
Abstract
Background
Beta-thalassemia is one of the most common genetic disorders in Thailand. Clinical phenotype ranges from silent carrier to clinically manifested conditions including severe beta-thalassemia major and mild beta-thalassemia intermedia.
Objective
This study aimed to characterize the spectrum of beta-globin gene mutations in pediatric patients who were followed-up in Phramongkutklao Hospital.
Patients and methods
Eighty unrelated beta-thalassemia patients were enrolled in this study including 57 with beta-thalassemia/hemoglobin E, eight with homozygous beta-thalassemia, and 15 with heterozygous beta-thalassemia. Mutation analysis was performed by multiplex amplification refractory mutation system (M-ARMS), direct DNA sequencing of beta-globin gene, and gap polymerase chain reaction for 3.4 kb deletion detection, respectively.
Results
A total of 13 different beta-thalassemia mutations were identified among 88 alleles. The most common mutation was codon 41/42 (-TCTT) (37.5%), followed by codon 17 (A>T) (26.1%), IVS-I-5 (G>C) (8%), IVS-II-654 (C>T) (6.8%), IVS-I-1 (G>T) (4.5%), and codon 71/72 (+A) (2.3%), and all these six common mutations (85.2%) were detected by M-ARMS. Six uncommon mutations (10.2%) were identified by DNA sequencing including 4.5% for codon 35 (C>A) and 1.1% initiation codon mutation (ATG>AGG), codon 15 (G>A), codon 19 (A>G), codon 27/28 (+C), and codon 123/124/125 (-ACCCCACC), respectively. The 3.4 kb deletion was detected at 4.5%. The most common genotype of beta-thalassemia major patients was codon 41/42 (-TCTT)/codon 26 (G>A) or betaE accounting for 40%.
Conclusion
All of the beta-thalassemia alleles have been characterized by a combination of techniques including M-ARMS, DNA sequencing, and gap polymerase chain reaction for 3.4 kb deletion detection. Thirteen mutations account for 100% of the beta-thalassemia genes among the pediatric patients in our study.
Introduction
Thalassemia is the most common inherited blood disorder in Southeast Asia and is caused by reduced or absent synthesis of the globin chains of hemoglobin (Hb) leading to imbalance of the globin chains.Citation1,Citation2 Beta-thalassemia is one of the major types of thalassemia and is caused by a mutation in the beta-globin gene (HBB) on chromosome 11. The clinical and hematological spectrum of beta-thalassemia ranges from silent carrier to clinically manifested conditions including severe transfusion dependent beta-thalassemia major and beta-thalassemia intermedia (TI).Citation3,Citation4 In Thailand, both beta-thalassemia and hemoglobin E (HbE) represent one of the most common forms distributed in all regions especially in Northeast Thailand. Most of the severe beta-thalassemia results from the interaction of beta-thalassemia and HbE.Citation5,Citation6
The molecular basis of thalassemia has been studied worldwide. More than 300 different beta-globin gene mutations have been characterized. Most of the beta-thalassemia mutations are caused by point mutations, small deletions or insertions within the coding regions and the exon-intron junctions. The types of the mutation are typically ethnic specific.Citation7–Citation9 In Thailand, the prevalence of beta-thalassemia carriers varies from 3%–9%.Citation10 To date, more than 30 different mutations have been identified.Citation11–Citation13 The heterogeneity of the mutations makes it difficult to identify the mutation in some beta-thalassemia patients. Various DNA analysis techniques such as dot blot analysis, reverse dot blot, allele specific amplification using amplification refractory mutation system (ARMS) or direct DNA sequencing have been widely used to identify beta-globin gene mutations.Citation14–Citation17
This study aimed to characterize the beta-globin gene mutations in 80 pediatric patients who carry beta-thalassemia mutations and were followed-up in Phramongkutklao Hospital, a tertiary care center for thalassemia patients from all regions, especially central Thailand.
Patients and methods
Patient selection
Eighty unrelated beta-thalassemia patients who attended the Hematology Clinic at the Department of Pediatrics, Phramongkutklao Hospital, Bangkok, Thailand, from January 2013 to December 2013 were enrolled in our study. The study protocol was approved by the Institutional Review Board of Phramongkutklao Hospital, Phramongkutklao College of Medicine, Thailand. Sixty-five patients had clinically manifested beta-thalassemia including 57 with beta-thalassemia/HbE and eight with homozygous or compound heterozygous beta-thalassemia. Fifteen patients had heterozygous beta-thalassemia. All patients were diagnosed at 18 years of age or less. Patients with homozygous beta-thalassemia and beta-thalassemia/HbE were clinically classified into severe transfusion dependent thalassemia major and mild TI based on criteria such as age at presentation, average Hb level at the steady state and transfusion frequency history, as previously described.Citation18
Mutation analysis
After informed consent was obtained, a total of 80 peripheral blood ethylenediaminetetraacetic acid (EDTA) samples from all individuals were collected. Genomic DNA was extracted from peripheral blood lymphocytes using an AxyPrep™ blood genomic DNA miniprep kit according to manufacturer’s protocol. The beta-globin gene mutations were first characterized using two sets of allele specific polymerase chain reaction (PCR) or multiplex amplification refractory mutation system (M-ARMS) to detect seven common mutations in Chinese and Southeast Asian populations including codon 41/42 (-TCTT), codon 17 (A>T), nucleotide -28 (A>G), IVS-II-654 (C>T), codon 71/72 (+A), IVS-I-1 (G>T) and IVS-I-5 (G>C) as previously described.Citation15,Citation17 These mutations have been found to be responsible for 80%–90% of beta-thalassemia alleles in this region.Citation19,Citation20 Unknown beta-thalassemia genes were further characterized by direct DNA sequencing of all coding regions and exon-intron boundaries to detect uncommon point mutations and small rearrangements in the beta-globin gene according to protocols previously described elsewhere.Citation16 Beta-thalassemia alleles that remained uncharacterized by the above M-ARMS and DNA sequencing methods were subsequently screened by gap-PCR to detect 3.4 kb deletion of the entire beta-globin gene, previously reported in Thai populations.Citation21
Results
A total of 88 beta-thalassemia alleles from eight homozygous or compound heterozygous beta-thalassemia patients, 57 beta-thalassemia/HbE patients, and 15 heterozygous beta-thalassemia individuals were included in our study. All 80 subjects were from unrelated families and 93.8% (75/80) of subjects lived in Bangkok and other provinces in central Thailand. Of these 65 patients who had clinically manifested beta-thalassemia, 92.3% (60/65) and 7.7% (5/65) of patients presented with beta-thalassemia major and TI, respectively. In the 60 beta-thalassemia major patients, 86.7% (52/60) of patients had beta-thalassemia/HbE whereas only 13.3% (8/60) of patients had homozygous or compound heterozygous beta-thalassemia. Alternatively, all patients with homozygous or compound heterozygous beta-thalassemia and 91.2% (52/57) of beta-thalassemia/HbE patients in this study presented with severe transfusion dependent beta-thalassemia major. Concerning genotype of beta-thalassemia major patients, the most common genotype was codon 41/42 (-TCTT)/codon 26 (G>A) or betaE accounting for 40%, and the second most common was codon 17 (A>T)/betaE accounting for 18.3% of all beta-thalassemia major genotypes ( and ).
Figure 1 Type of genotype in beta-thalassemia patients.
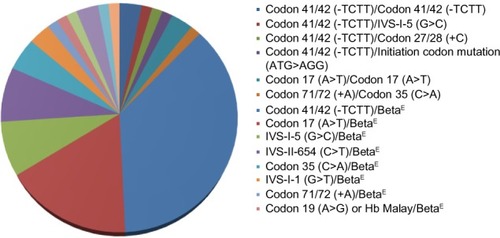
Table 1 Genotype of 65 clinically manifested beta-thalassemia patients
To elucidate the molecular basis of these 88 beta-thalassemia alleles, we first characterized each patient’s DNA by two sets of M-ARMS. Seventy-five alleles (85.2%) were identified by this method including 33 alleles (37.5%) of codon 41/42 (-TCTT), 23 alleles (26.1%) of codon 17 (A>T), seven alleles (8%) of IVS-I-5 (G>C), six alleles (6.8%) of IVS-II-654 (C>T), four alleles (4.5%) of IVS-I-1 (G>T) and two alleles (2.3%) of codon 71/72 (+A) (). Interestingly, nucleotide -28 (A>G) was not identified in our study. In 13 alleles (14.8%), neither set revealed beta-thalassemia mutation. Thus, direct DNA sequencing of all three coding exons and flanking exon-intron junctions in the beta-globin gene was the next step to characterize these 13 alleles. Six uncommon mutations were detected in nine alleles (10.2%) including four alleles (4.5%) of codon 35 (C>A) and one allele (1.1%) for initiation codon mutation (ATG>AGG), codon 15 (G>A), codon 19 (A>G), codon 27/28 (+C) and codon 123/124/125 (-ACCCCACC), respectively (). The remaining alleles, uncharacterized by M-ARMS and DNA sequencing, were further identified by gap-PCR to detect 3.4 kb deletion and this deletion was found in all four remaining alleles (4.5%).
Figure 2 Six uncommon mutations identified by direct DNA sequencing.
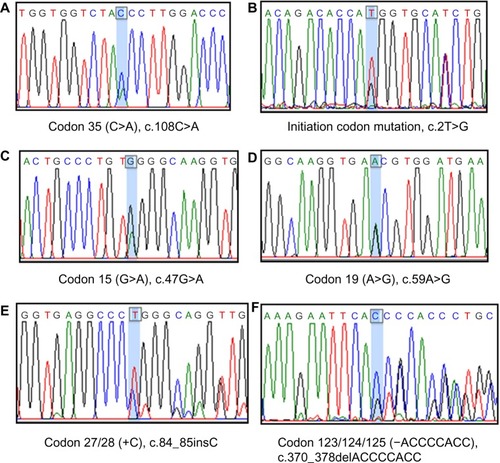
Table 2 The frequency of beta-thalassemia mutations in 88 alleles
In all, 100% of our 88 beta-thalassemia alleles were characterized by a combination of these techniques including two sets of M-ARMS, direct DNA sequencing and 3.4 kb deletion detection gap-PCR. Excluding the betaE-globin gene, 13 different beta-thalassemia mutations were encountered in the present study. The 4 bp deletion (-TCTT) in codons 41/42 was the most common mutation identified in our study and accounted for 37.5% of the alleles. The codon 17 (A>T) was the second most common and accounted for 26.1% of the alleles. Together, these two common mutations accounted for more than half (63.6%) of affected alleles. All other common mutations except nucleotide -28 (A>G) accounted for 21.6% of the alleles.
Discussion
This study provided useful information regarding the frequency distribution of beta-thalassemia mutations among pediatric patients especially in central Thailand since more than 90% of patients lived in Bangkok and other provinces in this region. In all, 92% of children who had clinically manifested beta-thalassemia had the beta-thalassemia major phenotype and more than 85% of pediatric patients with beta-thalassemia major had compound heterozygous beta- thalassemia and HbE, which is highly prevalent in Thailand.Citation5,Citation6,Citation8,Citation10 The frequency of the betaE-globin gene was not included in the analysis of beta-thalassemia mutation frequency since HbE can be easily detected by Hb electrophoresis.
Several methods can determine beta-thalassemia mutations.Citation14–Citation17 This study revealed that M-ARMS detected seven common mutations in Chinese and Southeast Asian populations and was able to detect 85.2% of the alleles as in previous reports.Citation19,Citation20 Since M-ARMS can detect only a given set of mutations specific to the primers employed, direct DNA sequencing is the next step to identify various point mutations and small rearrangements in the beta-globin gene. Six less frequent mutations were identified in 10.2% of the alleles. The disadvantage of DNA sequencing is that large deletions of the gene are undetectable. Thus, gap-PCR is the final step to detect 3.4 kb deletion, previously reported in Thai populations.Citation21 A combination of these techniques could identify beta-thalassemia mutations in all 88 alleles (100%) of pediatric patients in our study.
Excluding the betaE-globin gene, 13 different beta-thalassemia mutations were detected in 80 unrelated patients. The two most common mutations detected were codon 41/42 (-TCTT) and codon 17 (A>T), accounting for 37.5% and 26.1% of the affected alleles, respectively. All other common mutations detected by M-ARMS accounted for 21.6% of the alleles. In comparison with the previous study of beta-thalassemia mutations in Thailand, this revealed that the frequency of beta-globin gene mutations identified in our study was different. First, the nucleotide -28 (A>G) was not identified in our study. The nucleotide -28 (A>G) is a beta+-thalassemia mutation causing the mild beta-thalassemia phenotype; thus, this mutation has been observed only in TI patients.Citation22 This present study demonstrated that TI was the minority group (less than 10%) among pediatric patients with symptomatic beta-thalassemia. This may explain why this mutation was not observed in our study. Second, the frequency of other common mutations including codon 41/42 (-TCTT), codon 17 (A>T), IVS-II-654 (C>T), codon 71/72 (+A), IVS-I-1 (G>T) and IVS-I-5 (G>C) in our study was similar to other studies in central Thailand.Citation8,Citation10 However, allele frequency of this present study was different from studies in other parts of Thailand. Codon 41/42 (-TCTT) was the most common mutation identified in our population, who mainly lived in central Thailand. This frameshift mutation is also the most common mutation found in the other parts of Thailand, People’s Republic of China, and Southeast Asia.Citation20 While codon 17 (A>T) was the second most common mutated allele in central, northern, and northeast Thailand, IVS-I-5 (G>C) was the second most common in southern Thailand.Citation23 IVS-I-5 (G>C) was previously reported as the most common mutation among Thai Muslim patients in southern Thailand.Citation24 Additionally, codon 71/72 (+A) accounted for 2.3% in the present study while this mutation accounted for 6% in northCitation16 and 13.1% in northeastCitation13 Thailand indicating a higher frequency of this mutation in this area.
Six uncommon mutations including codon 35 (C>A), codon 15 (G>A), codon 19 (A>G) or Hb Malay, codon 27/28 (+C), initiation codon mutation (ATG>AGG) and codon 123/124/125 (-ACCCCACC) were identified by direct DNA sequencing. Although no new mutation was detected, two rare mutations in Thailand, initiation codon mutation and 8 bp deletion in exon 3, were found in our study. Initiation codon mutation (ATG>AGG) was first described in a Chinese beta-thalassemia patient in 1990.Citation25 In Thailand, this mutation was reported once in 2005 in two siblings with beta-thalassemia/HbE.Citation26 Although this mutation has seriously affected beta-globin chain synthesis and most likely causes beta0- thalassemia phenotype, all three reported patients had the mild phenotype, which can be explained by co-inheritance with heterozygous 3.7 kb deletion alpha-thal-2 genotype among Chinese patient and the association with C>T polymorphism at -158(G)gamma-globin gene in the two Thai siblings. Our patient’s phenotype was transfusion dependent beta-thalassemia major, diagnosed at 9 months of age, whereas the genotype was compound heterozygous between codon 41/42 (-TCTT) and initiation codon mutation. Both mutated alleles cause the beta0-thalassemia phenotype. Eight-base deletion in exon 3 or codon 123/124/125 (-ACCCCACC) was first characterized in northeast Thai children with the severe beta-thalassemia/HbE phenotype.Citation27 This deletion results in the synthesis of 135 amino acids betaX chain (beta-Khon Kaen), which are highly unstable and degraded soon after translation. Extensive alteration of amino acids at the codon 123 to 131 caused dominant inclusion body beta-thalassemia traits as in our patient.
Beta-thalassemia/HbE is a major thalassemia problem in Thailand and can be associated with various clinical phenotypes ranging from mild thalassemia intermedia to severe transfusion dependent thalassemia major.Citation5,Citation6,Citation8,Citation10 As in our study, 85% of beta-thalassemia major patients and 100% of TI patients had compound heterozygous beta-thalassemia and HbE. In beta-thalassemia major, the most common encountered genotype was codon 41/42 (-TCTT)/betaE (40%), followed by codon 17 (A>T)/betaE (18.3%), IVS-I-5 (G>C)/betaE (8.3%) and IVS-II-654 (C>T)/betaE (8.3%), which made up for 75% of all detected genotypes. Excluding the betaE-thalassemia mutation, all beta-thalassemia mutations in our symptomatic patients were classified as beta0 mutation except for codon 19 (A>G) or Hb Malay, classified as beta+ mutation and identified only in one patient.Citation28
Interestingly, our patient with codon 19 (A>G)/betaE had transfusion dependent thalassemia major manifestation instead of mild TI. On the other hand, compound heterozygotes among four beta0 mutations including codon 41/42 (-TCTT), codon 17 (A>T), 3.4 kb deletion, IVSI-1 (G>T) and betaE genotype manifested as TI instead of beta-thalassemia major phenotype. This phenomenon can be explained by several genetic and nongenetic factors, which may play roles in determining the variability of the disease.Citation5,Citation22,Citation29,Citation30 However, the additional genetic analysis (genetic modifiers) was not performed in our study.
The concept of pre-implantation genetic diagnosis is to allow transfer of embryos to the uterus in assisted reproduction procedures. This technique is rapid and suitable as a noninvasive clinical tool for identifying genetic disorders for the purpose of reducing selective miscarriages such as thalassemia major.Citation31,Citation32 Our study revealed different beta-thalassemia mutations maybe clinically applied to preimplantation genetic protocol, permitting molecular genetic analysis to amplify a specific region on the beta-globin gene for a couple.
In conclusion, the present study demonstrates the heterogeneity of molecular defects causing beta-thalassemia in Thai children. All of the beta-thalassemia alleles have been characterized by a combination of techniques including M-ARMS, direct DNA sequencing, and gap-PCR for 3.4 kb deletion detection. Thirteen mutations accounted for 100% of the beta-thalassemia genes in our study. The frequency obtained should represent the frequency of beta-globin gene mutations among pediatric patients, who mainly lived in central Thailand.
Acknowledgments
This study was approved by and received funding from the Phramongkutklao College of Medicine.
Disclosure
The authors declare no conflicts of interest in this work.
References
- WeatherallDJThe thalassemia syndromesTex Rep Biol Med1980403233337034274
- VichinskyEPChanging patterns of thalassemia worldwideAnn N Y Acad Sci20051054182416339647
- CaoAGalanelloRBeta-thalassemiaGenet Med2010122617620098328
- RundDRachmilewitzEBeta-thalassemiaN Engl J Med2005353111135114616162884
- FucharoenSWinichagoonPHaemoglobinopathies in southeast AsiaIndian J Med Res201113449850622089614
- FucharoenSKetvichitPPootrakulPSiritanaratkulNPiankijagumAWasiPClinical manifestation of beta-thalassemia/hemoglobin E diseaseJ Pediatr Hematol Oncol200022655255711132229
- Akhavan-NiakiHDerakhshandeh-PeykarPBanihashemiAA comprehensive molecular characterization of beta thalassemia in a highly heterogeneous populationBlood Cells Mol Dis2011471293221493114
- FucharoenSWinichagoonPHemoglobinopathies in Southeast Asia: molecular biology and clinical medicineHemoglobin19972142993199255610
- GiardineBvan BaalSKaimakisPHbVar database of human hemoglobin variants and thalassemia mutations: 2007 updateHum Mutat200728220617221864
- WasiPPootrakulSPootrakulPPravatmuangPWinichagoonPFucharoenSThalassemia in ThailandAnn N Y Acad Sci19803443523636156628
- FukumakiYFucharoenSFucharoenGMolecular heterogeneity of beta-thalassemia in ThailandSoutheast Asian J Trop Med Public Health199223Suppl 214211363706
- TheinSLWinichagoonPHeskethCThe molecular basis of beta-thalassemia in Thailand: application to prenatal diagnosisAm J Hum Genet19904733693752393018
- FucharoenSFucharoenGSriroongruengWMolecular basis of beta-thalassemia in Thailand: analysis of beta-thalassemia mutations using the polymerase chain reactionHum Genet198984141462606476
- MirasenaSShimbhuDSanguansermsriMSanguansermsriTDetection of beta-thalassemia mutations using a multiplex amplification refractory mutation system assayHemoglobin200832440340918654891
- BhardwajUZhangYHLoreyFMcCabeLLMcCabeERMolecular genetic confirmatory testing from newborn screening samples for the common African-American, Asian Indian, Southeast Asian, and Chinese beta-thalassemia mutationsAm J Hematol200578424925515795925
- SirichotiyakulSSaetungRSanguansermsriTAnalysis of beta-thalassemia mutations in northern Thailand using an automated fluorescence DNA sequencing techniqueHemoglobin2003272899512779270
- FucharoenSFucharoenGRatanasiriTJetsrisuparbAFukumakiYA simple non radioactive method for detecting beta-thalassemia/hbe disease: application to prenatal diagnosisSoutheast Asian J Trop Med Public Health199526Suppl 12782818629124
- HoPJHallGWLuoLYWeatherallDJTheinSLBeta-thalassaemia intermedia: is it possible consistently to predict phenotype from genotype?Br J Haematol1998100170789450794
- OldJMKhanSNVermaIA multi-center study in order to further define the molecular basis of beta-thalassemia in Thailand, Pakistan, Sri Lanka, Mauritius, Syria, and India, and to develop a simple molecular diagnostic strategy by amplification refractory mutation system-polymerase chain reactionHemoglobin200125439740711791873
- KazazianHHJrDowlingCEWaberPGHuangSLoWHThe spectrum of beta-thalassemia genes in China and Southeast AsiaBlood19866849649662875755
- SanguansermsriTPapeMLaigMHundrieserJFlatzGBeta zero-thalassemia in a Thai family is caused by a 3.4 kb deletion including the entire beta-globin geneHemoglobin19901421571682272839
- NuntakarnLFucharoenSFucharoenGSanchaisuriyaKJetsrisuparbAWiangnonSMolecular, hematological and clinical aspects of thalassemia major and thalassemia intermedia associated with Hb E-beta-thalassemia in Northeast ThailandBlood Cells Mol Dis2009421323518951049
- NopparatanaCPanichVSaechanVThe spectrum of beta-thalassemia mutations in southern ThailandSoutheast Asian J Trop Med Public Health199526Suppl 12292348629112
- LaosombatVNopparatanaCWongchanchailertMWiriyasateinkulAMolecular basis of beta-thalassemia in Thai Muslim patients in the the south of ThailandSoutheast Asian J Trop Med Public Health199728Suppl 31041059640609
- LamVMXieSSTamJWWooYKGuYLLiAMA new single nucleotide change at the initiation codon (ATG—AGG) identified in amplified genomic DNA of a Chinese beta-thalassemic patientBlood1990755120712082306523
- ViprakasitVChinchangWSuwantholLTanphaichitrVSCommon origin of a rare beta-globin initiation codon mutation (ATG–>AGG) in Asians. Clin Lab HaematolDec2005276409415
- FucharoenGFuchareonSJetsrisuparbAFukumakiYEight-base deletion in exon 3 of the beta-globin gene produced a novel variant (beta khon kaen) with an inclusion body beta-thalassemia traitBlood19917825375392070092
- YangKGKutlarFGeorgeEMolecular characterization of beta-globin gene mutations in Malay patients with Hb E-beta-thalassaemia and thalassaemia majorBr J Haematol198972173802736244
- OlivieriNFPakbazZVichinskyEHbE/beta-thalassemia: basis of marked clinical diversityHematol Oncol Clin North Am20102461055107021075280
- RundDFucharoenSGenetic modifiers in hemoglobinopathiesCur Mol Med200887600608
- NasriNWJamalARAbdullahNCRaziZRMokhtarNMPreimplantation genetic diagnosis for beta-thalassemia using single-cell DNA analysis for codons 17 and 26 of beta-globin geneArch Med Res20094011919064120
- HungCCChenSULinSYPreimplantation genetic diagnosis of beta-thalassemia using real-time polymerase chain reaction with fluorescence resonance energy transfer hybridization probesAnal Biochem20104001697720035706