
Abstract
Three international nosologies have been proposed for the diagnosis of Marfan syndrome (MFS): the Berlin nosology in 1988; the Ghent nosology in 1996 (Ghent-1); and the revised Ghent nosology in 2010 (Ghent-2). We reviewed the literature and discussed the challenges and concepts of diagnosing MFS in adults. Ghent-1 proposed more stringent clinical criteria, which led to the confirmation of MFS in only 32%–53% of patients formerly diagnosed with MFS according to the Berlin nosology. Conversely, both the Ghent-1 and Ghent-2 nosologies diagnosed MFS, and both yielded similar frequencies of MFS in persons with a causative FBN1 mutation (90% for Ghent-1 versus 92% for Ghent-2) and in persons not having a causative FBN1 mutation (15% versus 13%). Quality criteria for diagnostic methods include objectivity, reliability, and validity. However, the nosology-based diagnosis of MFS lacks a diagnostic reference standard and, hence, quality criteria such as sensitivity, specificity, or accuracy cannot be assessed. Medical utility of diagnosis implies congruency with the historical criteria of MFS, as well as with information about the etiology, pathogenesis, diagnostic triggers, prognostic triggers, and potential complications of MFS. In addition, social and psychological utilities of diagnostic criteria include acceptance by patients, patient organizations, clinicians and scientists, practicability, costs, and the reduction of anxiety. Since the utility of a diagnosis or exclusion of MFS is context-dependent, prioritization of utilities is a strategic decision in the process of nosology development. Screening tests for MFS should be used to identify persons with MFS. To confirm the diagnosis of MFS, Ghent-1 and Ghent-2 perform similarly, but Ghent-2 is easier to use. To maximize the utility of the diagnostic criteria of MFS, a fair and transparent process of nosology development is essential.
Introduction
Marfan syndrome (MFS) is a disorder of the connective tissue that is inherited in an autosomal dominant fashion and is caused by mutations in the gene coding for fibrillin-1 (FBN1). MFS has a low prevalence with similar frequency in both sexes and in all countries and races. MFS is a severe, chronic, and life-threatening disease with multiorgan involvement without availability of a curative therapy.Citation1 MFS is associated with chronic fatigue and pain, as well as with psychological despair that compromises the quality of life and imposes restrictions on the autonomy of affected persons.Citation2–Citation7
A classical study of life expectancy from the era prior to the availability of therapy documented poor survival of persons with MFS in the early 1970s.Citation1 The study documented 74 deaths in 257 affected persons with 50% of men and women dead at the age of 40 years and 48 years, respectively, which corresponded to a reduction of life expectancy by 30%–40% when compared to the normal population. The mean age of death was 32 years, whereby fatalities resulted from rupture of aortic aneurysm in 12 (16%) patients, dissection of aortic aneurysm in eight (11%), surgical death at attempted repair of the aorta in four (5%), insufficiency of the aortic valve in 14 (19%), congestive heart failure in eight (11%), bacterial endocarditis in two (3%), ventricular fibrillation after orthopedic surgery in one (1%), sudden unexplained death in three (5%), and suicide in one (1%) person, whereas the remaining patients died of miscellaneous reasons or of unknown causes.Citation1
The evolution of aortic surgery offered life-saving therapy for patients with acute aortic rupture and dissection, as well as prophylactic treatment to prevent patients with aortic root aneurysm from developing aortic rupture or dissection.Citation8 The various modifications of the technique of Bentall and De Bono, with complete replacement of the aortic root, became the operation of choice starting in the early 1970s.Citation9–Citation11 In addition, a classical open-label randomized trial of propranolol in adolescents and adults with MFS showed that prophylactic beta-adrenergic blockade (BAB) was effective in slowing the rate of aortic dilatation and reducing the development of aortic complications in some MFS patients.Citation12 Subsequently, a retrospective multicenter study documented that the life expectancy of patients with MFS had increased by over 25% since 1972, where benefits were documented from both cardiovascular surgery and medical therapy including BAB.Citation13 Since this study, the introduction of valve-sparing techniques for the replacement of the aortic root has resulted in a major improvement of life quality by obviating the risk for coagulation, where the technique of David yielded the best results.Citation14–Citation17 Alternatives for BAB have been tested in numerous, mostly small nonrandomized trials. These studies used angiotensin-converting enzyme inhibitors, calcium channel blockers, or angiotensin-receptor blockers.Citation18–Citation22 Results from these studies were conflicting. Very recently, a randomized double-blind trial in young patients with MFS showed equally beneficial effects with atenolol as compared to losartan, with no differences with regards to side effects, giving us the opportunity to choose either option.Citation23–Citation25
Once the diagnosis of MFS is suspected, care for patients should be coordinated in specialized centers. With the help of such centers, along with the support of regional Marfan patient organizations, affected persons can have a normal life expectancy with an acceptable quality of life.Citation26,Citation27 In this article, we review the literature and discuss the challenges and concepts of diagnosing MFS in adults.
Prevalence of MFS
A number of studies have assessed the prevalence of MFS. The earliest study comes from Northern Ireland and it reports 1.5 affected persons per 100,000 of the population, with a gene frequency of 0.729 per 100,000.Citation28 A second study that screened 29,067 children from 18 provinces and cities of the People’s Republic of China from 1951 to 1987 reported five children with MFS, giving a prevalence of 17.2 per 100,000 of the population, with a gene frequency of 8.61 per 100,000 inhabitants, and a penetrance of 71.69%.Citation29 A study of the population of Tayside and northeast Scotland identified 30 patients with MFS alive on prevalence day June 30, 1991, corresponding to an estimated prevalence of 1:14,691 or 6.81 per 100,000 inhabitants.Citation30 Based on 239 persons (122 males, 117 females) with MFS who were alive and living in Denmark by January 1, 1993, a fourth nation-wide study from Denmark identified a prevalence of 4.6 persons with MFS per 100,000 of the Danish population with an estimated average birth rate of 0.96 per 10,000 live born.Citation31 Our study of adults with MFS identified a prevalence of seven in 100,000 adults in the Hamburg metropolitan area.Citation32 Finally, a national cohort study from Taiwan identified 2.329 persons with MFS in a study period from 2000 to 2012 and calculated an average prevalence of 10.2 persons with MFS (95% confidence interval [CI]: 9.8–10.7) per 100,000 persons of the Taiwanese population, with a minimal birth rate incidence (estimated among those aged 20–29 years) of 23.3 per 100,000 (95% CI: 21.7–23.3) individuals.Citation33 provides an overview of the existing studies on the prevalence of MFS.
Table 1 Studies on the prevalence of MFS in the general population
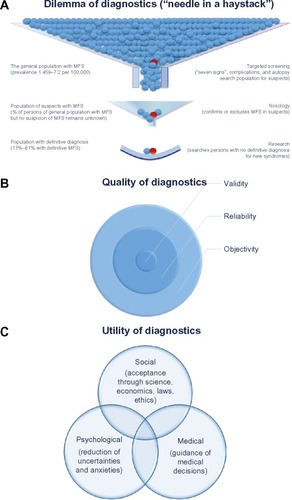
Taken together, all studies provide lower prevalence rates than do the widely cited estimate of one per 5,000 individuals.Citation34 Interestingly, these studies document a wide range from 1.5–17.2 per 100,000, or 0.075–0.86 per 5,000 individuals in the general population.Citation28–Citation33 However, all reported prevalence rates are far less than one in 2,000 citizens, and hence MFS complies with the European definition of a rare disease.Citation35 The wide range of the reported prevalence in the different studies reflects the usage of different diagnostic criteria of MFS, but it also seems likely that the medical workup in the different countries has led to the different estimates of population frequency. In fact, there is increasing evidence for the under-diagnosis of MFS in the general population, where the search for the “needle in the haystack” may pose a major diagnostic dilemma of MFS ().
Figure 1 Dilemma, quality and utility of diagnosis of Marfan syndrome.
Abbreviation: MFS, Marfan syndrome.
For instance, there is a marked delay in the diagnosis of MFS. The European Organisation for Rare Diseases (EURORDIS) performed a survey of patients with rare diseases in 18 European countries, where 682 families affected by MFS were included.Citation36 The time between the first clinical manifestations and diagnosis was 2 years for half of the patients with MFS, but it was as long as 4.5 years for 25% of patients. During the quest for diagnosis, more than five physicians were consulted by 38% of families and more than 20 physicians by 7% of families. Prior to obtaining the correct diagnosis of MFS, another diagnosis was given to 25% of patients, resulting in delayed diagnosis and inappropriate treatment in 81% of the patients. For 72% of families, the delay in diagnosis was considered responsible for deleterious consequences.Citation36 Similarly, a cohort study of all hospitalized patients with a diagnosis of MFS and related disorders in England still documents a high rate of cardiovascular events.Citation37 This study documents 159 patients with aortic dissection, 101 with stroke, and eleven with rupture of an abdominal aortic aneurysm in a total of 4,468 hospitalized MFS patients over a mean follow-up of 6.9 years.Citation37 Moreover, current data from autopsy studies suggest that the rate of MFS patients who die from aortic dissection outside the hospital is still as high as 7.8%.Citation38
Based on administrative data from 389 patients with MFS, RollCitation39 documented a negative linear relationship between the density of physicians in a given region and the probability of an immediate diagnosis of MFS, whereby a higher number of visits to physicians was associated with a significantly decreased probability of immediate diagnosis. Moreover, they found that the distance to medical health care centers was not a predictor of an immediate diagnosis.Citation39 The authors argue that diagnostic errors rarely occur because of a lack of knowledge about a rare disease, but rather because of cognitive errors in the process of gathering the right information about the patient, and in putting together heterogeneous information about the disease.Citation40 More importantly, they see structural deficiencies as a major cause of the delay in the diagnosis of MFS, and they discuss factors such as asymmetry of information among physicians, a behavioral pattern of “organized irresponsibility”, which might be facilitated through the availability of more physicians, a lack of long-term and sustained physician–patient relationships, and the lack of networking between physicians.Citation39,Citation41–Citation44
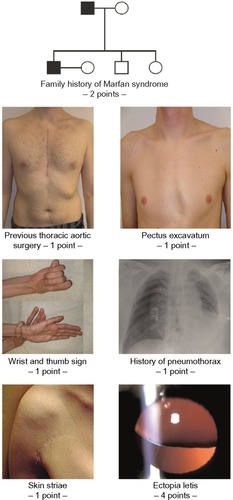
In summary, the identification of persons with MFS in the general population remains a diagnostic dilemma because it is not realistic to screen the entire population for a rare disease, and there is no simple way to define a target population. However, whenever a risk for MFS may be identified, targeted screening may be performed with tests like the “seven-signs” screening tool ( and ).Citation45–Citation48 In addition, young patients with aortic dissection, or family members of a person who died at a young age from aortic dissection, should also be considered for diagnostic testing for MFS.Citation49
Figure 2 “Seven-signs” of Marfan syndrome may be used for targeted screening of Marfan syndrome in adults of the general population.
Manifestations of MFS
MFS is a multiorgan disease affecting the skeleton, the cardiovascular system, the eyes, the lungs, the skin, and the nervous system.Citation50,Citation51 Individuals who have MFS do not exhibit all manifestations of MFS, and nosologies for diagnosing MFS select those manifestations as diagnostic criteria that are considered relevant. In this review, we propose five types of diagnostic contents, which together define the relevance of each sign, manifestation, or criterion of MFS (). The first is history: changes of diagnostic criteria should avoid large shifts in the population of people already labeled as having or not having MFS. Accordingly, the criteria of MFS should be largely congruent with the picture of MFS that evolved throughout the history of the syndrome. The second type of information is etiology and pathogenesis: demonstration of a causative FBN1 mutation or of pathogenetic changes that indicate progressive organ disease, such as increased transforming growth factor (TGF-β1) serum levels or aortic medial degeneration, can reduce diagnostic and prognostic uncertainty. Third, diagnostic triggers are manifestations that prompt suspicion of MFS in formerly undiagnosed subjects. Fourth, prognostic triggers are manifestations such as aortic dilatation or mitral valve prolapse that, fifth, may give rise to complications such as aortic dissection or mitral valve regurgitation ().
Table 2 Diagnostic content of various signs, manifestations, or criteria of MFS
History
provides a brief history of the initial descriptions of frequent or typical manifestations of MFS. This tabulation of their initial descriptions may be somewhat arbitrary, because the criteria used for defining both MFS and manifestations have varied since the initial description of the disease and, accordingly, descriptions of the history of MFS keep varying between authors. Nonetheless, some important lessons on the diagnosis of MFS may be derived from the analysis of history. First, initial descriptions of the syndrome relied on signs that can easily be observed, such as “spider-like” fingers and toes, chest wall deformities, and scoliosis. Accordingly, the syndrome was initially named after obvious presenting signs in the skeleton and musculature. Second, an essential step in the description of MFS was the description of major complications. Ironically, all early reports of such complications described atrial septal defects, patent ductus arteriosus, and aortic coarctation as congenital cardiovascular malformations of MFS, none of which are considered typical manifestations of MFS today. Third, ocular abnormalities, such as ectopic lenses, have been considered essential to the syndrome since their first description in 1912.Citation52,Citation53 MaumeneeCitation54 even speculated that the ophthalmologist E Williams might have provided the first description of MFS with his report of familial occurrence of dislocated lenses as early as June 1875. Ectopia lentis might be a classical example for a diagnostic sign that is both specific for MFS and clinically relevant because it can restrict the quality of life and the autonomy of patients. Finally, the heredity of the syndrome was recognized relatively soon after its discovery, which underpins the importance of the nature of a disease that affects whole families rather than isolated individuals.
Table 3 Historical perspective on advances in the diagnosis of MFS
Etiology and pathogenesis
Mutations in the FBN1 gene are today considered the exclusive cause of MFS.Citation55 Mutations in other genes were excluded as causes of MFS. Nonetheless, FBN1 mutation analysis has diagnostic limitations. First, a small portion of FBN1 mutations remains undetected in persons with MFS, most likely because of technical limitations.Citation56 Second, databases identify more than 1,500 different disease-causative FBN1 mutations, but there is no single FBN1 genotype feature that qualifies as a reliable predictor of disease evolution.Citation57,Citation58 Third, in some rare circumstances, mutations can present with phenotypes that do not fulfill clinical criteria of MFS, such as children with a purely skeletal phenotype, Shprintzen–Goldberg syndrome, or Weill–Marchesani syndrome.Citation56,Citation59–Citation61 Finally, an increasing number of nucleotide variants of the FBN1 gene have uncertain clinical significance and these introduce new uncertainties.Citation62,Citation63
FBN1 mutations initiate pathogenetic cascades that result in various manifestations and complications of MFS. One pathogenetic mechanism is structural weakness of the tissue, which presents as marked and premature degeneration of the medial layer of the aortic wall.Citation64 However, these changes are not specific to MFS and in clinical practice, they can only be diagnosed with invasive methods.Citation65 Indirect measurements of arterial stiffness parameters like pulse wave velocity or augmentation index can predict progression of aortic disease in MFS, but again this is not specific for MFS.Citation66–Citation69 Another major pathogenetic mechanism is the dysregulation of TGF-β1 that has a causal role in the evolution of various features of the Marfan. The causal role of TGF-β1 for the phenotypic features of MFS was identified in a mouse model, and humans with MFS exhibit increased serum levels of TGF-β1, which decrease with successful treatment of aortic disease with BAB, angiotensin-receptor blockers, or angiotensin-converting enzyme inhibitors.Citation18,Citation70–Citation72 However, such increases of TGF-β1 levels are not specific for aortic disease, but rather relate to the cumulative severity of organ involvement in MFS.Citation73 Moreover, increased TGF-β1 serum levels are not specific to MFS, but they also occur in bicuspid aortic valve disease or Loeys–Dietz syndrome.Citation73 In summary, exclusive FBN1 mutations currently qualify as the sensitive and specific causative or pathogenetic criteria of MFS.
Diagnostic triggers
In a series of 329 adults, general practitioners gave the following signs and manifestations as reasons for referral for diagnostic clarification of suspected MFS: a family history of Marfan features in 24%, aortic aneurysm or dissection both in 24%; skeletal features in 22%; MFS diagnosed in childhood in 10%; eye manifestations in 9%; miscellaneous reasons in 4%; nonaortic cardiovascular findings in 3%; and a history of pneumothorax, tissue weakness, and hypermobility in 2%. These reasons for referral reflect the perception of MFS as a familial disease of the aorta, which can be recognized through the presence of skeletal features and typical eye manifestations. Indeed, this perception is in accord with the time-honored historical descriptions of MFS. Interestingly, however, the percentage of confirmation of MFS was highest in persons who were referred for MFS already diagnosed in childhood (88%), for eye manifestations (83%), for a family history of Marfan features (52%), and for aortic aneurysm or dissection (50%). Conversely, the percentage of confirmation of MFS was low in persons who were referred for pneumothorax (43%), skeletal features (30%), tissue weakness and hypermobility (14%), and nonaortic cardiovascular findings in none.Citation48 In particular, tall stature, scoliosis, or joint hypermobility seem to be perceived as typical features of MFS. However, MFS was rarely confirmed in persons in whom MFS was suspected because of the presence of these signs.Citation48
highlights that very early in the history of MFS, clinicians felt the need to find methods that helped to rapidly and specifically identify persons with MFS in the general population. In 1966, Steinberg was the first to propose the thumb sign as a simple, quick screening test – as he put it – to truly distinguish lanky, thin persons who have spidery fingers, wear thick-lensed glasses, and appear prematurely old from those with MFS.Citation45 Since then, three other tools for targeted screening have been proposed to rapidly identify suspects of MFS based on a simple history and clinical signs ().Citation45–Citation48
Finally, provides a brief history of early systematic reviews and nosologies of MFS that had a major impact on the diagnosis and recognition of the syndrome. It is remarkable that as early as 1942, RadosCitation74 performed a systematic review of 204 patients published in the literature (101 males, 103 females), in which he described MFS as a heritable disease with a triad of skeletal, ocular, and cardiovascular manifestations. Since then, it has not been possible to identify a single definitive criterion to prove or refute MFS. Rather, common efforts were necessary to define an international consensus on the rules of how to diagnose MFS.
Prognostic triggers and complications
The risk for aortic dissection and rupture increases with growth of the aortic diameter, and hence the dilatation of the aortic root is a highly important prognostic trigger in MFS.Citation75 However, other manifestations of MFS can also trigger complications: dilatation of the thoracic or abdominal aorta and sleep-disordered breathing can lead to dissection or rupture; mitral valve prolapse to severe insufficiency or infective endocarditis; myocardial dysfunction to heart failure or sudden cardiac death; ectopia lentis or myopia to retinal detachment, glaucoma, or amaurosis; and apical pulmonary blebs to pneumothorax. Similarly, in some rare instances, dilatation of the main pulmonary artery can lead to dissection or rupture; skeletal malformations to impaired cardiorespiratory function, radiculopathy, or atlantoaxial or lumbal subluxation; osteopenia to fractures; and dural ectasia to intrapelvic meningocele and to dural leak with postural headache ().
The Ghent nosology (Ghent-1)
The first Ghent nosology was introduced in 1996 (Ghent-1), and it presented the revision of the criteria of the first international nosology, the so-called Berlin nosology (). In essence, Ghent-1 was the response to the new insights gained from the discovery of FBN1 gene mutations as the etiology of MFS.Citation76 The authors of the Ghent-1 nosology specified as reasons for their new nosology that the Berlin nosology relied completely on clinical criteria, and that the MFS phenotype had to be better separated from normal variation and from mild connective tissue phenotypes, such as the MASS phenotype (myopia, mitral valve prolapse, borderline and non-progressive aortic root dilatation, skeletal findings, and striae) and mitral valve prolapse syndrome (MVPS).Citation76 Of greatest concern were the misdiagnoses of relatives that arose by relying solely on the Berlin nosology after unequivocal diagnosis in a first-degree relative. Molecular evidence had shown that the criterion of a positive family history could produce a bias in favor of overdiagnosis. Accordingly, the authors stated as the most notable revisions the formulation of more stringent requirements for the diagnosis of MFS in relatives of an unequivocally affected individual; greater diagnostic weight on skeletal involvement, which constituted a major criterion if at least four of eight typical skeletal manifestations were present; contribution of molecular analysis, which the authors wanted neither to ignore nor to overemphasize; and delineation of the initial criteria for the diagnosis of other heritable conditions with partially overlapping phenotypes. The authors identified the diagnostic relevance of molecular data, especially in situations where clinical information concerning a relevant first-degree relative was unavailable, and both clinical and genotype data were available on a second-degree or more distant relative(s). On the other hand, they argued that since methods for the characterization of mutations in FBN1 were not entirely sensitive, and that the possibility of locus heterogeneity could not be completely ruled out, the inability to define an FBN1 mutation or the stringent documentation of recombination between FBN1 and the MFS phenotype did not constitute exclusion criteria.
Table 4 Comparison of the diagnostic criteria of MFS according to the Berlin versus Ghent-1 nosology
provides the entire list of criteria of MFS according to the Berlin and Ghent-1 nosologies. Ghent-1 mentions six organ systems with diagnostic relevance for MFS. For each of these organ systems, the nosology provides lists of manifestations that alone or in combination constitute major or minor criteria for MFS. In each organ system, the nosology distinguished between fulfillment of a major criterion, fulfillment of a minor criterion, and involvement of an organ system, where the combination of manifestations from the list of major criteria and from the list of minor criteria, or only from one of both lists was required. In addition, in the list of minor criteria of the skeletal system, three of five facial signs had to be present to fulfill one item in the list of minor criteria. For a final diagnosis, Ghent-1 distinguished between two diagnostic scenarios. First, for the index case: if the family or genetic history was not contributory, major criteria in at least two different organ systems and involvement of a third organ system were required for the diagnosis. If an FBN1 mutation known to cause MFS in others was detected, one major criterion in one organ system and involvement of a second organ system were required. Second, for a relative of an index case: The presence of a major criterion in the family history and one major criterion in an organ system, as well as involvement of a second organ system were required ().Citation76
Two studies compared the diagnostic performance of the Berlin and Ghent-1 nosology. The first study was only published as an abstract, and it found that 60 of 104 (58%) patients evaluated for suspected MFS were diagnosed as having MFS according to the Berlin nosology, of whom only 33 (32%) patients were confirmed by Ghent-1.Citation77 The second study found that 48 of 73 (66%) patients (aged 1 month to 62 years) evaluated for suspected MFS were diagnosed as having MFS according to the Berlin nosology, of whom only 39 (53%) patients were confirmed by Ghent-1.Citation78 Both studies, however, did not screen for FBN1 mutations in those 45% and 19% of patients who met the Berlin, but not the Ghent-1, criteria, respectively. The authors of the first study raised the concern that using the Ghent-1 criteria “in patient care [meant that] under-diagnosing is replacing over-diagnosing”.Citation77 Conversely, the authors of the second study concluded that Ghent-1 appropriately excluded some patients, but that further long-term follow-up or reliable molecular diagnostic techniques were necessary to establish the relative sensitivity and specificity of the Berlin and Ghent criteria.Citation78
In summary, Ghent-1 proposed more stringent clinical criteria that led to confirmation of MFS by Ghent-1 in only 32% and 53% of patients in the two studies available who had previously been diagnosed of MFS according to the Berlin nosology.
The revised Ghent nosology (Ghent-2)
The revised version of the Ghent nosology was introduced in 2010 (Ghent-2).Citation79 The authors of the Ghent-2 nosology listed the following as aims of the revision: first, identification of patients with a risk for aortic aneurysm or dissection; second, simplicity of use of diagnostic criteria; third, allowance for early diagnosis; fourth, consideration of availability and costs of diagnostic tests; fifth, better definition of entities such as familial ectopia lentis, the MASS phenotype, and MVPS; and, finally, delineation of triggers for alternative diagnoses such as Loeys–Dietz syndrome.Citation79 The authors listed five major changes, which comprised the following: 1) more diagnostic weight on aortic root aneurysm or dissection and ectopia lentis; 2) a more prominent role of molecular genetic testing; 3) complete removal of some clinical criteria, such as dilatation of the main pulmonary artery, dilatation or dissection of the descending thoracic or abdominal aorta, increased axial length of the globe and abnormally flat cornea, hypoplastic iris or hypoplastic ciliary muscle causing decreased miosis, joint hypermobility, spondylolisthesis, highly arched palate, and recurrent or incisional herniae, calcification of the mitral annulus, apical blebs of the lung, or mitigation of the diagnostic relevance of dural ectasia, or adding or modifying clinical criteria such as myopia >−3 diopters, hindfoot valgus, and thoracolumbar kyphosis; 4), provision of discriminating features of alternative diagnoses such as Loeys–Dietz syndrome; and 5) provision of context-specific recommendations for patient counseling and follow-up.Citation79
provides the entire list of criteria of MFS according to Ghent-2. In brief, in the absence of a family history of MFS, MFS is diagnosed in the presence of aortic root dilatation combined with ectopia lentis, or a causative FBN1 mutation, or a systemic score ≥7 points, or with the combination of ectopia lentis with an FBN1 mutation known to cause aortic dilatation. In the presence of a family history, MFS is diagnosed with the demonstration of ectopia lentis, or a systemic score ≥7 points, or aortic root dilatation (Z-scores ≥2 standard deviations [SD] above the mean with an age above 20 years, or Z-scores ≥3 SD above the mean with an age below 20 years).
Table 5 Diagnostic criteria of MFS according to the Ghent-2 nosology
lists five studies that performed a direct comparison of the diagnostic performance of Ghent-1 and Ghent-2.Citation80–Citation84 Faivre et alCitation80 concluded that if Ghent-2 criteria were applied with common sense and flexibility, they should help and support clinicians who are less experienced with the MFS phenotype. They recommended regular aortic follow-up in every patient with a diagnosis of ELS, MASS, or MVPS who does not reach the diagnosis of MFS. Radonic et alCitation81 found that the calculation of Z-scores according to the Ghent-2 criteria underestimated aortic root dilatation, especially in patients with a large body surface area, a problem that was fixed by the subsequent publication of improved nomograms.Citation85–Citation87 Aalberts et alCitation82 found that Ghent-2 led to a significant increase in the number of non-MFS diagnoses, which he found was due to lowering of the diagnostic threshold of MVPS. In addition, the authors concluded that Ghent-2 afforded a more straightforward diagnosis of MFS. Sheikhzadeh et alCitation83 found that the Ghent-1 criteria were useful when no genotyping was available, but that when gene sequencing is available, Ghent-2 could be assessed more easily and more rapidly than Ghent-1. Finally, Yang et alCitation84 reported that almost all the adult Korean patients who fulfilled the Ghent-1 criteria also fulfilled the Ghent-2 criteria.
Table 6 Comparison of diagnosis of MFS according to Ghent-1 versus Ghent-2
summarizes the Ghent-1 and Ghent-2 diagnoses of MFS in relation to the presence or absence of a causative FBN1 mutation. When not considering both early studies of the Ghent criteria, which exclusively included patients with FBN1 mutations and MFS, or with the FBN1 mutation MFS-like phenotype, our analysis documented that the Ghent-1 and Ghent-2 nosologies both yielded similar frequencies of MFS in persons with a causative FBN1 mutation (90% versus 92%) and in persons not having a causative FBN1 mutation (15% versus 13%).Citation80,Citation81 The study by Sheikhzadeh et alCitation83 documented that 245 of 300 persons with suspected MFS had an identical likelihood of exclusion or confirmation of MFS with both nosologies (82%). illustrates the diagnostic yield of Ghent-1 and Ghent-2 in studies of patients evaluated for suspected MFS, where MFS was diagnosed in 34% versus 35%, other syndromes in 26% versus 28%, and where no definitive diagnosis could be established in 40% versus 38%. Interestingly, only the proportion of diagnoses differed across the three studies, but not the results between Ghent-1 and Ghent-2. In summary, the review of the literature illustrates a similar diagnostic performance of Ghent-1 and Ghent-2 nosologies.
Table 7 Diagnostic yield in suspected MFS according to Ghent-1 versus Ghent-2
Prospects for reducing the inaccurate diagnoses of MFS
Classical criteria to estimate the quality of diagnosis comprise objectivity, reliability, and validity (). The diagnosis of MFS will be objective when diagnostic tests can be performed, analyzed, and interpreted in a standardized fashion that is independent of the individual clinician performing the analysis. Ghent-2 formulates diagnostic rules that are simpler and require both less diagnostic testing and less clinical expertise than Ghent-1. Similarly, Ghent-2 discards some “subjective qualifiers” of diagnostic features, and it introduces criteria for causality of FBN1 mutations. These changes indeed are likely to enhance objectivity. In contrast, Ghent-2 does not specify which of the many criteria for mitral valve prolapse and dural ectasia should be used for diagnosing MFS.Citation88–Citation91 This may have a negative impact on objectivity when assessing these signs, especially when clinicians use different criteria in the same patient. However, mitral valve prolapse and dural ectasia are features of the systemic score that only have a minor impact on the diagnosis of MFS.Citation80 Altogether, therefore, the objectivity of Ghent-2 is likely be higher than that of Ghent-1.
Clearly, objectivity is a prerequisite, but it may not necessarily lead to improved reliability. Until today, there is no study that compares intrasubject, intraobserver, and interobserver variation in the application of the Ghent-1 and Ghent-2 criteria. However, the overall diagnostic yield of MFS and alternative diagnoses is similar with both nosologies, which may argue for similar reliability (). However, there is no evidence that Ghent-2 has actually improved diagnostic reliability.
Finally, the diagnosis of MFS will be valid when MFS is correctly identified or excluded. However, to estimate the validity of the Ghent-1 or Ghent-2 criteria, we need an external diagnostic reference standard, which determines the “true” presence or absence of MFS, and would allow the sensitivity and specificity of nosologies to be calculated.Citation92 The defining criteria of a disease include etiology or clinical characteristics that allow for a clear delineation of the disease from both other diseases and the healthy population.Citation56,Citation59–Citation61 Similarly, none of the clinical characteristics of MFS mark a clear boundary to normality or to other diseases (), and none of the clinical signs of MFS is sufficient to prove or exclude MFS with both a sufficiently high positive likelihood ratio (>10) and a sufficiently low negative likelihood ratio (<0.1).Citation32 Thus, there is no external diagnostic reference standard of MFS and, hence, there is no stringent way to assess diagnostic validity by measuring the sensitivity, specificity, or accuracy of the Ghent-1 and Ghent-2 nosology.Citation93
In summary, the quality of diagnostic criteria of a specific nosology comprises objectivity, reliability, and validity, which have not been assessed in Ghent-1 or Ghent-2. Most notably, a nosology-based diagnosis of MFS lacks a diagnostic reference standard, and hence its sensitivity, specificity, or accuracy cannot be measured.
Perspectives from the clinic
The clinical perspective may be discussed in terms of the utility of the Ghent nosologies. In addition to objectivity, reliability, and validity, the utility of diagnostic criteria has not been described uniformly. However, the idea that all concepts have in common is to assess the usefulness of the criteria for doctors, patients, and other persons or groups. Whereas validity is an invariant and dichotomous quality of a diagnostic category, utility is gradable and context-dependent.Citation94 Based on the bio–social–psychological model of medicine, we will discuss the medical, social, and psychological dimension of the utility of diagnostic criteria using the example of the Ghent nosologies.Citation95
Medical utility
The diagnosis of MFS should provide information that is congruent with the historical criteria of MFS; and that informs about the etiology, pathogenesis, diagnostic triggers, prognostic triggers, and potential complications (). All these pieces of information together guide therapeutic decisions. Ideally, a single, easy, and quickly assessable criterion establishes the diagnosis of MFS which, at the same time, comprises all five types of information. As discussed earlier, this is not possible for MFS and, hence, diagnostic criteria compromise between feasibility and comprehensiveness. For instance, in contrast to Ghent-1, the Ghent-2 nosology does not list all diagnostic triggers. Indeed, this may not be necessary, because tests for targeted screening for MFS are available, and these may be used for the purpose of identifying patients in the general population ( and ).Citation45–Citation48 Similarly, dural ectasia and mitral valve prolapse have a high prevalence in MFS and they may give rise to complications. However, there are many other manifestations of MFS that can also cause complications, and these prognostic triggers need monitoring once the diagnosis of MFS is established (). In conclusion, the Ghent-2 nosology focused on establishing the diagnosis of MFS easily and quickly. Accordingly, Ghent-2 lists much fewer diagnostic criteria of MFS than does Ghent-1. At the same time, the diagnostic results are astonishingly congruent with the historical Ghent-2 criteria.
Molecular analysis of the FBN1 gene can significantly reduce diagnostic uncertainty in suspected MFS. In , we analyze how FBN1 analysis improves the diagnosis of MFS according to Ghent-2 criteria. First, in the absence of a family history of MFS, all diagnostic scenarios (1–5) with the isolated or combined presence of aortic root dilatation, ectopia lentis, or systemic score points ≥7 allow one to establish the diagnosis of MFS when a causative FBN1 mutation is present. Even in patients with a MASS phenotype or with isolated ectopia lentis, the presence of an FBN1 mutation indicates the possibility that the phenotypes progress towards MFS during follow-up.Citation32,Citation96 Exclusion of an FBN1 mutation also yields a significant gain of diagnostic certainty in all five scenarios, where the presence of alternative diseases becomes very likely (scenarios 1–3) or where MFS can at least be excluded with a higher degree of certainty (scenarios 4 and 5). In contrast, when the FBN1 gene is not analyzed, all diagnostic scenarios require extensive clinical and molecular analyses of many alternative syndromes (scenarios 1–3), or these scenarios remain diagnostically uncertain (scenarios 4 and 5). Second, Ghent-2 allows for the diagnosis of MFS with the isolated presence of ectopia lentis, systemic score points ≥7, or aortic root dilatation in all individuals with the presence of a family history of MFS (scenarios 6–8). However, when the FBN1 mutation status is unknown in the entire family with MFS, then the diagnostic uncertainty about MFS may remain as illustrated in scenarios 1–5. In contrast, when an FBN1 mutation is known, the family diagnosis of MFS is possible in all diagnostic scenarios (6–8), even without demonstrating the FBN1 mutation known in the family. However, testing for a specific nucleotide change in the FBN1 gene can be done with minimum of effort, time, and money, and these may be spent well, especially in subjects with an incomplete MFS phenotype ().
Table 8 Analysis of how FBN1 mutation status impacts the diagnosis of MFS according to current Ghent-2 diagnostic criteria
The definitive diagnosis of a specific genetic syndrome implies that there are different therapeutic approaches even to highly relevant manifestations, such as aortic root disease.Citation75,Citation93 Hence, one should avoid relying on a solely clinical diagnosis of MFS.Citation97,Citation98 Whenever MFS remains uncertain or diagnostic triggers of other diseases are observed, phenotypes should be explained by the presence of alternative syndromes, such as Loeys–Dietz syndrome. Ghent-2 clearly encourages the exclusion of related syndromes through clinical evaluation, as well as through molecular mutation analysis, and hence it may improve the guidance of therapy.Citation93
Social utility
Second, the social utility of the diagnostic criteria of MFS implies their acceptance by patients, patient organizations, the scientific community, doctors, hospital managers, and politicians. Thus, issues such as social labeling, identification with traditions, consistency with historical perceptions of MFS, scientific appropriateness, practicability, organizational needs, costs, social values, norms, and ethics come into play. Interestingly, both Ghent nosologies address most of these issues, however with a different emphasis. Ghent-2 mentions issues such as practicability and costs of diagnostics, consistency with historical perceptions of MFS, and consequences of misdiagnosis, such as restriction of career choices or access to insurance benefits, financial burden through frequent medical care, unfounded marital or reproductive decisions, and psychosocial stigmatization. The costs of clinical diagnostics based on Ghent-1 nosology for an outpatient visit in Germany are €389 per year.Citation99 Conversely, in Germany, the costs for conventional sequencing of the FBN1 gene are over €4,000, and these costs cannot be outweighed by a reduction in the clinical workup, as encouraged by Ghent-2.Citation100,Citation101
Psychological utility
Third, for patients, the psychological utility of the diagnostic criteria of MFS may imply a reduction of uncertainty, a mitigation of anxiety related to this uncertainty, and identification with a disease that has its own history; it also offers harborage for a community of patients, researchers, and doctors.Citation102 It may be important that the consensus process of nosology revision is a real joint effort that involves the entire community of patient organizations, researchers, and expert clinicians. In the absence of an absolute truth, the German sociologist Niklas Luhmann suggested legitimation by procedure.Citation103 For the quality and credibility of nosology criteria, rules of their development may be followed that are similar to those for official treatment guidelines, which may imply involvement of experts from all countries with Marfan centers, transparency of the process of expert selection, discussion, consensus, and disclosure of conflicts of interests.Citation104
In summary, in contrast to the reliability of diagnostic criteria, which is invariant and dichotomous, the utility of diagnostic criteria is graded and context-dependent. Hence, the task of optimizing the utility of diagnostic criteria can be maximized only through negotiation, compromise, and consensus ().Citation105,Citation106 Since utility depends on context, prioritization of the various diagnostic rules remains an issue of strategic decision making.
Conclusion
Screening tests for MFS should be used to identify persons with MFS. The Ghent-1 and Ghent-2 nosologies perform similarly, but Ghent-2 is much easier to use. Both nosologies are formal rules that may have to be applied “with common sense and flexibility”, as Faivre et alCitation80 have put it. We believe that one should not rely on a purely clinical diagnosis of MFS, but rather call for a molecular confirmation of MFS or alternative diagnoses. Similarly, regular aortic follow-up may be warranted in patients with a diagnosis of ectopia lentis syndrome, MASS phenotype, or MVPS.Citation80 Finally, to maximize the utility of diagnostic rules, a fair and transparent process of their development is essential.
Disclosure
The authors report no conflicts of interest in this work.
References
- MurdochJLWalkerBAHalpernBLKuzmaJWMcKusickVALife expectancy and causes of death in the Marfan syndromeN Engl J Med1972286158048085011789
- Rand-HendriksenSSørensenIHolmströmHAnderssonSFinsetAFatigue, cognitive functioning and psychological distress in Marfan syndrome, a pilot studyPsychol Health Med200712330531317510900
- BathenTVelvinGRand-HendriksenSRobinsonHSFatigue in adults with Marfan syndrome, occurrence and associations to pain and other factorsAm J Med Genet A2014164A81931193924719044
- PetersKFKongFHorneRFrancomanoCABieseckerBBLiving with Marfan syndrome I. Perceptions of the conditionClin Genet200160427328211683773
- PetersKFPetrillSAComparison of the background, needs, and expectations for genetic counseling of adults with experience with Down syndrome, Marfan syndrome, and neurofibromatosisAm J Med Genet A2011155A468469621344640
- Rand-HendriksenSJohansenHSembSOGeiranOStanghelleJKFinsetAHealth-related quality of life in Marfan syndrome: a cross-sectional study of Short Form 36 in 84 adults with a verified diagnosisGenet Med201012851752420613543
- Fusar-PoliPKlersyCStramesiFCallegariAArbustiniEPolitiPDeterminants of quality of life in Marfan syndromePsychosomatics200849324324818448780
- NienaberCAvon KodolitschYMeta-analysis of the prognosis of thoracic aortic dissection: changing mortality in the last four decadesHerz1992176398416 German1483629
- BentallHDe BonoAA technique for complete replacement of the ascending aortaThorax19682343383395664694
- GottVLGreenePSAlejoDEReplacement of the aortic root in patients with Marfan’s syndromeN Engl J Med1999340171307131310219065
- BernhardtAMTreedeHRybczynskiMComparison of aortic root replacement in patients with Marfan syndromeEur J Cardiothorac Surg20114051052105721435894
- ShoresJBergerKRMurphyEAPyeritzREProgression of aortic dilatation and the benefit of long-term beta-adrenergic blockade in Marfan’s syndromeN Engl J Med199433019133513418152445
- SilvermanDIBurtonKJGrayJLife expectancy in the Marfan syndromeAm J Cardiol19957521571607810492
- SarsamMAYacoubMRemodeling of the aortic valve anulusJ Thorac Cardiovasc Surg199310534354388445922
- DavidTEFeindelCMAn aortic valve-sparing operation for patients with aortic incompetence and aneurysm of the ascending aortaJ Thorac Cardiovasc Surg19921034617621 discussion 6221532219
- BenedettoUMelinaGTakkenbergJJRoscitanoAAngeloniESinatraRSurgical management of aortic root disease in Marfan syndrome: a systematic review and meta-analysisHeart2011971295595821228428
- KallenbachKBarakiHKhaladjNAortic valve-sparing operation in Marfan syndrome: what do we know after a decade?Ann Thorac Surg2007832S764S768 discussion S785–S79017257923
- AhimastosAAAggarwalAD’OrsaKMEffect of perindopril on large artery stiffness and aortic root diameter in patients with Marfan syndrome: a randomized controlled trialJAMA2007298131539154717911499
- PhomakayVHuettWGGossettJMTangXBornemeierRACollinsRT2ndβ-Blockers and angiotensin converting enzyme inhibitors: comparison of effects on aortic growth in pediatric patients with Marfan syndromeJ Pediatr2014165595195525109242
- Rossi-FoulkesRRomanMJRosenSEPhenotypic features and impact of beta blocker or calcium antagonist therapy on aortic lumen size in Marfan syndromeAm J Cardiol19998391364136810235096
- BrookeBSHabashiJPJudgeDPPatelNLoeysBDietzHC3rdAngiotensin II blockade and aortic-root dilation in Marfan’s syndromeN Engl J Med2008358262787279518579813
- RadonicTde WittePBaarsMJZwindermanAHMulderBJGroeninkMCOMPARE study groupLosartan therapy in adults with Marfan syndrome: study protocol of the multi-center randomized controlled COMPARE trialTrials201011320067609
- Attenhofer JostCHGreutmannMConnollyHMMedical treatment of aortic aneurysms in Marfan syndrome and other heritable conditionsCurr Cardiol Rev201410216117124527681
- BowenJMConnollyHMOf Marfan’s syndrome, mice, and medicationsN Engl J Med2014371222127212825405389
- LacroRVDietzHCSleeperLAPediatric Heart Network InvestigatorsAtenolol versus losartan in children and young adults with Marfan’s syndromeN Engl J Med2014371222061207125405392
- von KodolitschYRybczynskiMTrivicVHofmannTMeinertzTIn Kompetenzzentren behandeln: Lebensqualität und Lebenser-wartung beim Marfan-Syndrom verbessernKlinikarzt200231201206 German
- RadkeRMBaumgartnerHDiagnosis and treatment of Marfan syndrome: an updateHeart2014100171382139124777611
- LynasMAMarfan’s syndrome in Northern Ireland; an account of thirteen familiesAnn Hum Genet195822428930913559881
- SunQBZhangKZChengTOMarfan syndrome in China: a collective review of 564 cases among 98 familiesAm Heart J199012049349482220547
- GrayJRBridgesABFaedMJAscertainment and severity of Marfan syndrome in a Scottish populationJ Med Genet199431151548151638
- FuchsJMarfan syndrome and other systemic disorders with congenital ectopia lentis. A Danish national surveyActa Paediatr19978699479529343273
- RybczynskiMBernhardtAMRehderUThe spectrum of syndromes and manifestations in individuals screened for suspected Marfan syndromeAm J Med Genet A2008146A243157316619012347
- ChiuHHWuMHChenHCKaoFYHuangSKEpidemiological profile of Marfan syndrome in a general population: a national database studyMayo Clin Proc2014891344224388020
- MilewiczDMDietzHCMillerDCTreatment of aortic disease in patients with Marfan syndromeCirculation200511111e150e15715781745
- European Organisation for Rare Diseases (EURORDIS)Rare Diseases: Understanding this Public Health PriorityParis, FranceEuropean Organisation for Rare Diseases (EURORDIS)2005
- KoleAFauissonFThe Voice of 12,000 Patients: Experiences and Expectations of Rare Disease Patients on Diagnosis and Care in Europe2009 Available from: http://www.eurordis.org/IMG/pdf/voice_12000_patients/EURORDISCARE_FULLBOOKr.pdfAccessed July 2, 2014
- PitcherAGoldacreRGoldacreMJEstimation of the risk of cardiovascular complications in Marfan syndrome: A national cohort study with 30,000 patient years follow-up [abstract]Paper presented at: 9th International Research Symposium on Marfan Syndrome and Related DisordersSeptember 25–27, 2014Paris
- PrakashSKHaden-PinneriKMilewiczDMSusceptibility to acute thoracic aortic dissections in patients dying outside the hospital: an autopsy studyAm Heart J2011162347447921884863
- RollKThe influence of regional health care structures on delay in diagnosis of rare diseases: the case of Marfan SyndromeHealth Policy20121052–311912722420917
- BernerESGraberMLOverconfidence as a cause of diagnostic error in medicineAm J Med20081215 SupplS2S2318440350
- BeckAEBeck’s sociology of risk: a critical assessmentSociology2002362293315
- WeissLJBlusteinJFaithful patients: the effect of long-term physician-patient relationships on the costs and use of health care by older AmericansAm J Public Health19968612174217479003131
- LiuKYKingMBearmanPSSocial influence and the autism epidemicAJS201011551387143420503647
- KalkbrennerAEDanielsJLEmchMMorrisseyJPooleCChenJCGeographic access to health services and diagnosis with an autism spectrum disorderAnn Epidemiol201121430431021376278
- SteinbergIA simple screening test for the Marfan syndromeAm J Roentgenol Radium Ther Nucl Med1966971118124
- WalkerBAMurdochJLThe wrist sign. A useful physical finding in the Marfan syndromeArch Intern Med197012622762775433066
- MuellerGCStarkVSteinerKWeilJvon KodolitschYMirTSThe Kid-Short Marfan Score (Kid-SMS) – an easy executable risk score for suspected paediatric patients with Marfan syndromeActa Paediatr20131022e84e8923110520
- SheikhzadehSKuschMLRybczynskiMA simple clinical model to estimate the probability of Marfan syndromeQJM2012105652753522301820
- RippergerTTrögerHDSchmidtkeJThe genetic message of a sudden, unexpected death due to thoracic aortic dissectionForensic Sci Int20091871–31519285815
- von KodolitschYRybczynskiMCardiovascular aspects of the Marfan syndrome: a systematic reviewRobinsonPNGodfreyMMarfan Syndrome: A Primer for Clinicians and ScientistsNew York, NYKluwer Academic/Plenum Publishers2004
- PyeritzREChapter 153: Marfan syndrome and related disordersRimoinDKorfRPEmery and Rimoin’s Principles and Practice of Medical Genetics6th edOxford, UKAcademic Press2013152
- BörgerFÜber zwei Fälle von ArachnodaktylieZ Kinder-Heilk1914122–3161184 German
- von PfaundlerMArachnodaktylieMünchener Medizinische Wochenschrift191461280 German
- MaumeneeIHThe eye in the Marfan syndromeBirth Defects Orig Artic Ser19821865155246983370
- DietzHCCuttingGRPyeritzREMarfan syndrome caused by a recurrent de novo missense mutation in the fibrillin geneNature199135263333373391852208
- LoeysBDe BackerJVan AckerPComprehensive molecular screening of the FBN1 gene favors locus homogeneity of classical Marfan syndromeHum Mutat200424214014615241795
- GillisEVan LaerLLoeysBLGenetics of thoracic aortic aneurysm: at the crossroad of transforming growth factor-β signaling and vascular smooth muscle cell contractilityCirc Res2013113332734023868829
- FaivreLCollod-BeroudGLoeysBLEffect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international studyAm J Hum Genet200781345446617701892
- MilewiczDMGrossfieldJCaoSNKieltyCCovitzWJewettTA mutation in FBN1 disrupts profibrillin processing and results in isolated skeletal features of the Marfan syndromeJ Clin Invest1995955237323787738200
- SoodSEldadahZAKrauseWLMcIntoshIDietzHCMutation in fibrillin-1 and the Marfanoid-craniosynostosis (Shprintzen-Goldberg) syndromeNat Genet19961222092118563763
- FaivreLGorlinRJWirtzMKIn frame fibrillin-1 gene deletion in autosomal dominant Weill-Marchesani syndromeJ Med Genet2003401343612525539
- ten BoschJRGrodyWWKeeping up with the next generation: massively parallel sequencing in clinical diagnosticsJ Mol Diagn200810648449218832462
- Lerner-EllisJPAldubayanSHHernandezALThe spectrum of FBN1, TGFβR1, TGFβR2 and ACTA2 variants in 594 individuals with suspected Marfan Syndrome, Loeys-Dietz Syndrome or Thoracic Aortic Aneurysms and Dissections (TAAD)Mol Genet Metab2014112217117624793577
- SchlatmannTJBeckerAEPathogenesis of dissecting aneurysm of aorta. Comparative histopathologic study of significance of medial changesAm J Cardiol19773912126831424
- LarsonEWEdwardsWDRisk factors for aortic dissection: a necropsy study of 161 casesAm J Cardiol19845368498556702637
- BaumgartnerDBaumgartnerCMátyásGDiagnostic power of aortic elastic properties in young patients with Marfan syndromeJ Thorac Cardiovasc Surg2005129473073915821637
- LaurentSCockcroftJVan BortelLEuropean Network for Noninvasive Investigation of Large ArteriesExpert consensus document on arterial stiffness: methodological issues and clinical applicationsEur Heart J200627212588260517000623
- MortensenKAydinMARybczynskiMAugmentation index relates to progression of aortic disease in adults with Marfan syndromeAm J Hypertens200922997197919574960
- MortensenKBaulmannJRybczynskiMAugmentation index and the evolution of aortic disease in marfan-like syndromesAm J Hypertens201023771672420395939
- MattPSchoenhoffFHabashiJGenTAC ConsortiumCirculating transforming growth factor-beta in Marfan syndromeCirculation2009120652653219635970
- FrankenRden HartogAWde WaardVCirculating transforming growth factor-β as a prognostic biomarker in Marfan syndromeInt J Cardiol201316832441244623582687
- NeptuneERFrischmeyerPAArkingDEDysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndromeNat Genet200333340741112598898
- HillebrandMMillotNSheikhzadehSTotal serum transforming growth factor-β1 is elevated in the entire spectrum of genetic aortic syndromesClin Cardiol2014371167267925113270
- RadosAMarfan’s syndrome: arachnodactyly coupled with dislocation of the lensArch Ophthalmol1942273477538
- von KodolitschYRobinsonPBergerJWhen should surgery be performed in Marfan syndrome and other connective tissue disorders to protect against type A dissection?BonserRSPaganoDHaverichAMascaroJControversies in Aortic Dissection and Aneurysmal DiseaseLondon, UKSpringer20141747
- De PaepeADevereuxRBDietzHCHennekamRCPyeritzRERevised diagnostic criteria for the Marfan syndromeAm J Med Genet19966244174268723076
- KousseffBGJenningsJWRanellsJDThe revised diagnostic criteria of Marfan syndrome: a clinical analysis [abstract]Am J Hum Genet199965A36
- RosePSLevyHPAhnNUA comparison of the Berlin and Ghent nosologies and the influence of dural ectasia in the diagnosis of Marfan syndromeGenet Med20002527828211399208
- LoeysBLDietzHCBravermanACThe revised Ghent nosology for the Marfan syndromeJ Med Genet201047747648520591885
- FaivreLCollod-BeroudGAdèsLThe new Ghent criteria for Marfan syndrome: what do they change?Clin Genet201281543344221564093
- RadonicTde WittePGroeninkMCritical appraisal of the revised Ghent criteria for diagnosis of Marfan syndromeClin Genet201180434635321332468
- AalbertsJJThioCHSchuurmanAGDiagnostic yield in adults screened at the Marfan outpatient clinic using the 1996 and 2010 Ghent nosologiesAm J Med Genet A2012158A598298822461464
- SheikhzadehSKadeCKeyserBAnalysis of phenotype and genotype information for the diagnosis of Marfan syndromeClin Genet201282324024721883168
- YangJHHanHJangSYA comparison of the Ghent and revised Ghent nosologies for the diagnosis of Marfan syndrome in an adult Korean populationAm J Med Genet A2012158A598999522162372
- RomanMJDevereuxRBKramer-FoxRO’LoughlinJTwo-dimensional echocardiographic aortic root dimensions in normal children and adultsAm J Cardiol19896485075122773795
- PyeritzRELoeysBThe 8th international research symposium on the Marfan syndrome and related conditionsAm J Med Genet A2012158A1424922140025
- DevereuxRBde SimoneGArnettDKNormal limits in relation to age, body size and gender of two-dimensional echocardiographic aortic root dimensions in persons ≥15 years of ageAm J Cardiol201211081189119422770936
- LundbyRRand-HendriksenSHaldJKDural ectasia in Marfan syndrome: a case control studyAJNR Am J Neuroradiol20093081534154019461064
- SheikhzadehSSondermannCRybczynskiMComprehensive analysis of dural ectasia in 150 patients with a causative FBN1 mutationClin Genet201486323824523991918
- RybczynskiMMirTSSheikhzadehSFrequency and age-related course of mitral valve dysfunction in the Marfan syndromeAm J Cardiol201010671048105320854973
- RybczynskiMTreedeHSheikhzadehSPredictors of outcome of mitral valve prolapse in patients with the Marfan syndromeAm J Cardiol2011107226827421211604
- KnottnerusJAvan WeelCMurisJWEvaluation of diagnostic proceduresBMJ2002324733547748011859054
- von KodolitschYRybczynskiMBernhardtAMarfan syndrome and the evolving spectrum of heritable thoracic aortic disease: do we need genetics for clinical decisions?Vasa2010391173220186673
- KendellRJablenskyADistinguishing between the validity and utility of psychiatric diagnosesAm J Psychiatry2003160141212505793
- EngelGLThe need for a new medical model: a challenge for biomedicineScience19771964286129136847460
- PepeGLapiniIEvangelistiLIs ectopia lentis in some cases a mild phenotypic expression of Marfan syndrome? Need for a long-term follow-upMol Vis2007132242224718087243
- CookJRRamirezFClinical, diagnostic, and therapeutic aspects of the Marfan syndromeAdv Exp Med Biol2014802779424443022
- CookJRCartaLGalatiotoJRamirezFCardiovascular manifestations in Marfan syndrome and related diseases; multiple genes causing similar phenotypesClin Genet2015871112024867163
- ManowMLPaulsenNRybczynskiMAnalysis of costs and profits of ambulatory care of Marfan patients after initiation of a novel German legal directive (116 b SGB V)Med Klin (Munich)20101058529537 German20824410
- AchelrodDBlankartCRLinderRvon KodolitschYStargardtTThe economic impact of Marfan syndrome: a non-experimental, retrospective, population-based matched cohort studyOrphanet J Rare Dis201499024954169
- von KodolitschYBlankartCRVoglerMKallenbachKRobinsonPNGenetics and prevention of genetic aortic syndromes (GAS) and of the Marfan syndromeBundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz2015582146153 German25446311
- De NobeleSCaring for Marfan patients from a patient’s perspective – where do we go from here? [abstract]Paper presented at: 9th International Research Symposium on Marfan Syndrome and Related DisordersSeptember 25–27, 2014Paris, France
- LuhmannNLegitimation durch Verfahren [Legitimation by Procedure]Frankfurt am Main, GermanySuhrkamp2001
- BaumannMHLewisSZGuttermanDAmerican College of Chest PhysiciansACCP evidence-based guideline development: a successful and transparent approach addressing conflict of interest, funding, and patient-centered recommendationsChest200713231015102417540835
- von KodolitschYvon KodolitschKOverlackCwebpage on the InternetThe art of medicine – a task of strategyHamburg, GermanyInternationales Clausewitz – Zentrum (ICZ)2014 Available from: http://www.clausewitznetzwerk.de/forschung/publikationen/2014/Accessed October 14, 2014
- von KodolitschYOverlackCvon KodolitschKStrategisches Denken als Schlüssel zu chirurgischer Exzellenz. Ärztlicher Denkstil in der Tradition von Kant und Clausewitz [Strategic thinking as the key to surgical excellence. Medical thought style in the tradition of Kant and Clausewitz]Zeitschrift für Herz-, Thorax- und Gefäßchirurgie201327282289 German
- FrankenRRadonicTden HartogAWCOMPARE study groupThe revised role of TGF-β in aortic aneurysms in Marfan syndromeNeth Heart J201523211612125342281
- RobertsWCHonigHSThe spectrum of cardiovascular disease in the Marfan syndrome: a clinico-morphologic study of 18 necropsy patients and comparison to 151 previously reported necropsy patientsAm Heart J198210411151357046406
- NagashimaHSakomuraYAokaYAngiotensin II type 2 receptor mediates vascular smooth muscle cell apoptosis in cystic medial degeneration associated with Marfan’s syndromeCirculation200110412 Suppl 1I282I28711568070
- TrotterSEOlsenEGMarfan’s disease and Erdheim’s cystic medionecrosis. A study of their pathologyEur Heart J199112183872009899
- LedouxMBeauchetAFermanianCBoileauCJondeauGSaiagPA case-control study of cutaneous signs in adult patients with Marfan disease: diagnostic value of striaeJ Am Acad Dermatol201164229029521112669
- BehanWMLongmanCPettyRKMuscle fibrillin deficiency in Marfan’s syndrome myopathyJ Neurol Neurosurg Psychiatry200374563363812700307
- AttanasioMPratelliEPorcianiMCDural ectasia and FBN1 mutation screening of 40 patients with Marfan syndrome and related disorders: role of dural ectasia for the diagnosisEur J Med Genet201356735636023684891
- FattoriRNienaberCADescovichBImportance of dural ectasia in phenotypic assessment of Marfan’s syndromeLancet1999354918291091310489951
- ChowKPyeritzRELittHIAbdominal visceral findings in patients with Marfan syndromeGenet Med20079420821217438384
- MulderBJThe distal aorta in the Marfan syndromeNeth Heart J2008161138238619065277
- PyeritzREWappelMAMitral valve dysfunction in the Marfan syndrome. Clinical and echocardiographic study of prevalence and natural historyAm J Med19837457978076837604
- AlpenduradaFWongJKiotsekoglouAEvidence for Marfan cardiomyopathyEur J Heart Fail201012101085109120861133
- De BackerJFDevosDSegersPPrimary impairment of left ventricular function in Marfan syndromeInt J Cardiol2006112335335816316698
- RybczynskiMKoschykDHAydinMATissue Doppler imaging identifies myocardial dysfunction in adults with Marfan syndromeClin Cardiol2007301192417262773
- SavolainenANisulaLKetoPLeft ventricular function in children with the Marfan syndromeEur Heart J19941556256308056001
- AydinAAdsayBASheikhzadehSObservational cohort study of ventricular arrhythmia in adults with Marfan syndrome caused by FBN1 mutationsPLoS One2013812e8128124349050
- HoffmannBARybczynskiMRostockTProspective risk stratification of sudden cardiac death in Marfan’s syndromeInt J Cardiol201316762539254522738784
- KarpmanCAughenbaughGLRyuJHPneumothorax and bullae in Marfan syndromeRespiration201182321922421252480
- CorsicoAGGrossoATriponBPulmonary involvement in patients with Marfan SyndromePanminerva Med201456217718224994580
- SheikhzadehSDe BackerJGorganNThe main pulmonary artery in adults: a controlled multicenter study with assessment of echocardiographic reference values, and the frequency of dilatation and aneurysm in Marfan syndromeOrphanet J Rare Dis20149120325491897
- GiampietroPFRaggioCDavisJGMarfan syndrome: orthopedic and genetic reviewCurr Opin Pediatr2002141354111880731
- MouraBTubachFSulpiceMMultidisciplinary Marfan Syndrome Clinic GroupBone mineral density in Marfan syndrome. A large case-control studyJoint Bone Spine200673673373517056292
- BrautbarALeMaireSAFrancoLMCoselliJSMilewiczDMBelmontJWFBN1 mutations in patients with descending thoracic aortic dissectionsAm J Med Genet A2010152A241341620082464
- MimounLDetaintDHamrounDDissection in Marfan syndrome: the importance of the descending aortaEur Heart J201132444344921147864
- KühneKKeyserBGroeneEFFBN1 gene mutation characteristics and clinical features for the prediction of mitral valve disease progressionInt J Cardiol2013168295395923176764
- KnosallaCWengYGHammerschmidtROrthotopic heart transplantation in patients with Marfan syndromeAnn Thorac Surg20078351691169517462381
- SavolainenAKupariMToivonenLKaitilaIViitasaloMAbnormal ambulatory electrocardiographic findings in patients with the Marfan syndromeJ Intern Med199724132212269104435
- HallJRPyeritzREDudgeonDLHallerJAJrPneumothorax in the Marfan syndrome: prevalence and therapyAnn Thorac Surg19843765005046732339
- PatiPKGeorgePVJoseJVGiant pulmonary artery aneurysm with dissection in a case of Marfan syndromeJ Am Coll Cardiol201361668523391202
- StreetenEAMurphyEAPyeritzREPulmonary function in the Marfan syndromeChest19879134084123816319
- FrantzFWIndications and guidelines for pectus excavatum repairCurr Opin Pediatr201123448649121670676
- VoermansNCHosmanAJvan AlfenNRadicular dysfunction due to spinal deformities in Marfan syndrome at older age: three case reportsEur J Med Genet2010531353919879983
- MacKenzieJMRankinRSudden death due to atlantoaxial subluxation in marfan syndromeAm J Forensic Med Pathol200324436937014634478
- DomanIKövérFIllésTDócziTSubluxation of a lumbar vertebra in a patient with Marfan syndrome. Case reportJ Neurosurg2001941 Suppl15415711147854
- VoyvodicFScroopRSandersRRAnterior sacral meningocele as a pelvic complication of Marfan syndromeAust N Z J Obstet Gynaecol199939226226510755797
- DiazJHEpidemiology and outcome of postural headache management in spontaneous intracranial hypotensionReg Anesth Pain Med200126658258711707800
- TeohPCBronchiectasis and spontaneous pneumothorax in Marfan’s syndromeChest1977725672673913156
- ParidaSKKrissVMHallBDHiatus/paraesophageal hernias in neonatal Marfan syndromeAm J Med Genet19977221561589382135
- MarfanAB-JUn cas de déformation congènital des quatre membres plus pronouncée aux extrémitiés charactérisée par l’allongement des os avec un certain degré d’amonassesmentBull Mem Soc Med Hop (Paris) Ser. 3189613220226 French
- MarfanAB-JLa dolichoste´nome´lie (dolichome´lie arachnodactylie)Ann Med193844529 French
- AchardMCArachnodactylieBull Mem Soc Med Hop Paris190219834840
- SalleVÜber einen Fall von angeborener abnormer Größe der Extremitäten mit einem an Akromegalie erinnernden SymptomenkomplexJahrbuch Kinderheilkunde191275540550 German
- PiperRKIrvine-JonesEArachnodactylia and its association with congenital heart disease: Report of a case and review of the literatureAmerican Journal of Diseases of Children1926316832839
- ApertELes formes frustes du syndrome dolichosténomélique de MarfanNourrisson1938261 French
- McKusickVAThe cardiovascular aspects of Marfan’s syndrome: a heritable disorder of connective tissueCirculation195511332134214352380
- WeveHÜber Arachnodakylie (Dystrophia mesodermalis congenita, Typus Marfan)Arch. Augenheilkd1931104146
- JéquierPMObservations sur le syndrome de Marfan (dolichosténomelié ou arachnodactylie)Helvetica Medica Acta194310233236
- BaerRWTaussingHBOppenheimerEHCongenital aneurysmal dilatation of the aorta associated with arachnodactylyBull Johns Hopkins Hosp194372309331
- EtterLEGloverLPArachnodactyly complicated by dislocated lens and death from rupture of dissecting aneurysm of aortaJAMA194312328889
- TungHLLiebowAAMarfan’s syndrome; observations at necropsy: with special reference to medionecrosis of the great vesselsLab Invest19521338240614955976
- NelsonJDThe Marfan syndrome, with special reference to congenital enlargement of the spinal canalBr J Radiol19583137056156413584748
- KacheleGEThe embryogenesis of ectopia lentis: an uncommon dermatological manifestationArch Ophthalmol19606413513914408308
- McKusickVAThe Marfan syndrome: from clinical delineation to mutational characterization, a semiautobiographic accountRobinsonPNGodfreyMMarfan Syndrome: A Primer for Clinicians and ScientistsNew York, NYKluwer Academic/Plenum Publishers2004112
- BowdenDHFavaraBEDonahoeJLMarfan’s syndrome: accelerated course in childhood associated with lesions of mitral valve and pulmonary arteryAm Heart J196569969914253110
- BealsRKHechtFCongenital contractural arachnodactyly. A heritable disorder of connective tissueJ Bone Joint Surg Am19715359879935557609
- HechtFBealsRK“New” syndrome of congenital contractural arachnodactyly originally described by Marfan in 1896Pediatrics19724945745794552107
- McKusickVAHeritable Disorders of Connective Tissue4th edSt Louis, MOMosby19566871
- BeightonPde PaepeADanksDInternational nosology of heritable disorders of connective tissue, Berlin, 1986Am J Med Genet19882935815943287925