
Abstract
Inflammatory bowel disease (IBD), namely Ulcerative Colitis (UC) and Crohn’s Disease (CD), is believed to be due to a dysregulation of the innate immune response. The complexity of the immune cascade offers both a challenge and an opportunity to researchers seeking out new treatments for IBD, as various points along the inflammatory pathways can be targeted for interruption. Sphinogosine-1-phosphate (S1P) is a phospholipid molecule with wide ranging biological effects caused by binding five known S1P receptor subtypes. Ozanimod is a small molecule drug that selectively targets S1P receptors 1 and 5 which play a crucial role in lymphocyte trafficking. In clinical trials for both UC and CD, it has been shown to induce a reversible lymphopenia which correlates with response to therapy. Reported adverse events include infection, anemia, and elevated liver enzymes. Rare instances of bradycardia, heart block, and macular edema were also reported. As a newly available therapy approved for UC patients, we aim to summarize ozanimod’s novel mechanism of action, pre-clinical and clinical trial results, and to give context to this newly available drug that gastroenterologists may utilize in their treatment algorithm.
Introduction
Inflammatory bowel disease (IBD), mainly divided between Crohn’s disease (CD) and ulcerative colitis (UC), is a chronic inflammatory disorder of the gastrointestinal tract. Though the search for a specific cause for IBD continues, much progress has been made in defining the abnormal pathophysiology, and associated genetic abnormalities, underlying the spontaneous and chronic inflammation that characterizes these disorders. Broadly, current disease models regard IBD as an inappropriate overexpression and impaired downregulation of the body’s inflammatory defense response.Citation1 For many patients, this constitutive activation of the body’s immune system begins with a compromised gut mucosal barrier exposing and eliciting an immune response to native bacterial components. It is notable that some of the over 200 single nucleotide polymorphisms identified using genome-wide association studies in the IBD population are found in those genes that are implicated in the body’s identification and response to pathogens.Citation2–4
In addition to this expanded understanding of the root causes of IBD, advances in identifying and targeting the chemical and cellular pathways of inflammation have led to a proliferation of treatment options for IBD patients over the past twenty years. Consistent with these mechanisms, medications that suppress or modify immune activity form the core of effective treatment. Immune therapy began with non-targeted immune suppressants such as corticosteroids, thiopurines (mercaptopurine and azathioprine), and methotrexate, but these were plagued with adverse drug reactions and need for close monitoring. In 1998, when infliximab was approved for the treatment of CD, the era of more targeted “biological therapies” began a revolution in the management of IBD. Infliximab, a monoclonal antibody against the proinflammatory cytokine tumor necrosis factor alpha (TNF-α) was the first targeted therapy approved for the treatment of IBD and unique in that it was produced by living cell lines, rather than the traditional chemical processes of small molecule/drug production.Citation5 Infliximab was followed by other anti-TNF-α monoclonal antibodies including adalimumab, certolizumab, and golimumab. Given the favorable outcomes of the early biologics, new generation biologics with unique mechanisms of action have been developed after the initial TNF-α inhibitor class. Natalizumab and vedolizumab, target the α4β7 integrin on white blood cell surfaces, inhibiting their migration to target tissues.Citation6 Ustekinumab targets the p40 subunit of the proinflammatory cytokine IL12/23Citation7 and Risankizumab even more specifically targets the p40 subunit of IL23.Citation8 The rapid proliferation of biologic drugs suggested that the future of IBD management would be based on monoclonal antibody therapy. However, despite the revolutionary nature of these drugs, efficacy and durability of treatment was not universal. Additionally, the large protein structure of biologics necessitated either parenteral or subcutaneous routes of administration. Hence, there was a gap for safe and effective oral therapies for IBD. The development and approval of oral therapies targeting the JAK-STAT pathway was the first sign of a renaissance in “small molecule” therapies. Tofacitinib and upadacitinib have provided further treatment options for UC, but significant infectious, hematologic, and cardiac risks have mandated their use to second-line therapy after failure of TNF-α inhibitors.Citation9 The next FDA approved small molecule therapy with a new mechanism of action via S1P modulation was ozanimod. The aim of the following review is to highlight the mechanism, clinical development, recent approval, and practical application of ozanimod.
Discussion
Sphingosine-1-Phosphate and It’s Receptors
Sphingosine-1-phosphate (S1P) is a membrane-derived phospholipid molecule. Sphingomyelin, a major component in the mammalian cell membrane, is first broken down to ceramide and phosphorylcholine via acid sphingomyelinase. Ceramide is then converted to sphingosine and a fatty acid residue chain via ceramidase and sphingosine is then phosphorylated by sphingosine kinase to yield S1PCitation10 ().Citation11 S1P, found mainly in plasma and lymph, is predominantly produced by erythrocytes,Citation12 vascular endothelial cells,Citation13 and lymphatic endothelial cells.Citation14
Figure 1 S1P Synthesis. Adapted from Nitric Oxide Synthesis 1, by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates.Citation11
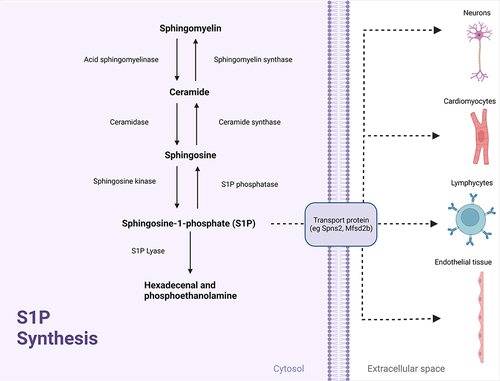
After being synthesized intracellularly, S1P is transferred to the extracellular space via specialized S1P transporter channels. It is in the extracellular space where S1P exerts its biologic effect by binding to an S1P receptor on the same cell or neighboring cells, in an autocrine or paracrine fashion, respectively.Citation15,Citation16 S1P receptors are transmembrane proteins, which when bound by S1P activate a downstream cascade of molecular reactions affecting nuclear transcription and other vital cellular processes across a broad array of cell types. There are currently 5 known subtypes of the S1P receptor, known simply as S1P1-5.Citation17
S1P1 is expressed on endothelial surfaces, smooth muscle cells, cardiac tissue, lymphocytes, and neurons, affecting vascular development and permeability, vascular tone and blood pressure, heart rate, B and T lymphocyte migration, and neuronal migration and function, respectively. S1P2 is also expressed on endothelial surfaces and cardiac tissue. It is also thought to play a role in the formation of pulmonary fibrosis via lung fibroblast stimulation. S1P3 is similar to S1P2 in that it is also expressed on endothelial surfaces, smooth muscle cells, cardiac tissue, and lung fibroblasts. It also shares characteristics of S1P1 through its effect on neuronal migration and function. S1P4 has mainly been found to be active on lymphocytes and dendritic cells while S1P5 is expressed on natural killer cells and oligodendrocytes.Citation18,Citation19 Given the currently accepted IBD model of immune system dysregulation,Citation1 the interaction between S1P and lymphocyte receptors appeared to offer a potential mechanism by which to influence immune activity through modulation of B and T lymphocyte migration to sites of inflammation.
The S1P Gradient and Immune Regulation
The synthesis of S1P and its concentration in various tissue types is a result of balanced synthesis and degradation via a host of metabolically active enzymes.Citation20 Early research has indicated that S1P is in high concentration in blood and lymph and, conversely, in low concentration intracellularly and in interstitial spaces including immune organs such as the spleen, thymus, and lymph nodes.Citation21 This differential in S1P concentrations across different tissue types has become known as the S1P gradient which plays a key role in the trafficking of lymphocytes and other leukocytes from immune organs to sites of inflammation, thus affecting the overall immune response.
How is this S1P gradient created and maintained? It was originally thought that platelets in the blood stream were responsible for the high plasma levels of S1P because they lack the S1P degrading enzyme S1P lyaseCitation22 and express the Mfsd2b transporter protein needed to shuttle S1P from inside the erythrocyte, across the lipid membrane, and into the extracellular environment.Citation23 However, this theory was rejected by a study in which mice that lacked platelets still maintained baseline levels of plasma S1P.Citation12 Erythrocytes were then analyzed as the cells possibly responsible for plasma levels of S1P because, they too lack multiple S1P degrading enzymesCitation24 and express the Mfsd2b transport protein needed by S1P to traverse the lipid membrane.Citation23 This theory was confirmed in rodent studies in which plasma S1P levels of sphingosine kinase deficient mice were restored after introduction of wild-type erythrocytes.Citation12 In lymph, endothelial cells which express the transport protein Spns2 are believed to be responsible for producing and maintaining the S1P gradient. This theory is supported by a study which demonstrated that endothelial cells that lacked Spns2 had approximately 60% less circulating S1P in the surrounding lymph.Citation25
The current model of understanding lymphocyte and other leukocyte migration throughout their life cycle includes this S1P gradient and the ability for S1P to bind S1P1. From their initial maturation in primary lymphoid tissues, lymphocytes move on to peripheral lymph nodes, and, finally, to sites of immune response where they exert their biological effect. T cells in the thymus and T and B cells in the spleen highly express S1P1 on their cell surfaces but due to the low interstitial level of S1P, these receptors do not have substrate to bind with. For S1P1 possessing lymphocytes to bind S1P, the cells are chemically drawn via a chemotactic effect into the bloodstream where the S1P concentration is high.Citation26 Although postulated,Citation27,Citation28 this mechanism has not been fully established as a mode for B cells migrating from bone marrow where they mature.
After crossing into the bloodstream from primary lymphoid organs, the binding of S1P to S1P1 causes the receptor to be internalized and degraded.Citation29 The receptor components are then recycled to produce a new receptor that can be swiftly sent back to the cell surface. Lymphocytes then travel through the blood stream and, via a complex “migration” mechanism,Citation30 enter the peripheral lymph nodes where they undergo immune maturation. Inside the lymph nodes where S1P concentrations are low, the expression of S1P1 on the cell surface increases once again.Citation26 Lymphocytes will then remain in the lymph nodes until they are stimulated to exit toward sites of inflammation. Critically, S1P production is triggered by the pro inflammatory cytokines interleukin-1 (IL-1), TNF-α, and vascular endothelial growth factor (VEGF), increasing S1P concentration in efferent lymphatic vessels.Citation31 This increase of the S1P gradient between the lymph node and the efferent lymphatic vessels during periods of inflammation causes lymphocytes to exit the lymph node via chemotaxis, re-enter the blood stream, be mobilized to the site of inflammation, and perpetuate the inflammatory response ().Citation32 It is this innate mechanism which became the basis for the development of S1P modulation as a therapeutic target for the treatment of IBD.
Figure 2 S1P Gradient. Adapted from Stimulated T Cells Migrate Out of Lymph Nodes and Enter Inflamed Tissue, by BioRender.com; 2022. Retrieved from: https://app.biorender.com/biorender-templates.Citation32
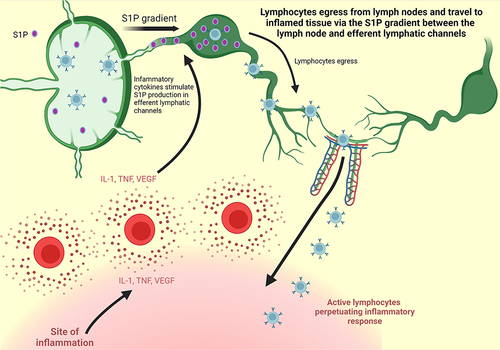
Evidence for this mechanism has been obtained from lab studies that disturb the binding of S1P to S1P1 by artificially reducing either component. By decreasing circulating S1P via selective enzyme inhibition, T cells were markedly decreased in both blood and lymph, but the number of T cells inside lymphoid organs was unaffected,Citation12,Citation29 supporting the notion that the S1P gradient is necessary for T cells to exit lymphoid tissues. Additionally, S1P1 deficient T cells that were transferred into wild-type mice were found to be able to circulate through the bloodstream into peripheral lymphoid organs but then remained sequestered there,Citation33 seemingly unable to exit via the efferent lymphatic vessels. Together, these studies show that both circulating S1P and S1P1 are required for successful lymphocyte egress from lymph nodes.
S1P Receptor Modulators
The first drug in the S1P modulator class was fingolimod, a mostly nonspecific S1P modulator that affects all S1P receptors except S1P2.Citation34 After randomized clinical trials demonstrated that fingolimod decreased the rate of disease relapse and disability progression in patients with MS,Citation35 it received United States (US) Food and Drug Administration (FDA) approval in 2010 for adults with certain forms of Multiple Sclerosis (MS)Citation36 with approval extended in 2018 to include children starting at age 10 as well.Citation37 The mechanism of action of fingolimod is postulated to be twofold.Citation31 By artificially binding S1P1 on lymphocytes, fingolimod exerts an antagonistic effect,Citation29,Citation33,Citation38 causing the S1P receptors to be internalized and degraded, limiting their availability for further S1P binding and subsequent lymphocyte mobilization. As discussed above, multiple S1P receptors are found on endothelial surfaces and play a role in regulating endothelial function and stabilization. Specifically, stimulation of S1P1 is thought to enhance cellular junctions while stimulation of S1P2 and S1P3 disrupts those same junctions.Citation31 Given that fingolimod affects S1P1 to a greater affect S1P3,Citation31 it is thought to act as an agonist on endothelial S1P1 and induce “tightening” of cell junctions, thereby further restricting lymphocyte egress to the periphery.Citation38–40 The net result of these dual modulator effects is a transient decrease in circulating lymphocytes which is reversible upon medication discontinuation.
While fingolimod’s main therapeutic effect is due to S1P1 modulation, its broad range of activity on all S1P receptors except S1P2 is likely responsible for the, albeit rare, adverse events associated with its use. Specifically, the modulation of S1P3 on cardiac cells is likely responsibleCitation41 for the few reported cases of bradycardia and heart block observed in fingolimod trialsCitation35,Citation42 and prolongation of corrected QT (QTc) intervalCitation43 reported by the FDA in their 2010 review of fingolimod. The modulation of S1P1 and S1P3 on vascular smooth muscle cells is also likely responsible for hypertensionCitation44 which was also reported in the fingolimod trials.Citation35,Citation42 Cases of macular edema are also thought to be due to S1P3 modulation and its effect on the orbital vascular endothelial junctions. Lymphopenia, defined by an absolute lymphocyte count (ALC) of <200 cells/μL, infection, and elevated liver enzymes were also reported.Citation35
The next S1P modulator to be FDA approved in 2019, also for the treatment of MS, was siponimodCitation45 which is a more selective drug, effecting only S1P1 and S1P5,Citation46 theoretically decreasing its side effect profile. Indeed, the risk of bradycardia and sustained lymphopenia was lower with siponimod than fingolimod.Citation46 However, siponimod is currently not being investigated for benefit in IBD.
Ozanimod, which was approved for MS in 2020,Citation47 is specific to S1P1 and S1P5.Citation48 After achieving statistically significant results treating MS, further trials were conducted to assess its efficacy for IBD.Citation49–55 Ponesimod, which was approved for some forms of MS in 2021,Citation56 and etrasimod, which is currently in Phase 3 trials for UC,Citation57 are another two selective S1P modulators which are in the pipeline of IBD therapies.
Ozanimod, originally designated RPC1063, was investigated for its potential role in treating immune-mediated diseases. In vitro studies assessing RPC1063ʹs affinity for all 5 known S1P receptors showed that its greatest affinity was for S1P1 and S1P5 specifically.Citation48 However, its affinity for S1P1 was found to be 27 times greater than for S1P5. These studies also showed that RPC1063ʹs affinity for S1P1 over S1P2,3,4 was greater than 10,000‐fold, leaving RPC1063 as one of the most selective S1P modulators under development. Due to its high specificity for S1P1, ozanimod likely exerts its affect via the proposed dual mechanism of action described above for fingolimod ().Citation32
Figure 3 Ozanimod mechanism of action. Adapted from Stimulated T Cells Migrate Out of Lymph Nodes and Enter Inflamed Tissue, by BioRender.com; 2022. Retrieved from: https://app.biorender.com/biorender-templates.Citation32
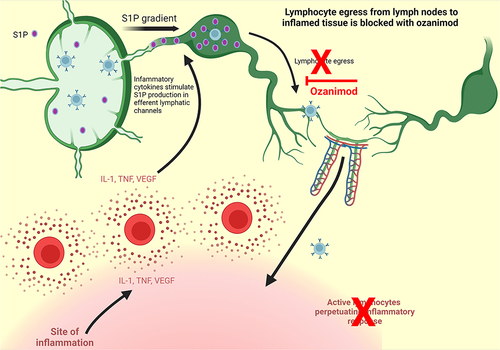
Following the initial in vitro studies, RPC1063 was tested using three animal studies, one simulating MS and two models to simulate IBD in the human colon, including the trinitrobenzene sulfonate (TNBS) induced colitisCitation58 and T cell transfer induced colitisCitation59 models. Scott et alCitation48 demonstrated that in both IBD models, RPC1063 was efficacious as a treatment for the artificially induced colitis in rodents.
Clinical Trials
The clinical trials presented in this review were chosen by conducting a PubMed search of published papers with keywords “ozanimod” and “inflammatory bowel disease” and a search with keywords “ozanimod” and “safety”, which yielded 70 results and 60 results, respectively. The articles were then individually reviewed, including only those publications reporting randomized controlled trials and/or pharmacokinetic and safety outcomes, yielding a total of 8 unique articles which are summarized in .
Table 1 Key Clinical Trials for Ozanimod and Its Efficacy in IBD
Phase One
The Phase 1, single‐center, randomized, double‐blind, placebo‐controlled trial of ozanimod included 88 healthy subjects split between single ascending‐dose (SAD) cohorts, multiple ascending‐dose cohorts for 7 days (MAD‐7) or 28 days (MAD‐28), and a dose‐escalation (DE) cohort.Citation60
Drug safety was evaluated by monitoring for treatment‐emergent adverse events (TEAEs), changes from baseline laboratory studies, electrocardiogram (ECG), and vital signs. Given the theoretical and previously observed side effects of fingolimod, special attention was given to cardiac, pulmonary, infectious, hepatic, ophthalmologic, and hematologic events.
The most common drug-related TEAEs in the ozanimod group were headache (13.2%), somnolence (8.8%), nausea (8.8%), dizziness (7.4%), fatigue (5.9%). While all 88 subjects, including those who received placebo, experienced an expected physiologic reduction in heart rate associated with circadian rhythm in the first 24 hours initial dosing, those who received ozanimod had a greater decrease in heart rate compared to the placebo group. Furthermore, reductions in heart rate in the ozanimod group appeared to be dose dependent. Twenty hours after dosing, subjects who received 1mg or 2mg fixed dose of ozanimod experienced a mean decrease in heartrate of 20–25 beats per minute (BPM) vs those who received a dose escalated fashion of 1mg or 2mg ozanimod who experienced a mean decrease in heartrate of approximately 15 BPM. Four patients experienced cardiac TEAEs within the first 6 hours after receiving the initial dose. Two (2.9%) experienced an asymptomatic two-second sinus pause during periods of bradycardia and one (1.5%) experienced asymptomatic intermittent sinus bradycardia. These patients were in the 0.3-mg SAD cohort. One patient (1.5%) experienced a second‐degree Mobitz type 1 atrioventricular (AV) block after receiving the 1st dose of 1.5mg of ozanimod. All four cases resolved without intervention. No clinically significant changes in blood pressure were recorded.Citation60 Another phase 1, randomized, double-blind, placebo-controlled trialCitation61 specifically analyzing the effect of ozanimod on QTc found no clinically relevant effect on QTc even at supratherapeutic dosing up to 2mg.
In the initial phase 1 trial discussed above,Citation60 two patients experienced pulmonary TEAEs with mild, asymptomatic, and transient decreases in pulmonary function tests (forced expiratory volume in 1 second [FEV1] and forced vital capacity [FVC]) (PTFs) that self-resolved one week after discontinuing the medication. Two subjects in the ozanimod group developed oral herpes. Liver enzyme levels and ophthalmologic examination were unchanged for all subjects.
The effect on ALC was also measured as part of the safety profile. After 28 days of 1mg dosing, a 65% reduction in ALC was achieved. Higher dosing of 1.5mg daily only showed an additional 3% further decrease in ALC, suggesting a plateau effect at 1mg. While the decrease in ALC observed with ozanimod was similar to that observed with other S1P modulators, the recovery of ALC back to the normal range occurred in 2–3 days with 1mg ozanimod compared to within 6 weeks with 0.5mg of fingolimodCitation62 (the approved therapeutic dose).
This favorable time period for ALC recovery to baseline is due to the optimal pharmacokinetics reported in initial human RPC1063 testing.Citation60 The half-life of RPC1063 in humans is 17–21 hours, allowing a steady state to be achieved in only 3–4 days of initiating therapy and effective drug elimination within just a few days after drug discontinuation, enabling ALC recovery. However, some downstream metabolites of ozanimod have been found to have longer half-lives,Citation63,Citation64 contributing to the recommendation to stop ozanimod at least 3 months before planned pregnancy or administering live vaccines.Citation65 RPC1063 also achieved high oral bioavailability and volume of distribution,Citation60 optimizing its use as a once daily oral therapy and avoiding the need of parenteral administration of most newly available IBD therapy. Finally, in a phase 1, randomized, open-label study of 24 subjects, ozanimod pharmacokinetics have been shown to be unaffected by food consumption,Citation66 allowing patients more flexibility for medication administration without regard to meals.
Clinical Trials: Crohn’s Disease
Phase Two
The Phase 2 “Stepstone” trialCitation38 was an uncontrolled, international, multicenter trial of ozanimod as induction therapy for adults with moderate to severe Crohn’s Disease, defined by a Crohn’s Disease Activity Index (CDAI) score of 220–450, with a Simple Endoscopic Score for Crohn’s Disease (SES-CD) of 6 or greater (or in isolated ileum disease SES-CD ≥4) and an average daily stool score of 4 or more points or an average daily abdominal pain score of 2 or more points, or both. The trial consisted of 69 patients evaluated for induction therapy for a period of 12 weeks. To minimize the risk of bradycardia, patients received ozanimod in a dose escalated fashion during the first week, with goal dosing of 1mg daily, which was achieved at week 1 and continued for an additional 11 weeks.
The primary outcome was the change in SES-CD from baseline to week 12. The proportions of patients with endoscopic response (≥50% decrease in SES-CD) and endoscopic remission (SES-CD ≤4 points and SES-CD decrease ≥2 points with no SES-CD subscore >1 point) at week 12 were also assessed. Secondary endpoints were the change in CDAI score from baseline to week 12, proportion of patients with clinical remission (CDAI score of <150) at week 12, and proportion of patients with clinical response (CDAI reduction from baseline of ≥100 points) at week 12. Patient reported outcomes (PRO) were evaluated as the change from baseline two-item PRO (PRO2) score to week 12. The proportions of patients with clinical remission (average daily stool score ≤3 points and average daily abdominal pain score ≤1 point) and clinical response (PRO2 decrease ≥50%) were also assessed at week 12. Finally, changes from baseline histologic samples, C-reactive protein (CRP), and fecal calprotectin were also assessed at week 12.
At 12 weeks, the mean reduction in SES-CD from baseline of 13.3 was 2.2 points. Endoscopic response and endoscopic remission were reported in 23.2% and 10.1% of patients, respectively. Biologic-naive patients experienced >9% higher rates of both endoscopic response and remission than biologic-experienced patients. The mean reduction in CDAI from baseline of 320.8 was 130.4 points. Clinical response and clinical remission were reported in 56.5% and 39.1% of patients, respectively. Biologic-naive patients experienced an 8.5% higher rate of clinical response and a 26.1% higher rate of clinical remission than biologic-experienced patients. The mean reduction in PRO2 at week 12 was 66.1 points. Using PRO2 to assess clinical response and remission, the rates were 33.3% and 24.6%, respectively. Biologic-naive patients experienced an 25.3% higher rate of clinical response and a 24% higher rate of clinical remission than biologic-experienced patients.
Histologic analysis at week 12 using both the Global Histologic Disease Activity Score (GHAS) and Robarts Histopathology Index (RHI) showed improvement in disease severity from baseline with similar rate of change in both biologic-naïve and biologic-experienced patients.
Patients with elevated baseline CRP that were measured at weeks 8 and 12 experienced a decrease of 36.2% and 34.7%, respectively. Patients with elevated baseline fecal calprotectin that were measured at weeks 8 and 12 experienced a decrease of 52.4% and 55.9%, respectively.
The most common TEAEs reported were CD flare (26%), abdominal pain (15%), lymphopenia (13%), arthralgia (13%), and nausea (12%). Elevated alanine aminotransferase (ALT) and aspartate aminotransferase (AST) of ≥3 times the upper limit of normal were found in 4% and 3% of patients, respectively. Fistula complications were reported in three patients (4.3%), two of which had prior history of fistulas. One of these patients eventually developed sepsis and died and was considered to be possibly treatment related by the investigators. Abdominal abscess and intestinal obstruction were reported in two patients (2.9%) each. One case each of anal abscess, diverticulitis, pancreatitis, and pancreatic carcinoma were reported. Herpes-zoster and campylobacter infection was reported in two and one patients, respectively. No significant bradycardia or AV blocks were recorded on cardiac monitoring. No ophthalmologic or pulmonary complications were reported in this study.
Phase 3 trials of ozanimod for adults with moderate to severe CD are currently ongoing.Citation53–55
Clinical Trials: Ulcerative Colitis
Phase Two
The phase two “Touchstone” trialCitation49 of ozanimod for induction and maintenance therapy for moderate to severe UC was a multi-center, double-blind, placebo-controlled trial consisting of 197 adults with moderate-to-severe UC, determined by a Mayo Clinic score of 6–12 and endoscopic sub-score of 2–3. Patients were randomly assigned, in equal ratios, to receive ozanimod at a dose of 0.5 mg or 1 mg or placebo once daily. The trial consisted of induction until week 8, maintenance from week 8 to 32, and an open label extension period after week 32.
The primary outcome of the study was clinical remission, defined as a Mayo Clinic score ≤2, with no sub-score >1, at 8 weeks. Secondary outcomes evaluated at week 8 included clinical response (defined as a reduction in the Mayo Clinic score of ≥3 points and ≥30% from baseline, with a decrease in the rectal bleeding subscore of ≥1 point or a subscore of ≤1), change from baseline Mayo Clinic score, and mucosal healing (endoscopy subscore ≤1).
At 8 weeks, clinical remission was achieved in 16% of patients who received 1mg of ozanimod, 14% of those who received 0.5mg ozanimod, and 6% of patients who received placebo. The 1mg ozanimod group (P = 0.048), but not the 0.5mg ozanimod group (P = 0.14) was found to be superior to placebo. Given that remission in the 0.5mg ozanimod group compared to placebo was not statistically significant, all further comparison data for secondary outcomes were deemed exploratory. Additional observations included a clinical response at 8 weeks for 57% of those receiving 1 mg of ozanimod and 54% of those receiving 0.5 mg, as compared with 37% of those receiving placebo. At week 32, the rate of clinical remission was 21% in the group that received 1 mg of ozanimod, 26% in the group that received 0.5 mg, and 6% in the group that received placebo; the rate of clinical response at week 32 was 51%, 35%, and 20%, respectively.
Adverse events were similar in all three study groups. The most common TEAEs were UC flare, anemia, headache, nausea, pyrexia, arthralgia, and elevated ALT which occurred in ≥4 patients across all three groups. Interestingly, adverse cardiac events occurred in the placebo and 0.5mg ozanimod groups but not in the 1mg ozanimod group. UC flare, pyrexia, and elevated ALT were the most common adverse events in the 1mg ozanimod group with 4% of patients experiencing each adverse outcome.
The open label extension period consisted of 170 patients receiving ozanimod 1mg daily for 200 weeks.Citation51 Clinical remission and response were evaluated using partial, three-component (stool frequency, rectal bleeding, and endoscopic findings), and four component (stool frequency, rectal bleeding, endoscopic findings, and physician global assessment), Mayo scores and reported descriptively. Endoscopic, histologic, and inflammatory biomarker changes were also assessed. TEAEs and discontinuation rates throughout the study period were also tracked and reported.
Partial Mayo clinical response and remission were 71.2% and 54.7%, respectively, at week 56. Three-component Mayo score clinical response and remission were 35.3% and 21.2%, respectively, at week 56. Four-component Mayo score clinical response and remission were 39.4% and 18.8%, respectively, at week 56. Endoscopic improvement, endoscopic remission, and mucosal healing at week 56 was 22.9%, 5.9% and 2.4%, respectively. Partial Mayo clinical response and remission were 41.2% and 36.5%, respectively, at week 200.
The percentage of patients with histologic remission, defined as a Geboes index score of <2.0, at week 56 was 18.2%.Citation51 A post hoc analysis of tissue samples obtained during the phase 2 Touchstone study analyzed 4 histologic indices and found that they can all be reliably used to assess general disease activity of UC and to also assess response to treatment. However, there was a poorer correlation between histologic scores and endoscopic scores, suggesting that longer duration of therapy may be required to achieve histologic remission.Citation67
In the extension period,Citation51 median CRP concentration was reduced by 35% from baseline to week 8 and this reduction was generally maintained through week 200. The median fecal calprotectin concentration was reduced by 68% from baseline to week 8 and by 63% through week 200.
The most common TEAEs (>5%) were UC flare (6.5%), hypertension (5.9%), upper respiratory infection (5.9%), and increased gamma glutamyl-transferase (5.3%). Nine patients (5.3%) developed reduced ALC or lymphopenia, two patients (1.2%) developed macular edema, and one patient (0.6%) developed herpes zoster. TEAEs considered potentially related to study treatment in one patient each included adenocarcinoma of unknown origin, ascites, pneumococcal pneumonia, pneumonia, hyperbilirubinemia, and hemolytic anemia with resultant jaundice. One case of spontaneous abortion in a 29-year-old woman occurred and was deemed to be “possibly” related to study medication.
Three patients had a serious infection during the open label extension, but none were associated with grade 4 lymphopenia of ALC <200 cells/μL. One death was reported due to the above-mentioned mucinous adenocarcinoma of unknown origin with metastasis to the liver in a patient who had been in the study for more than 2 years. No significant bradycardia, AV blocks, or decrease in PFTs were reported. No clinically significant elevations in AST or ALT were reported.
Discontinuation rates were 28% at year 1 and 15–18% annually through year 4 which is superior to reported discontinuation rates for the TNF-α inhibitors infliximab and adalimumab.Citation68
Phase Three
The phase 3 “True North” trialCitation50 was a multicenter, randomized, double-blind, placebo-controlled trial of ozanimod as induction and maintenance therapy in adult patients with moderate to severe UC (defined as a Mayo Clinic score of 6–12, with an endoscopy subscore of ≥2, a rectal bleeding subscore of ≥1, and a stool-frequency subscore of ≥1). The trial consisted of induction until week 10, maintenance from weeks 10–52, and an optional open label extension period after 52 weeks which is still ongoing at the time of the writing of this paper. In total, 1012 patients were split into two cohorts. Cohort one patients were randomly assigned to receive 1mg ozanimod or placebo daily. Cohort two patients received open label ozanimod 1mg daily. At 10 weeks, patients with a clinical response (defined as a reduction in the total Mayo score of ≥3 points and ≥30% from baseline or in the three-component Mayo score of ≥2 points and ≥35% from baseline, as well as a reduction in the rectal-bleeding subscore of ≥1 point or an absolute rectal bleeding subscore of ≤1 point) to ozanimod in either cohort were randomized again to receive 1mg ozanimod or placebo for the maintenance period of up to 52 weeks.
The primary endpoint for both the induction and maintenance periods was clinical remission, assessed using the three-component Mayo score. Clinical remission was defined as a rectal-bleeding subscore of 0, a stool-frequency subscore of 1 or less (with a decrease of at least 1 point from baseline), and an endoscopy subscore of 1 or less. Secondary endpoints at week 10 were clinical response, endoscopic improvement, and mucosal healing. Secondary endpoints at week 52 were maintenance of clinical remission, glucocorticoid-free remission, and durable clinical remission.
At 10 weeks, clinical remission was higher among patients who received ozanimod vs those who received placebo (18.4% vs 6.0%, P < 0.001). The rate of successful maintenance of clinical remission was also significantly higher among patients who received ozanimod than among those who received placebo during the maintenance period (37.0% vs 18.5% [among patients who achieved clinical response at week 10], P < 0.001). The incidence of clinical response was also higher for ozanimod than placebo both during induction (47.8% vs 25.9%, P < 0.001) and maintenance (60.0% vs 41.0%, P < 0.001). The rate of mucosal healing for ozanimod vs placebo was also found to statistically significant during both induction (12.6% vs 3.7%, P < 0.001) and maintenance (29.6% vs 14.1%, P < 0.001). All other key secondary end points showed statistically significant improvement with ozanimod as compared with placebo at weeks 10 and 52. The significant findings of endoscopic improvement and mucosal healing with ozanimod are of particular interest given recent guidelines that support a “treat to target” strategy for IBD.Citation69,Citation70 These guidelines are based on data which have shown improved long-term outcomes in patients who achieve endoscopic mucosal healing,Citation71 with some even suggesting histologic improvement as a treatment target as well.Citation72,Citation73
The overall incidence of adverse events was similar in the ozanimod and placebo group during induction, but higher for ozanimod during the maintenance period. The frequency of serious infections was less than 2% in each group. The overall incidence of infection, including nasopharyngitis, upper respiratory tract infection (URI), and herpes-zoster with ozanimod therapy was equal to placebo in the induction period (11.6%) but higher during the maintenance period (23% in ozanimod vs 11.9% in placebo group). Patients in the trial were required to have a documented presence of varicella–zoster virus IgG antibody or complete varicella–zoster vaccination. Herpes zoster occurred in 0.4% of ozanimod patients during the induction period and in 2.2% of ozanimod patients during maintenance. Herpes zoster infection did not occur in any placebo treated patient. One patient (in cohort 2) with a history of ischemic cardiomyopathy and prolonged tobacco use died from influenza complicated by acute respiratory distress syndrome.
As observed in earlier trials, ALC decreased by a mean of approximately 54% from baseline at week 10. During the induction period, ALC of <200 cells/μL occurred in a total of 1.5% of patients who received ozanimod and in none of patients who received placebo. During the maintenance period, ALC of <200 cells/μL occurred in a total of 2.2% of patients who received ozanimod and in none of patients who received placebo. All patients with an ALC of <200 cells/μL at any point during the trial subsequently improved to ≥200 cells/μL during ozanimod treatment. No patients with a serious or opportunistic infection had an absolute lymphocyte count of <200 cells/μL.
Bradycardia occurred more frequently with ozanimod therapy than with placebo during the induction period (0.6% vs 0% in placebo group). No cases of bradycardia were reported in either group during the maintenance period. No cases of second-degree type 2 AV block or third-degree AV block occurred. One patient receiving ozanimod had a hypertensive crisis on day 1 of the induction period which spontaneously resolved. During the maintenance period, hypertensive crisis occurred in 1 patient each in the ozanimod group and the placebo group. General hypertension was more common with ozanimod therapy than with placebo during induction period (1.6% vs 0%) and during the maintenance period (1.7% vs 1.3%).
ALT levels ≥2 times the upper limit of normal were more common with ozanimod than with placebo during induction period (5.4% vs 0.9%) and maintenance period (13.9% vs 5.3%). However, none of these patients developed severe liver injury. Macular edema occurred in 0.25% of patients receiving ozanimod during induction, and in 0.4% during maintenance. No cases of macular edema were reported in the placebo group during induction or maintenance. All cases resolved with treatment cessation.
A basal-cell carcinoma was diagnosed in 1 patient (0.13%) receiving ozanimod during the induction period. No other malignancy was reported during the induction period. During the maintenance period, a basal-cell carcinoma and rectal adenocarcinoma were diagnosed in 2 separate patients (0.4% each) who received ozanimod during both the induction and maintenance periods. A colon adenocarcinoma and breast cancer were diagnosed in 2 different patients (0.4% each), both which received ozanimod during the induction period and placebo during the maintenance period.
Ulcerative Colitis Prescribing Considerations
The FDA granted approval for the use of ozanimod for the treatment of moderate to severely active UC in adults in May of 2021.Citation74 Current prescribing guidelines do not limit therapy for those with prior biologic or immune modulator failure. Given the adverse event profile observed in the ozanimod clinical trials and those for other S1P modulators, several steps are suggested before and during treatment to help mitigate the occurrence and severity of these events.Citation65
Prior to initiating ozanimod therapy, a baseline complete blood count (CBC) with lymphocyte count, liver transaminase, and bilirubin levels should be obtained. Patients should also undergo a screening ECG to assess for baseline bradycardia or AV blocks. Cardiology evaluation should be requested in patients with current significant QT prolongation (QTc >450 msec in males, >470 msec in females), ischemic heart disease, or heart failure. Patients taking a beta blocker in conjunction with calcium channel blocker or taking class 1a or class 3 antiarrhythmic drugs should also be evaluated by a cardiologist prior to initiating ozanimod therapy. Cardiology evaluation should also be sought for patients with a history of cardiac arrest, myocardial infarction, cerebrovascular disease, uncontrolled hypertension, second-degree Mobitz type 2 or higher AV block, sick sinus syndrome, or sinoatrial block. As an additional precaution against medication-induced bradycardia or conduction abnormalities, ozanimod is prescribed in titrated doses over a one-week period before reaching the full therapeutic dose.
Although changes in PFTs were reported in the phase 1 trial, later trials did not report any incidence of pulmonary adverse events. Nevertheless, baseline PFTs should be considered in patients with severe obstructive or restrictive pulmonary disease. Baseline ophthalmologic evaluation is recommended for patients with diabetes mellitus, uveitis, or macular edema. It is also recommended to test all patients for varicella zoster virus (VZV) antibodies who do not have either a health-care professional–confirmed history of varicella (chickenpox) or without documentation of a full course of vaccination against VZV. If negative, VZV vaccination is recommended at least one month prior to initiating ozanimod therapy.
Currently listed contraindications to ozanimod use in the U.S.Citation65 include monoamine oxidase inhibitor (MAOI) use and severe, untreated, sleep apnea. Ozanimod is also contraindicated in patients with current second-degree Mobitz type 2 AV block, third-degree AV block, sick sinus syndrome, or sinoatrial block, unless the patient has a functioning pacemaker. Finally, ozanimod is contraindicated in patients who experienced a myocardial infarction, stroke, transient ischemic attack, unstable angina, decompensated heart failure requiring hospitalization, or class III or IV heart failure in the last 6 months.
Ozanimod use is not recommended in patients with active infection or hepatic impairment.
CBC with lymphocyte count, liver transaminase, and bilirubin levels should be monitored periodically during therapy. Treatment should be reevaluated if infection or significant liver injury develops. Any change in vision should prompt an urgent ophthalmology consult and treatment discontinuation should be considered if macular edema develops.
It is also recommended to avoid the use of live vaccines during and up to 3 months after stopping ozanimod therapy. Finally, though data are limited, ozanimod use during pregnancy or within three months of planned pregnancy is discouraged. It is currently unknown if ozanimod is present in breastmilk.
Conclusion
Small molecule therapy with ozanimod, a first in class S1P modulator for IBD, offers an oral treatment option for the treatment of UC. Ozanimod demonstrated efficacy and a favorable side effect profile, providing a unique option among the currently available therapies. Though there are no restrictions to ozanimod as a first-line therapy, its correct positioning in the therapeutic algorithm for the treatment of UC remains to be defined, and its potential role for the treatment of CD remains investigational.
Ozanimod’s development also appears to harken back to a prior era in IBD management, with a shift back to oral therapy. Though the large molecule biologics are likely to remain a mainstay of therapy for a long time to come, the convenience of a once-daily pill has obvious appeal. Beyond that, the oral route may prove to be cost saving, especially compared to infusion biologics with their necessary infusion team and infrastructure. Looking into the near future, it is also likely that ozanimod’s distinct mechanism of action and favorable side effect profile would lend itself to clinical trials as part of combination therapy with other IBD treatments such as the biologics. Certainly, past experience has shown that the combination of multiple IBD therapies can be more effective than monotherapy,Citation75 and this is being further explored by trials of other IBD treatment combinations currently in progress.Citation76 As is true for all medications, the information obtained from even the best designed clinical trials will still need to be followed up with close observation and real world data.
Disclosure
Dr Arun Swaminath reports advisory board from BMS and BI, grants from Janssen, grants from Takeda, outside the submitted work. The authors report no other conflicts of interest in this work.
References
- Xu XR, Liu CQ, Feng BS, Liu ZJ. Dysregulation of mucosal immune response in pathogenesis of inflammatory bowel disease. World J Gastroenterol. 2014;20(12):3255–3264. doi:10.3748/wjg.v20.i12.3255
- Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448(7152):427–434. doi:10.1038/nature06005
- Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411(6837):599–603. doi:10.1038/35079107
- Mirkov MU, Verstockt B, Cleynen I. Genetics of inflammatory bowel disease: beyond NOD2. Lancet Gastroenterol Hepatol. 2017;2(3):224–234. doi:10.1016/S2468-1253(16)30111-X
- Nightingale SL. From the food and drug administration. JAMA. 1998;280(13):1128. doi:10.1001/jama.280.13.1128-JFD80009-2-1
- Feuerstein JD, Ho EY, Shmidt E, et al. AGA clinical practice guidelines on the medical management of moderate to severe luminal and perianal fistulizing crohn’s disease. Gastroenterology. 2021;160(7):2496–2508. doi:10.1053/j.gastro.2021.04.022
- Feuerstein JD, Isaacs KL, Schneider Y, et al. AGA clinical practice guidelines on the management of moderate to severe ulcerative colitis. Gastroenterology. 2020;158(5):1450–1461. doi:10.1053/j.gastro.2020.01.006
- D’Haens G, Panaccione R, Baert F, et al. Risankizumab as induction therapy for Crohn’s disease: results from the phase 3 ADVANCE and MOTIVATE induction trials. Lancet. 2022;399(10340):2015–2030. doi:10.1016/S0140-6736(22)00467-6
- Administration USFaD. FDA requires warnings about increased risk of serious heart-related events, cancer, blood clots, and death for JAK inhibitors that treat certain chronic inflammatory conditions. FDA. Available from: https://www.fda.gov/drugs/drug-safety-and-availability/fda-requires-warnings-about-increased-risk-serious-heart-related-events-cancer-blood-clots-and-death. Accessed May 25, 2022.
- Proia RL, Hla T. Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J Clin Invest. 2015;125(4):1379–1387. doi:10.1172/JCI76369
- Biorender.com. “Nitric Oxide Synthesis 1”, by BioRender.com; 2022. Available from: https://app.biorender.com/biorender-templates. Accessed August 25, 2022.
- Pappu R, Schwab SR, Cornelissen I, et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science. 2007;316(5822):295–298. doi:10.1126/science.1139221
- Venkataraman K, Lee YM, Michaud J, et al. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ Res. 2008;102(6):669–676. doi:10.1161/CIRCRESAHA.107.165845
- Pham TH, Baluk P, Xu Y, et al. Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J Exp Med. 2010;207(1):17–27. doi:10.1084/jem.20091619
- Rosen H, Goetzl EJ. Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nat Rev Immunol. 2005;5(7):560–570. doi:10.1038/nri1650
- Alvarez SE, Milstien S, Spiegel S. Autocrine and paracrine roles of sphingosine-1-phosphate. Trends Endocrinol Metab. 2007;18(8):300–307. doi:10.1016/j.tem.2007.07.005
- Chun J, Hla T, Lynch KR, Spiegel S, Moolenaar WH. International union of basic and clinical pharmacology. LXXVIII. Lysophospholipid receptor nomenclature. Pharmacol Rev. 2010;62(4):579–587. doi:10.1124/pr.110.003111
- Peyrin-Biroulet L, Christopher R, Behan D, Lassen C. Modulation of sphingosine-1-phosphate in inflammatory bowel disease. Autoimmun Rev. 2017;16(5):495–503. doi:10.1016/j.autrev.2017.03.007
- Argollo M, Furfaro F, Gilardi D, et al. Modulation of sphingosine-1-phosphate in ulcerative colitis. Expert Opin Biol Ther. 2020;20(4):413–420. doi:10.1080/14712598.2020.1732919
- Nielsen OH, Li Y, Johansson-Lindbom B, Coskun M. Sphingosine-1-phosphate signaling in inflammatory bowel disease. Trends Mol Med. 2017;23(4):362–374. doi:10.1016/j.molmed.2017.02.002
- Hla T, Venkataraman K, Michaud J. The vascular S1P gradient-cellular sources and biological significance. Biochim Biophys Acta. 2008;1781(9):477–482. doi:10.1016/j.bbalip.2008.07.003
- Serra M, Saba JD. Sphingosine 1-phosphate lyase, a key regulator of sphingosine 1-phosphate signaling and function. Adv Enzyme Regul. 2010;50(1):349–362. doi:10.1016/j.advenzreg.2009.10.024
- Vu TM, Ishizu AN, Foo JC, et al. Mfsd2b is essential for the sphingosine-1-phosphate export in erythrocytes and platelets. Nature. 2017;550(7677):524–528. doi:10.1038/nature24053
- Ito K, Anada Y, Tani M, et al. Lack of sphingosine 1-phosphate-degrading enzymes in erythrocytes. Biochem Biophys Res Commun. 2007;357(1):212–217. doi:10.1016/j.bbrc.2007.03.123
- Mendoza A, Bréart B, Ramos-Perez WD, et al. The transporter Spns2 is required for secretion of lymph but not plasma sphingosine-1-phosphate. Cell Rep. 2012;2(5):1104–1110. doi:10.1016/j.celrep.2012.09.021
- Obinata H, Hla T. Sphingosine 1-phosphate and inflammation. Int Immunol. 2019;31(9):617–625. doi:10.1093/intimm/dxz037
- Allende ML, Tuymetova G, Lee BG, Bonifacino E, Wu YP, Proia RL. S1P1 receptor directs the release of immature B cells from bone marrow into blood. J Exp Med. 2010;207(5):1113–1124. doi:10.1084/jem.20092210
- Pereira JP, Xu Y, Cyster JG. A role for S1P and S1P1 in immature-B cell egress from mouse bone marrow. PLoS One. 2010;5(2):e9277. doi:10.1371/journal.pone.0009277
- Schwab SR, Pereira JP, Matloubian M, Xu Y, Huang Y, Cyster JG. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science. 2005;309(5741):1735–1739. doi:10.1126/science.1113640
- Hampton HR, Chtanova T. Lymphatic migration of immune cells. Front Immunol. 2019;10:1168. doi:10.3389/fimmu.2019.01168
- Brinkmann V. Sphingosine 1-phosphate receptors in health and disease: mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol Ther. 2007;115(1):84–105. doi:10.1016/j.pharmthera.2007.04.006
- Biorender.com. “Stimulated T Cells Migrate Out of Lymph Nodes and Enter Inflamed Tissue”, by BioRender.com; 2022. Available from: https://app.biorender.com/biorender-templates. Accessed August 25, 2022.
- Matloubian M, Lo CG, Cinamon G, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427(6972):355–360. doi:10.1038/nature02284
- Brinkmann V, Davis MD, Heise CE, et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem. 2002;277(24):21453–21457. doi:10.1074/jbc.C200176200
- Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401. doi:10.1056/NEJMoa0909494
- Sharma S, Mathur AG, Pradhan S, Singh DB, Gupta S. Fingolimod (FTY720): first approved oral therapy for multiple sclerosis. J Pharmacol Pharmacother. 2011;2(1):49–51. doi:10.4103/0976-500X.77118
- Administration USFaD. FDA expands approval of Gilenya to treat multiple sclerosis in pediatric patients. Available from: https://www.fda.gov/news-events/press-announcements/fda-expands-approval-gilenya-treat-multiple-sclerosis-pediatric-patients. Accessed May 28, 2022.
- Brinkmann V, Cyster JG, Hla T. FTY720: sphingosine 1-phosphate receptor-1 in the control of lymphocyte egress and endothelial barrier function. Am J Transplant. 2004;4(7):1019–1025. doi:10.1111/j.1600-6143.2004.00476.x
- Mandala S, Hajdu R, Bergstrom J, et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296(5566):346–349. doi:10.1126/science.1070238
- Singer II, Tian M, Wickham LA, et al. Sphingosine-1-phosphate agonists increase macrophage homing, lymphocyte contacts, and endothelial junctional complex formation in murine lymph nodes. J Immunol. 2005;175(11):7151–7161. doi:10.4049/jimmunol.175.11.7151
- Sanna MG, Vincent KP, Repetto E, et al. Bitopic sphingosine 1-phosphate receptor 3 (S1P3) antagonist rescue from complete heart block: pharmacological and genetic evidence for direct S1P3 regulation of mouse cardiac conduction. Mol Pharmacol. 2016;89(1):176–186. doi:10.1124/mol.115.100222
- Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–415. doi:10.1056/NEJMoa0907839
- Administration USFaD. Center for drug evaluation and research. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022527Orig1s000medr.pdf. Accessed July 28, 2022.
- Chaudhry BZ, Cohen JA, Conway DS. Sphingosine 1-phosphate receptor modulators for the treatment of multiple sclerosis. Neurotherapeutics. 2017;14(4):859–873. doi:10.1007/s13311-017-0565-4
- Al-Salama ZT. Siponimod: first global approval. Drugs. 2019;79(9):1009–1015. doi:10.1007/s40265-019-01140-x
- Scott LJ. Siponimod: a review in secondary progressive multiple sclerosis. CNS Drugs. 2020;34(11):1191–1200. doi:10.1007/s40263-020-00771-z
- Lamb YN. Ozanimod: first Approval. Drugs. 2020;80(8):841–848. doi:10.1007/s40265-020-01319-7
- Scott FL, Clemons B, Brooks J, et al. Ozanimod (RPC1063) is a potent sphingosine-1-phosphate receptor-1 (S1P1) and receptor-5 (S1P5) agonist with autoimmune disease-modifying activity. Br J Pharmacol. 2016;173(11):1778–1792. doi:10.1111/bph.13476
- Sandborn WJ, Feagan BG, Wolf DC, et al. Ozanimod Induction and Maintenance Treatment for Ulcerative Colitis. N Engl J Med. 2016;374(18):1754–1762. doi:10.1056/NEJMoa1513248
- Sandborn WJ, Feagan BG, D’Haens G, et al. Ozanimod as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2021;385(14):1280–1291. doi:10.1056/NEJMoa2033617
- Sandborn WJ, Feagan BG, Hanauer S, et al. Long-term efficacy and safety of ozanimod in moderately to severely active ulcerative colitis: results from the open-label extension of the randomized, phase 2 TOUCHSTONE Study. J Crohns Colitis. 2021;15(7):1120–1129. doi:10.1093/ecco-jcc/jjab012
- Feagan BG, Sandborn WJ, Danese S, et al. Ozanimod induction therapy for patients with moderate to severe Crohn’s disease: a single-arm, phase 2, prospective observer-blinded endpoint study. Lancet Gastroenterol Hepatol. 2020;5(9):819–828. doi:10.1016/S2468-1253(20)30188-6
- U.S. National Library of Medicine NIoH. Induction Study #2 of oral ozanimod as induction therapy for moderately to severely active crohn’s disease. Available from: https://clinicaltrials.gov/ct2/show/NCT03440385. Accessed May 28, 2022.
- U.S. National Library of Medicine NIoH. Induction Study #1 of oral ozanimod as induction therapy for moderately to severely active crohn’s disease. Available from: https://clinicaltrials.gov/ct2/show/NCT03440372. Accessed May 28, 2022.
- U.S. National Library of Medicine NIoH. An extension study of oral ozanimod for moderately to severely active crohn’s disease. Available from: https://clinicaltrials.gov/ct2/show/NCT03467958. Accessed May 28, 2022.
- Markham A. Ponesimod: first Approval. Drugs. 2021;81(8):957–962. doi:10.1007/s40265-021-01523-z
- U.S. National Library of Medicine NIoH. A phase 3 study of etrasimod in subjects with moderately to severely active ulcerative colitis. Available from: https://clinicaltrials.gov/ct2/show/NCT04176588. Accessed May 28, 2022.
- Antoniou E, Margonis GA, Angelou A, et al. The TNBS-induced colitis animal model: an overview. Ann Med Surg. 2016;11:9–15. doi:10.1016/j.amsu.2016.07.019
- Ostanin DV, Bao J, Koboziev I, et al. T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. Am J Physiol Gastrointest Liver Physiol. 2009;296(2):G135–46. doi:10.1152/ajpgi.90462.2008
- Tran JQ, Hartung JP, Peach RJ, et al. Results from the first-in-human study with ozanimod, a novel, selective sphingosine-1-phosphate receptor modulator. J Clin Pharmacol. 2017;57(8):988–996. doi:10.1002/jcph.887
- Tran JQ, Hartung JP, Olson AD, et al. Cardiac safety of ozanimod, a novel sphingosine-1-phosphate receptor modulator: results of a thorough QT/QTc Study. Clin Pharmacol Drug Dev. 2018;7(3):263–276. doi:10.1002/cpdd.383
- Francis G, Kappos L, O’Connor P, et al. Temporal profile of lymphocyte counts and relationship with infections with fingolimod therapy. Mult Scler. 2014;20(4):471–480. doi:10.1177/1352458513500551
- Surapaneni S, Yerramilli U, Bai A, et al. Absorption, metabolism, and excretion, in vitro pharmacology, and clinical pharmacokinetics of ozanimod, a novel sphingosine 1-phosphate receptor modulator. Drug Metab Dispos. 2021;49(5):405–419. doi:10.1124/dmd.120.000220
- Tran JQ, Zhang P, Ghosh A, et al. Single-dose pharmacokinetics of ozanimod and its major active metabolites alone and in combination with Gemfibrozil, Itraconazole, or Rifampin in Healthy Subjects: a Randomized, Parallel-Group, Open-Label Study. Adv Ther. 2020;37(10):4381–4395. doi:10.1007/s12325-020-01473-0
- Squibb BM. Zeposia ® (ozanimod) Full Prescribing Information. Available from: https://packageinserts.bms.com/pi/pi_zeposia.pdf. Accessed August 8, 2022.
- Tran JQ, Hartung JP, Tompkins CA, Frohna PA. Effects of high- and low-fat meals on the pharmacokinetics of ozanimod, a novel sphingosine-1-phosphate receptor modulator. Clin Pharmacol Drug Dev. 2018;7(6):634–640. doi:10.1002/cpdd.409
- Jairath V, Peyrin-Biroulet L, Zou G, et al. Responsiveness of histological disease activity indices in ulcerative colitis: a post hoc analysis using data from the TOUCHSTONE randomised controlled trial. Gut. 2019;68(7):1162–1168. doi:10.1136/gutjnl-2018-316702
- Null KD, Xu Y, Pasquale MK, et al. Ulcerative colitis treatment patterns and cost of care. Value Health. 2017;20(6):752–761. doi:10.1016/j.jval.2017.02.005
- Peyrin-Biroulet L, Sandborn W, Sands BE, et al. Selecting Therapeutic Targets in Inflammatory Bowel Disease (STRIDE): determining therapeutic goals for treat-to-target. Am J Gastroenterol. 2015;110(9):1324–1338. doi:10.1038/ajg.2015.233
- Turner D, Ricciuto A, Lewis A, et al. STRIDE-II: an update on the Selecting Therapeutic Targets in Inflammatory Bowel Disease (STRIDE) Initiative of the International Organization for the Study of IBD (IOIBD): determining therapeutic goals for treat-to-target strategies in IBD. Gastroenterology. 2021;160(5):1570–1583. doi:10.1053/j.gastro.2020.12.031
- Shah SC, Colombel JF, Sands BE, Narula N. Mucosal healing is associated with improved long-term outcomes of patients with ulcerative colitis: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2016;14(9):1245–1255.e8. doi:10.1016/j.cgh.2016.01.015
- Mosli MH, Feagan BG, Sandborn WJ, et al. Histologic evaluation of ulcerative colitis: a systematic review of disease activity indices. Inflamm Bowel Dis. 2014;20(3):564–575. doi:10.1097/01.MIB.0000437986.00190.71
- Peyrin-Biroulet L, Bressenot A, Kampman W. Histologic remission: the ultimate therapeutic goal in ulcerative colitis? Clin Gastroenterol Hepatol. 2014;12(6):929–34.e2. doi:10.1016/j.cgh.2013.07.022
- Squibb BM. U.S. food and drug administration approves Bristol Myers squibb’s Zeposia® (ozanimod), an oral treatment for adults with moderately to severely active ulcerative colitis. Available from: https://news.bms.com/news/details/2021/U.S.-Food-and-Drug-Administration-Approves-Bristol-Myers-Squibbs-Zeposia-ozanimod-an-Oral-Treatment-for-Adults-with-Moderately-to-Severely-Active-Ulcerative-Colitis1/default.aspx. Accessed May 27, 2022.
- Colombel JF, Sandborn WJ, Reinisch W, et al. Infliximab, azathioprine, or combination therapy for Crohn’s disease. N Engl J Med. 2010;362(15):1383–1395. doi:10.1056/NEJMoa0904492
- U.S. National Library of Medicine NIoH. A study of efficacy and safety of combination therapy with guselkumab and golimumab in participants with moderately to severely active ulcerative colitis (VEGA). Available from: https://clinicaltrials.gov/ct2/show/NCT03662542. Accessed July 28, 2022.