
Abstract
Antisense-mediated splicing modulation is a tool that can be exploited in several ways to provide a potential therapy for rare genetic diseases. This approach is currently being tested in clinical trials for Duchenne muscular dystrophy and spinal muscular atrophy. The present review outlines the versatility of the approach to correct cryptic splicing, modulate alternative splicing, restore the open reading frame, and induce protein knockdown, providing examples of each. Finally, we outline a possible path forward toward the clinical application of this approach for a wide variety of inherited rare diseases.
Introduction
Genetic diseases are generally rare diseases caused by mutations in specific genes. Sometimes mutations in different genes can give rise to similar phenotypes, eg, there have been dozens of genes identified in patients suffering from muscular dystrophies.Citation1 However, mutations in a single gene can also give rise to multiple diseases with varying phenotypes. Probably the most notorious example is the LMNA gene, in which mutations are associated with multiple phenotypes, including Emery–Dreifuss muscular dystrophy, familial partial lipodystrophy, limb girdle muscular dystrophy, dilated cardiomyopathy, Charcot–Marie–Tooth disease, restrictive dermopathy, and Hutchinson–Gilford progeria syndrome (HGPS).Citation2 For a significant number of patients with, or suspected of having a genetic disease, the mutation in the causative gene has not yet been identified. However, with the rapid advances made by “next generation sequencing”, it is anticipated that mutations will soon be identified for almost all patients with genetic disorders; the International Rare Disease Research Consortium (IRDiRC) has set itself an ambitious goal of having the means available to diagnose most rare genetic diseases by 2020 (http://www.irdirc.org).
With the availability of next generation sequencing, it is now possible to perform a more in-depth analysis of candidate genes. In the past, generally only exons and the donor and acceptor splice sites (ie, the first and last two base pairs of an intron, respectively) were analyzed. However, it is now feasible to analyze complete introns and there are multiple publications that report deep intronic mutations that activate cryptic splice sites and thus disrupt normal transcript processing.Citation3 In parallel, antisense-mediated splicing modulation has been developed from preclinical cell and animal models into the clinical trial phase for Duchenne muscular dystrophy (DMD) and spinal muscular atrophy (SMA).Citation4–Citation8 This approach makes use of antisense oligonucleotides (ASOs, small pieces of chemically modified DNA or RNA) and would offer a potential treatment for genetic disorders caused by mutations that disrupt splicing, but can also be exploited in other ways to address genetic mutations.Citation9 ASOs need to be chemically modified to improve stability to nucleases and provide favorable pharmacokinetic and pharmacodynamic characteristics for in vivo use. The most commonly used ASO chemistries are the 2′-O-methyl RNA phosphorothioate chemistry, which is negatively charged, and the charge neutral phosphorodiamidate morpholino oligomer. For more background on ASO chemistry, we refer the reader to a recent book chapter.Citation10 This review will describe different ways antisense-mediated splicing modulation can be used to treat genetic diseases. Since there are already numerous reviews describing exon skipping for DMD and SMA,Citation11–Citation14 we have chosen to discuss these neuromuscular applications only briefly and to focus on the potential of the exon skipping approach for other genetic diseases. For each approach we have chosen a few illustrative examples, focusing on those where data from animal models is available.
Cryptic splicing
Cryptic splice sites are sequences in the pre-messenger RNA (pre-mRNA) that are not normally used as splice sites, but which are activated by mutations that either inactivate the canonical splice sites or create splice sites where one did not exist before.Citation15 The classical example of this phenomenon is in the β-globin gene, where mutations in the canonical splice sites cause activation of cryptic splice sites, leading to the inclusion of aberrant exons, resulting in nonfunctional transcripts, defective β-globin expression, and consequently β-thalassemia.Citation16 For the majority of patients, the disease is caused by mutations that activate cryptic splice sites.Citation17 In the early 1990s, ASOs were developed by Dominski and Kole using this paradigm (): by targeting the cryptic splice sites with 2′-O-methyl RNA ASOs to block the access of the spliceosome to these sites, their group was able to restore normal β-globin splicing in cellsCitation18 and normal hemoglobin expression, to some extent, in a thalassemia mouse model.Citation19 The restoration of normal splicing by suppressing cryptic splicing, therefore, represents the very earliest manifestation of ASOs being used to modulate splicing for therapy. Even despite this early progress, clinical applications of this technology for the treatment of β-thalassemia have not materialized, likely as a result of the need to generate, validate, and trial specific ASOs for each type of mutation in patients, and the fact that ASO therapy only leads to a transient correction of splicing. As a result, most gene therapy research in the field has concentrated on viral-mediated transduction of β-globin into hematopoietic stem cells as a means of achieving durable and long-term correction of β-globin expression, with one patient achieving independence from transfusions, the usual treatment for β-thalassemia.Citation20
Figure 1 Schematic depiction of antisense-mediated splicing modulation approaches.
Abbreviations: APOB, apolipoprotein B100; ASOs, antisense oligonucleotides; LDL, low-density lipoprotein; mRNA, messenger RNA; STAT3, signal transducer and activator of transcription 3; USH1C, Usher syndrome type 1C gene.
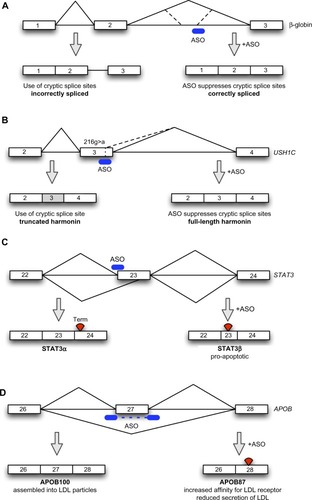
Even though in β-thalassemia, ASO-mediated correction is being side-lined in favor of other approaches, for other diseases targets that could be amenable to ASO-mediated cryptic splice site suppression continue to be identified. A recent example is Usher syndrome type 1C, characterized by congenital deafness, retinitis pigmentosa, and vestibular dysfunction (). This is caused by defects in the USH1C gene that encodes harmonin. Harmonin is an actin-binding scaffold protein that, together with myosin VIIa and cadherin 23, are required to ensure that the stereocilia in the inner ear cohere together.Citation21,Citation22 The USH1C216G>A mutation, found in all Louisiana Acadian cases of Usher syndrome type 1C, creates a cryptic 5′ splice site within exon 3 of the gene, which is used preferentially over the canonical 5′ splice site, and the creation of a frame-shift and premature truncation of the harmonin protein.Citation23 In a collaborative project, Lentz et al were able to show that an ASO targeted to the cryptic 5′ splice site was able to correct the splicing of USH1C transcript, restore harmonin expression, and rescue cochlear hair cells, vestibular function, and hearing for at least 6 months when given to an animal model of USH1C216G>A.Citation24
Alternative splicing
In humans, a myriad of proteins is responsible for most of the functions that form the basis of life. However, this array of different proteins is coded by a much (3–4-fold) smaller number of genes. This puzzled the field at first,Citation25 but can be explained by the occurrence of alternative splicing, a mechanism that allows the production of several proteins from the same gene by means of the exclusion, inclusion, or trimming of specific exons. In fact, 95% of human pre-mRNA is alternatively spliced.Citation26 Alternative splicing is responsible for generating proteins with very different, sometimes even antagonistic roles. Alternative splicing can also introduce stop codons that cause nonsense-mediated decay, and therefore, suppress the expression of the gene.Citation27 In healthy individuals, this flexibility contributes to the clockwork functioning of human biology. However, aberrant alternative splicing can also be the origin of disease, eg, a shift from an antiproliferative isoform to a proproliferative isoform can underlie tumor formation. From the therapeutic viewpoint, this provides researchers the opportunity to use ASOs as tools to knowingly shift alternative splicing to treat disease.
A first example involves lamin A, a nuclear envelope protein involved in the regulation of gene expression, nuclear stability, and chromatin structure that is encoded by the LMNA gene. A point mutation in this gene leads to a shift in alternative splicing,Citation28 and increased production of progerin, a truncated form of lamin A that is present in small amounts in normal aging, but which accumulates in high levels in HGPS. This leads to accelerated aging and shortened life span due to growth impairment, lipodystrophy, dermal and bone abnormalities, and cardiovascular alterations.Citation29 This shift in the standard splicing of the LMNA gene favoring the truncated progerin protein represents an ideal example of how the usually firmly kept balance can be disturbed by the presence of this cryptic splice site: the new protein is only 50 amino acids shorter than the original one, but lacks a vital target sequence for protein cleavage,Citation30 and this in turn leads to the deleterious effects experienced by HGPS patients. A phosphorodiamidate morpholino oligomer ASO masking the progerin splice site has been successfully used in fibroblast cultures from HGPS patients to restore normal splicing and correct nuclear abnormalities and mislocalization of other nuclear envelope proteins.Citation31 Following these advances, in a recent study on a murine model that recapitulates the main clinical signs of HGPS, vivo-morpholino ASOs were able to reverse many of the phenotypical alterations seen in these mice.Citation32
Signal transducer and activator of transcription 3 (STAT3) is involved in the activation of several oncogenic pathways. It is usually spliced to the isoform STAT3α, but an alternative acceptor site in exon 23 leads to the expression of isoform STAT3β, which lacks the C-terminal transactivation domain. Overexpression of STATβ can induce apoptosis and inhibit tumor growth. Zammarchi et al developed an ASO targeting a splicing enhancer site that was able to shift the balance of STAT3 expression to favor STAT3β expression ().Citation33 This approach has been tested in cell and animal models and has served a double purpose: on one hand it allowed a detailed study of the precise functions of STAT3β in its physiological environment. On the other hand, ASOs that shift splicing to the STAT3β isoform have antitumor applications.
Knockdown
Exon-skipping ASOs can also be used to knockdown the function of an undesired gene, by inducing exon skipping and creating mRNA isoforms that encode nonfunctional proteins or trigger nonsense-mediated decay of the mRNA,Citation34 to inhibit the expression of the undesired gene; or encode alternative isoforms with desired physiological or therapeutic functions that could modify or antagonize the effect of an undesired gene. The use of exon-skipping ASOs for the first purpose is not generally widespread except in the special case of zebrafish genetics.Citation35 Other methods, such as RNA interference,Citation36 ASOs that trigger RNase H-mediated degradation of mRNAs,Citation37 or translation inhibition,Citation38 are more generally used if simple knockdown is desired. An example of this is the knockdown of apolipoprotein B100 (APOB100) as a means of reducing low-density lipoprotein (LDL) secretion and cholesterol levels in familial hypercholesterolemia.Citation39 Both RNase H “gap-mer” ASOs and RNA interference have been used for this purpose, reducing LDL cholesterol in vivo.Citation40,Citation41 These simple knockdown approaches, however, have the undesired effect of knocking down the APOB48 isoform, and therefore reduce chylomicron secretion.Citation42 This will interfere with fat transport from the intestine and malabsorption of fat-soluble vitamins such as vitamin K.Citation43
Exon-skipping ASOs offer a distinct advantage over simple knockdown, as engineered alternative isoforms may possess their own therapeutic properties, inhibit the function of a normal isoform, and/or avoid side effects of knockdown of the normal isoform. Capitalizing on this advantage, exon-skipping exon 27 of the APOB gene using ASOs causes a reduction of LDL particle secretion and an increase in LDL affinity for the LDL receptor ().Citation44 These two effects combine to cause a powerful reduction of LDL cholesterol when the “APO-skip” ASOs are introduced into hypercholesterolemic mice transgenic for human APOB, validating the therapeutic concept.Citation45 Importantly, exon 27 skipping does not affect APOB48 expression, as the RNA editing necessary for APOB48 expression occurs in exon 26.Citation45 In this example, therefore, exon-skipping technology enables the reengineering of gene expression to produce a desired outcome with clear therapeutic advantages over other approaches to the same problem.
Reading frame correction
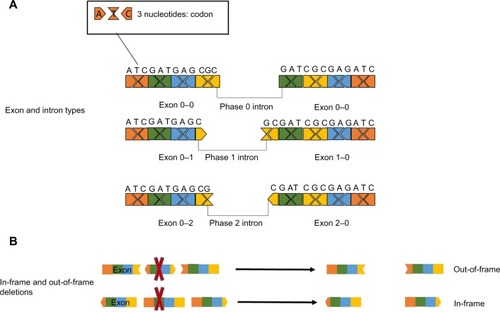
The coding sequence for proteins is dispersed over exons, and the location where introns interrupt the code can be in three phases in relation to the coding sequence: either between complete codons (phase 0 introns), interrupting after the first nucleotide of the codon (phase 1 introns), or interrupting after the second nucleotide of the codon (phase 2 introns). As this will apply to both the beginning and the end of each exon, each exon will have two jigsaw-like ends that will require a complementary exon before and after to maintain the open reading frame (ORF).Citation46 Therefore, a symmetrical exon can be deleted without interrupting the ORF, but others, if deleted, would cause a disruption in the ORF (). For most genes, the deletion of an exon would result in a nonfunctional protein regardless of whether the ORF is disrupted by the deletion, since the majority of proteins do not have redundant domains. However, there are some exceptions where deletions that maintain the ORF allow the production of internally deleted but partially functional proteins. This concept is best illustrated in the neuromuscular disorder DMD, an early onset, severely progressive muscle-wasting disorder.Citation47 In the majority of DMD patients, deletions or duplications of one or more exons cause a disruption of the ORF and the deficit of its encoded protein, dystrophin.Citation48 In a stark contrast to DMD, another disorder in which one or more exons are also deleted from the DMD gene, Becker muscular dystrophy, has a much milder progression. The difference is that in Becker patients, the deletions do not disrupt the ORF and a shorter, but partially functional dystrophin protein is produced.Citation48 The existence of Becker muscular dystrophy is the conceptual basis for the model of exon skipping: ASOs are used to “mask” specific exons to the splicing machinery to restore the ORF and partial dystrophin function for DMD patients.
Figure 2 Effect of exon deletions on the open reading frame.
From early proof of concept experiments to the latest clinical trials, the developments in the use of ASOs as a potential therapeutic agent in DMD have been fast: restoration of dystrophin has been achieved following intramuscularCitation49,Citation50 and systemicCitation4,Citation5,Citation51 injections of ASOs targeting DMD exon 51, and these and several other ASOs targeting other exons are progressing through clinical trials.Citation52
SMA, the most common genetic cause of death in infants under the age of 2 years, is caused by deletions and mutations of SMN1, and consequently low levels of SMN protein, a ubiquitously expressed protein involved in RNA processing.Citation53 Although a duplicate of SMN1, SMN2 also exists within the genome and also encodes SMN; it is usually inefficiently spliced due to minor differences in and around exon 7. As a result, only 20% of SMN2 mRNA transcripts include exon 7, which is necessary for the stability of SMN protein.Citation54 In healthy individuals, this is irrelevant, but in SMA patients, increasing the amount of exon 7 inclusion from SMN2 transcripts could allow the generation of sufficient amounts of SMN protein to ameliorate their disease. In fact, several studies using ASOs have already demonstrated in vitro and in vivo that this is possible,Citation6,Citation55–Citation58 and clinical trials are underway.
Toward clinical application
From the above, it is clear that antisense-mediated splicing modulation may offer a potential treatment for rare inherited diseases, and as such, can be seen as a platform approach. For DMD and SMA, the approach has made it to the clinical trial phase, whereas other genetic diseases are still in preclinical development. While each of these diseases will have their own quirks and challenges (eg, delivery to the target tissue and dosing regimens will vary between diseases), there will be common challenges and hurdles as well, since they all use the exon-skipping platform. Furthermore, there is no reason why people embarking on developing exon skipping for their disease of choice should have to repeat mistakes others have made in the past. Therefore, networking between scientists involved in this endeavor is warranted. The European Cooperation of Science and Technology (COST) provides funding for networking to overcome challenges such as these, and has funded the Action “Networking towards clinical application of antisense-mediated exon skipping for rare diseases” (BM1207, see http://www.cost.eu and http://www.exonskipping.eu).Citation59 Within this Action, regular workshops are organized to discuss common challenges, such as delivery to the target tissue, ASO chemistry and safety, and to align preclinical work. Furthermore, the Action focuses on the development of new regulatory models and has recently had a workshop on translational and regulatory challenges with representatives from patient organizations, academia, and regulatory agencies,Citation60 following a larger workshop organized by the Translational Research in Europe – Assessment and Treatment of Neuromuscular Diseases (TREAT-NMD) network and hosted by the European Medicines Agency that focused on the particulars of exon skipping for DMD.Citation61
One of the key factors for clinical development will be the harmonization of outcome measures. While the regulators will provide market approval only when clinically relevant benefit has been convincingly shown, biochemical outcome measures can provide additional information, eg, by confirming mechanism of action of ASOs. Lessons could be learned from the case of DMD: when the first clinical trials using ASOs were outlined, methods to measure dystrophin restoration relied heavily on standard clinical diagnostic techniques and academic protocols, and each research center used their own version of the methods.Citation4,Citation5,Citation49,Citation51,Citation52 The most commonly used method was immunofluorescent staining, but the large size and low expression level of the protein made accurate quantification technically challenging, especially when the amount of restored dystrophin is relatively small and it needs to be distinguished from preexisting dystrophin-positive revertant fibers and residual trace dystrophin expression.Citation52,Citation62 To overcome some of those difficulties, refinements developed, in particular using better software analysis.Citation63,Citation64 A group of laboratories from academia and industry have embarked on an effort to provide a data-driven reproducible methodology for dystrophin quantification that could be used in future trials. This group has revised methodologies, validated them among their laboratories, and proposed a standard operating procedure.Citation52,Citation65
Conclusion
ASO-mediated splicing modulation holds potential for treating rare genetic diseases as outlined in this review, and as corroborated by the advances made for DMD and SMA. International networks on exon skipping aim to help scientists developing this approach for other diseases benefit from the experience gained from the DMD field, to jointly overcome challenges and further this approach toward clinical application for as many rare genetic diseases as possible.
Acknowledgments
Authors gratefully acknowledge funding of the COST Action BM1207 by the Cooperation of Science and Technology (COST). Virginia Arechavala-Gomeza is funded by a Miguel Servet Fellowship (Spanish Health Institute Carlos III) and a Marie Curie Career Integration Grant (European Commission).
Disclosure
Dr Khoo discloses being co-inventor on UK Patent GB1117880.3. Dr Aartsma-Rus discloses being employed by the Leiden University Medical Center (LUMC) which has patents on exon skipping. Dr Aartsma-Rus is co-inventor on some of these patents and as such is entitled to a share of potential royalties. Dr Arechavala-Gomeza has nothing to declare.
References
- KaplanJCHamrounDThe 2014 version of the gene table of monogenic neuromuscular disorders (nuclear genome)Neuromuscul Disord201323121081111124446552
- BertrandATChikhaouiKYaouRBBonneGClinical and genetic heterogeneity in laminopathiesBiochem Soc Trans20113961687169222103508
- LewandowskaMAThe missing puzzle piece: splicing mutationsInt J Clin Exp Pathol20136122675268224294354
- CirakSArechavala-GomezaVGuglieriMExon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation studyLancet2011378979159560521784508
- GoemansNMTuliniusMvan den AkkerJTSystemic administration of PRO051 in Duchenne’s muscular dystrophyN Engl J Med2011364161513152221428760
- HuaYSahashiKHungGAntisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse modelGenes Dev201024151634164420624852
- ZanettaCNizzardoMSimoneCMolecular therapeutic strategies for spinal muscular atrophies: current and future clinical trialsClin Ther201436112814024360800
- YokotaTNakamuraANagataTExtensive and prolonged restoration of dystrophin expression with vivo-morpholino-mediated multiple exon skipping in dystrophic dogsNucleic Acid Ther201222530631522888777
- van Roon-MomWMAartsma-RusAOverview on applications of antisense-mediated exon skippingMethods Mol Biol2012867799622454056
- SalehAFArzumanovAAGaitMJOverview of alternative oligonucleotide chemistries for exon skippingMethods Mol Biol201286736537822454073
- DouglasAGWoodMJSplicing therapy for neuromuscular diseaseMol Cell Neurosci20135616918523631896
- FosterHPopplewellLDicksonGGenetic therapeutic approaches for Duchenne muscular dystrophyHum Gene Ther201223767668722647146
- Aartsma-RusAAntisense-mediated modulation of splicing: therapeutic implications for Duchenne muscular dystrophyRNA Biol20107445346120523110
- Arechavala-GomezaVAnthonyKMorganJMuntoniFAntisense oligonucleotide-mediated exon skipping for Duchenne muscular dystrophy: progress and challengesCurr Gene Ther201212315216022533380
- RocaXKrainerAREperonICPick one, but be quick: 5′ splice sites and the problems of too many choicesGenes Dev201327212914423348838
- WieringaBMeyerFReiserJWeissmannCUnusual splice sites revealed by mutagenic inactivation of an authentic splice site of the rabbit beta-globin geneNature1983301589538436296682
- BusslingerMMoschonasNFlavellRABeta + thalassemia: aberrant splicing results from a single point mutation in an intronCell1981272 Pt 12892986895866
- DominskiZKoleRRestoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotidesProc Natl Acad Sci U S A19939018867386778378346
- SvastiSSuwanmaneeTFucharoenSRNA repair restores hemoglobin expression in IVS2-654 thalassemic miceProc Natl Acad Sci U S A200910641205121019164558
- Cavazzana-CalvoMPayenENegreOTransfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemiaNature2010467731331832220844535
- BoedaBEl-AmraouiABahloulAMyosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundleEMBO J200221246689669912485990
- BahloulAMichelVHardelinJPCadherin-23, myosin VIIa and harmonin, encoded by Usher syndrome type I genes, form a ternary complex and interact with membrane phospholipidsHum Mol Genet201019183557356520639393
- LentzJSavasSNgSSAthasGDeiningerPKeatsBThe USH1C 216G–>A splice-site mutation results in a 35-base-pair deletionHum Genet2005116322522715578223
- LentzJJJodelkaFMHinrichAJRescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafnessNat Med201319334535023380860
- PennisiEWhy do humans have so few genes?Science200530957318015994526
- JohnsonJMCastleJGarrett-EngelePGenome-wide survey of human alternative pre-mRNA splicing with exon junction microarraysScience200330256532141214414684825
- StoilovPDaoudRNaylerOStammSHuman tra2-beta1 autoregulates its protein concentration by influencing alternative splicing of its pre-mRNAHum Mol Genet200413550952414709600
- ErikssonMBrownWTGordonLBRecurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndromeNature2003423693729329812714972
- HennekamRCHutchinson-Gilford progeria syndrome: review of the phenotypeAm J Med Genet A2006140232603262416838330
- GruenbaumYMargalitAGoldmanRDShumakerDKWilsonKLThe nuclear lamina comes of ageNat Rev Mol Cell Biol200561213115688064
- ScaffidiPMisteliTReversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndromeNat Med200511444044515750600
- OsorioFGNavarroCLCadinanosJSplicing-directed therapy in a new mouse model of human accelerated agingSci Transl Med20113106106ra107
- ZammarchiFdeSEBournazouEAntitumorigenic potential of STAT3 alternative splicing modulationProc Natl Acad Sci U S A201110843177791778422006329
- MaquatLENonsense-mediated mRNA decay: splicing, translation and mRNP dynamicsNat Rev Mol Cell Biol200452899915040442
- DraperBWMorcosPAKimmelCBInhibition of zebrafish fgf8 pre-mRNA splicing with morpholino oligos: a quantifiable method for gene knockdownGenesis200130315415611477696
- McManusMTSharpPAGene silencing in mammals by small interfering RNAsNat Rev Genet200231073774712360232
- GrunwellerAWyszkoEBieberBJahnelRErdmannVAKurreckJComparison of different antisense strategies in mammalian cells using locked nucleic acids, 2′-O-methyl RNA, phosphorothioates and small interfering RNANucleic Acids Res200331123185319312799446
- McCaffreyAPMeuseLKarimiMContagCHKayMAA potent and specific morpholino antisense inhibitor of hepatitis C translation in miceHepatology200338250350812883495
- MarksDThorogoodMNeilHAHumphriesSEA review on the diagnosis, natural history, and treatment of familial hypercholesterolaemiaAtherosclerosis2003168111412732381
- ZimmermannTSLeeACAkincARNAi-mediated gene silencing in non-human primatesNature2006441708911111416565705
- RaalFJSantosRDBlomDJMipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trialLancet20103759719998100620227758
- SoutschekJAkincABramlageBTherapeutic silencing of an endogenous gene by systemic administration of modified siRNAsNature2004432701417317815538359
- CuchelMMeagherEAdu ToitTHEfficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 studyLancet20133819860404623122768
- KhooBRocaXChewSLKrainerARAntisense oligonucleotide-induced alternative splicing of the APOB mRNA generates a novel isoform of APOBBMC Mol Biol20078317233885
- DistererPAl-ShawiREllmerichSExon skipping of hepatic APOB pre-mRNA with splice-switching oligonucleotides reduces LDL cholesterol in vivoMol Ther201321360260923319054
- KolkmanJAStemmerWPDirected evolution of proteins by exon shufflingNat Biotechnol200119542342811329010
- EagleMBaudouinSVChandlerCGiddingsDRBullockRBushbyKSurvival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilationNeuromuscul Disord2002121092692912467747
- MuntoniFTorelliSFerliniADystrophin and mutations: one gene, several proteins, multiple phenotypesLancet Neurol200321273174014636778
- KinaliMArechavala-GomezaVFengLLocal restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept studyLancet Neurol200981091892819713152
- van DeutekomJCJansonAAGinjaarIBLocal dystrophin restoration with antisense oligonucleotide PRO051N Engl J Med2007357262677268618160687
- MendellJRRodino-KlapacLRSahenkZEteplirsen for the treatment of Duchenne muscular dystrophyAnn Neurol201374563764723907995
- Aartsma-RusADystrophin analysis in clinical trialsJ Neuromuscul Dis201414153
- LefebvreSBurglenLReboulletSIdentification and characterization of a spinal muscular atrophy-determining geneCell19958011551657813012
- MonaniURLorsonCLParsonsDWA single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2Hum Mol Genet1999871177118310369862
- RigoFChunSJNorrisDAPharmacology of a central nervous system delivered 2’-o-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primatesJ Pharmacol Exp Ther20143501465524784568
- PassiniMABuJRichardsAMAntisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophySci Transl Med201137272ra18
- HuaYVickersTAOkunolaHLBennettCFKrainerARAntisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic miceAm J Hum Genet200882483484818371932
- HuaYVickersTABakerBFBennettCFKrainerAREnhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exonPLoS Biol200754e7317355180
- Aartsma-RusAAntisense-mediated exon skipping: networking to meet opportunities and to overcome challengesNucleic Acid Ther20142411324506778
- Aartsma-RusAFerliniAGoemansNTranslational and regulatory challenges for exon skipping therapiesHum Gene Ther2014251088589225184444
- MuntoniFMeeting Steering Committee and TREAT-NMD NetworkThe development of antisense oligonucleotide therapies for Duchenne muscular dystrophy: report on a TREAT-NMD workshop hosted by the European Medicines Agency (EMA), on September 25th 2009Neuromuscul Disord201020535536220347306
- Arechavala-GomezaVKinaliMFengLRevertant fibres and dystrophin traces in Duchenne muscular dystrophy: implication for clinical trialsNeuromuscul Disord201020529530120395141
- Arechavala-GomezaVKinaliMFengLImmunohistological intensity measurements as a tool to assess sarcolemma-associated protein expressionNeuropathol Appl Neurobiol201036426527420002311
- TaylorLEKaminohYJRodeschCKFlaniganKMQuantification of dystrophin immunofluorescence in dystrophinopathy muscle specimensNeuropathol Appl Neurobiol201238659160122243335
- AnthonyKArechavala-GomezaVTaylorLEDystrophin quantification: Biological and translational researcher implicationsNeurology Epub20141029