndrome has been thoroughly described in the medical literature, this is the first time that a peculiar urine odor has been reported. Given the rarity and thus the relatively limited data on the condition, this finding should not be disdained and, until proven otherwise, we cautiously conclude that odorous urine may be a new feature to add to the clinical presentation of Dubowitz syndrome. More data and descriptive presentation are required to improve the diagnosis and the quality of care for individuals with Dubowitz syndrome.
Acknowledgments
Parental consent has been obtained for the use of patient photos.
Disclosure
The authors report no conflicts of interest in this work.
References
- DarcyDCRosenthalSWallersteinRJChromosome deletion of 14q32.33 detected by array comparative genomic hybridization in a patient with features of dubowitz syndromeCase Rep Genet2011201130607223074674
- HuberRSHoulihanDFilterKDubowitz syndrome: a review and implications for cognitive, behavioral, and psychological featuresJ Clin Med Res20113414715522121397
- PascualJCBetllochIBañulsJVergaraGWhat syndrome is this? Dubowitz syndromePediatr Dermatol200522548048116191007
- TsukaharaMOpitzJMDubowitz syndrome: review of 141 cases including 36 previously unreported patientsAm J Med Genet19966312772898723121
- YueJLuHLanSIdentification of the DNA repair defects in a case of Dubowitz syndromePLoS ONE201381e5438923372718
- Clinical Growth ChartsCenters of Disease Control and Prevention Available from: http://www.cdc.gov/growthcharts/clinical_chartsAccessed July 7, 2013
- Al-NemriARKilaniRASalihMAAl-AjlanAAEmbryonal rhabdomyosarcoma and chromosomal breakage in a newborn infant with possible Dubowitz syndromeAm J Med Genet200092210711010797433
- DubowitzVFamilial low birthweight dwarfism with an unusual facies and a skin eruptionJ Med Genet196521121714296916
- VielufDKortingHCBraun-FalcoOWaltherJUDubowitz syndrome: atopic dermatitis, low birth weight dwarfism and facial dysmorphismDermatologica199018042472492358105
- MartinezFJLeeJHLeeJEWhole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz-like syndromeJ Med Genet201249638038522577224
- SunkuAJGomezMRKlassDWEpileptic seizures, EEG abnormalities, and neuronal heterotopia in the Dubowitz syndromeAm J Electroneurodiagnostic Technol1998383156163
- StruthersSScanlonJParkerKGoddardJHallettRParental reporting of smelly urine and urinary tract infectionArch Dis Child200388325025212598394
- EnnsGMPackmanSDiagnosing inborn errors of metabolism in the newborn: clinical featuresNeoReviews200128e183e191
- YeşilkayaEKaraerKBideciACamurdanOPerçinEFCinazPDubowitz syndrome: a cholesterol metabolism disorder?Genet Couns200819328729018990984
- AhmadAAmalfitanoAChenYTKishnaniPSMillerCKelleyRDubowitz syndrome: a defect in the cholesterol biosynthetic pathway?Am J Med Genet199986550350410508998
- Harvard Medical SchoolUrine Color and Odor ChangesHarvard Women’s Health Watch. Cambridge, MAHarvard University2010 Available from: http://www.health.harvard.edu/newsletters/Harvard_Womens_Health_Watch/2010/June/urine-color-and-odor-changesAccessed July 7, 2013