Abstract
Purpose
The primary aim of the present study was to assess the pharmacokinetic bioequivalence between a generic formulation of meloxicam 15 mg tablets (Meloxicam Hexal) and its respective brand product (Mobic), in order to verify whether the generic product conforms to the regulatory standards of bioequivalence in the postmarketing setting. As a secondary exploratory aim, the pharmacodynamic effects of the two formulations were also evaluated by means of rating scales following hyperalgesia induced by cutaneous freeze injury.
Subjects and methods
A single 15 mg dose of generic or branded meloxicam tablets was administered to 24 healthy male volunteers in a crossover fashion. Plasma samples, collected for 24 hours after dosing, were assayed for meloxicam concentration by a validated highperformance liquid chromatography method.
Results
The analysis of pharmacokinetic parameters did not show any significant difference between the two meloxicam formulations: the 90% confidence intervals fell within the acceptance range of 80%–125% (0.84–1.16 for area under the curve [0–24], and 0.89–1.23 for peak concentration). No difference in the pharmacodynamic end point was observed between the two groups.
Conclusion
The pharmacokinetic profiles of the two meloxicam formulations confirm the regulatory criteria for bioequivalence; pharmacodynamic data indicate a similar antihyperalgesic effect. The two formulations can be used interchangeably in the clinical setting.
Introduction
The purpose of assessing bioequivalence is to demonstrate equivalence in biopharmaceutic quality between the generic medicinal product and the reference branded formulation, in order to allow bridging of preclinical tests and clinical trials associated with the patented original product. Accordingly, pharmaceutical companies, involved in the development of generic drugs, are allowed to follow abbreviated procedures to demonstrate bioequivalence between brand and generic formulations, without the need of repeating a complete, and expensive program of pharmacological and clinical development.Citation1
In the Italian setting, open access to preregistration documentation of generic drugs is not allowed, and, on the basis of European Medicines Agency (EMA) guidance, an immediate-release oral formulation of a generic copy (including nonsteroidal anti-inflammatory drugs) might be approved as a biowaiver, by means of in vitro dissolution tests, without performing any clinical investigation.Citation1 It is also being increasingly acknowledged that the regulatory criteria for assuming bioequivalence between branded and generic drugs might be insufficient at times, and this is an important issue deserving consideration, in order to ensure full therapeutic equivalence in the postmarketing period. Therefore, performing postmarketing investigations on generic drugs appears to be important to control the quality of marketed formulations. Of note, some postmarketing studies are raising doubts on the actual therapeutic interchangeability between branded and generic drugs available in the pharmaceutical market. For instance, in a postmarketing pharmacokinetic (PK) crossover trial on two generic amoxicillin formulations, a lack of interchangeability was found for one generic product.Citation2 Moreover, some authorsCitation3–Citation5 argued that switching from brand antiepileptics to generic copies might result in an increased risk of therapeutic failure (breakthrough seizures) or adverse reactions. Other examples of treatment failure, with marketed antibiotic,Citation6 anticoagulant,Citation7 proton-pump inhibitor,Citation8,Citation9 antiarrhythmic,Citation10 or antidepressantCitation11 generic products, have also been published. Thus, the validity of current criteria for assessing interchangeability of generic and branded drugs is being questioned, at least in some instances, and it is becoming evident that inadequate programs for postmarketing quality control of medicinal products might increase the risk of therapeutic failures and/or safety concerns in patients.Citation12 Moreover, generic drug use is steadily increasing in Italy, particularly in Tuscany, where the regional health authority strongly supports the prescription of such products in order to reduce health-care costs.
Based on the above considerations, our Clinical Pharmacology Centre for Drug Experimentation has implemented a research program to perform clinical studies aimed at verifying the quality of generic drugs available in the Italian pharmaceutical market, owing to specific scientific interest on the quality of generic drugs prescribed in current clinical practice.
Since nonsteroidal anti-inflammatory drugs represent one of the most prescribed pharmaceutical classes in Italy, we selected Mobic (Boehringer Ingelheim GmbH, Ingelheim, Germany) as a branded meloxicam formulation and randomly selected Meloxicam Hexal among the generic compounds, both marketed in Italy, in order to evaluate whether the generic formulation fulfills the criteria for clinical PK bioequivalence versus its reference branded product.
The choice to indicate both the originator (Mobic) and generic (Meloxicam Hexal) meloxicam brand names was due to transparency reasons and the lack of any conflict of interest between the respective drug companies and the authors. Moreover, we deemed it interesting to perform an exploratory pharmacodynamic (PD) study on the two meloxicam formulations, in order to assess their analgesic effects and compare their PD/PK relationship. In this regard, the PD test was carried out by estimating the mechanical pain threshold of cutaneous freeze injury with a von Frey device.Citation13
Materials and methods
Subjects
Twenty-four healthy male volunteers were recruited at the Clinical Pharmacology Centre for Drug Experimentation of the University Hospital of Pisa (Italy). The exclusion of women from participating in the present study was due to sex-dependent differences in the PK of meloxicam, as previously reported by Meineke and Türck,Citation14 who demonstrated that both age and sex significantly affected meloxicam clearance.
In addition, this study investigated the relationship between the PK and PD of meloxicam in healthy volunteers, and therefore the exclusion of females was justified by the known hormonal influence on pain tolerance and threshold in menstruating women.Citation15–Citation18 At the same time, the exclusion of women might be regarded as a limitation, since a more heterogeneous study population might have increased the generalization of our findings. Eligible subjects were aged 18–50 years, had a body mass index (BMI) between 19 and 25 kg/m2, did not smoke, and had an unremarkable clinical history. Subjects with history or evidence of renal, gastrointestinal, hepatic, or hematologic abnormalities, any acute or chronic disease, or any drug allergy were excluded. Clinical evaluations and tests included: medical history, physical examination, height, weight, BMI, vital signs (heart rate, systolic and diastolic blood pressure, and body temperature), electrocardiogram, renal and liver-function tests, urinalysis, and urine drug and alcohol testing. Volunteers were requested to report any abnormality occurring throughout and after the study. Subjects were required to be negative for human immunodeficiency virus (HIV) and hepatitis B and C viruses.
At the time of enrollment, the volunteers were informed of the purpose, duration, and risks of the study, and they were requested to sign a written informed consent. They were not allowed to consume alcohol, or beverages and foods containing caffeine from 48 hours prior to drug administration until the end of the study. All participants were requested to fast for 10 hours before drug dosing, in order to minimize the PK variability and allow appropriate comparisons with data available in the literature.Citation19–Citation22 The dietary regimen was the same for all subjects in both trial periods, and consisted of two standard meals served 12 hours before and 5 hours and 9 hours after dosing. No additional food intake was allowed throughout the study period. Liquid consumption was allowed ad libitum from 2 hours after dosing. Subjects were reimbursed for their time and transportation expenses, irrespective of whether they completed the study or not, in accordance with standard operating procedures implemented at our Clinical Pharmacology Centre.Citation23 The study was performed according to the rules of Good Clinical PracticeCitation24 and the Declaration of Helsinki;Citation25 the study protocol was approved by the Ethics Committee of Pisa University Hospital.
Study design and drug administration
This was a single-dose, randomized two-treatment, crossover, single-blind PK/PD evaluation designed to compare generic 15 mg tablet Meloxicam Hexal (test product) with branded Mobic 15 mg tablets (reference product). The enrolled volunteers were randomized into two groups of twelve subjects each, and each group received the two drug treatments at two different times, with an intervening 2-week washout period. Meloxicam tablets were administered with 250 mL of water at 8 am after overnight fasting. Venous blood samples of 5 mL were collected through an indwelling cannula placed on the forearm, into Vacutainer™ (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) tubes (containing sodium heparin) at preset time intervals of 0 (predose), 0.5, 1, 2, 4, 6, 8, 12, and 24 hours after dosing. Blood samples were centrifuged at 900 g for 15 minutes; plasma samples were transferred into Vacutainer™ tubes (no additive) and stored at −80°C until subsequent analysis.
Analytical method
Measurement of meloxicam concentrations in plasma samples was performed by validated high-performance liquid chromatography (HPLC) with ultraviolet detection,Citation26 with some modifications. Briefly, for each sample, 200 μL of plasma was added with 20 μL of piroxicam 10 μg/mL (internal standard) plus 200 μL of acetonitrile acidified with H3PO4 0.1% (v/v), vortexed for 20 seconds, and centrifuged for 5 minutes at 11,000 rpm in a Heraeus Biofuge 15 (Thermo Fisher Scientific, Waltham, MA, USA) centrifuge. The clear supernatants (approximately 150 μL) were collected in vials and transferred into the autosampler. The Waters Breeze HPLC system (Waters, Milford MA, USA), equipped with a Waters 2476 dual-wavelength ultraviolet detector set at 355 nm, was used. The mobile phase, consisting of potassium phosphate buffer (K2HPO4) 20 mM (pH 3.5) and acetonitrile (70:30, vol/vol), was isocratically pumped at a flow rate of 1.2 mL/minute through an Alltima CN chromatographic column, 150 × 4.6 mm, 5 μm (Alltech Italia, Milan, Italy). Plasma from untreated healthy subjects was used to reconstitute calibration and quality standards. In particular, for calibration standards, meloxicam was added to plasma to obtain final concentrations of 50, 10, 5, 2.5, 1.25, 0.625, 0.313, 0.156, 0.078, and 0.039 mg/L in a final volume of 200 μL. Quality controls were prepared independently from calibration-standard samples at drug concentrations of 5, 0.625, and 0.078 mg/L. Samples were prepared and extracted as described above. Calibration curves of standard meloxicam were generated by plotting analyte peak areas versus drug concentrations, and linearity was estimated on at least three calibration curves obtained on 3 consecutive days. Calibration standards were also used to evaluate the parameters listed below.
Determination of meloxicam plasma concentration
Under the aforementioned chromatographic conditions, meloxicam and piroxicam had retention times of 5.93 minutes ± 0.03 minutes and 4.53 minutes ± 0.02 minutes, and their recovery accounted for 91% and 85%, respectively. The background noise (defined as the mean amplitude of baseline oscillations) was 5 μV, and the limit of detection -the concentration of analytes generating a signal three times higher than baseline noise – was estimated to be 0.005 mg/L on the basis of standard calibration curves. For meloxicam, the limit of quantification, defined as a sample concentration for which the method was still in the linear range, was equal to 0.039 mg/L, which represented the lowest concentration among the calibration standards. The linearity of calibration curves ranged between 0.039 mg/L and 5 mg/L, with a mean value of correlation coefficient for meloxicam equal to 0.989 ± 0.003. The linear range was calculated by means of linear regression analysis, obtaining a standard deviation of the residuals that accounted for 0.11 mg/L ± 0.09 mg/L. Precision and accuracy values were calculated as reported in a previous study.Citation27 In particular, accuracy values accounted for 88.03%–114.66% over the whole range of the calibration standard. Precision values ranged from 2.01% up to 7.00% over the entire range, with the exception of the lowest calibration standard (0.039 mg/L), the precision of which accounted for 17.90%. For quality controls, accuracy and precision values were in the ranges of 87.00%–102.64% and 2.98%–13.38%, respectively.
Pharmacokinetic evaluations
The actual times of sample collection were used for PK analysis of branded and generic meloxicam formulations. Area under the curve (AUC) from time 0 to 24 hours (AUC0–24) and AUC from time 0 to 5 hours (AUC0–5) were estimated by the linear trapezoidal method, while AUC from time 0 to infinity (AUC0–inf) was obtained by adding to the AUC0–24 value the C24/kel ratio value, where C24 and kel represent the plasma concentration of meloxicam at 24 hours postdose and the elimination constant, respectively. Maximum concentration (Cmax) and the time to achieve Cmax (Tmax) were obtained from direct visual inspection of plasma concentration versus time curves.
PK parameters were calculated by a noncompartmental method using Stata version 10.0 (StataCorp, College Station, TX, USA). Bioequivalence comparison was carried out by the statistical software SAS version 8.2 (SAS Institute, Cary, NC, USA).
Pharmacodynamic evaluation
Twenty-two hours before the first day of each experimental session, a small skin area (designed as Z1) on the anterior glabrous region of the forearm was superficially frozen. For this purpose, the tip (1.8 cm2) of a cylindrical copper bar, previously cooled to −28°C, was applied to induce a first-degree burn injury. Interindividual variability in the intensity of the hyperemic response was low and blisters were never observed, in agreement with previous results from Chassaing et al.Citation13 Subsequently, sensory testing (ie, the assessment of baseline values of the first session) was performed 22 hours after the induction of cold injury. In the present study, mechanical hyperalgesia, as defined in the Textbook of Pain,Citation28 was assessed by punctate-evoked pain with a von Frey device, as previously validated by Chassaing et al.Citation13 Briefly, the quantification of pain, both at baseline and after treatment with meloxicam, was obtained by application of punctate stimuli with a constant slope of increasing punctate pressure up to the subjective perception of hyperalgesia. Mechanical pain threshold (MPT) was defined as the lowest pressure that produced a sensation of pain. Each MPT value was averaged from five separate consecutive measurements at different points within the test areas. A normal skin area (designed as Z0) on the contralateral arm was used as control. Both basal and postdosing tests were performed by the same investigator, who was blind to drug allocation. The punctate stimulation was started well outside the hyperalgesic area, where no sensation of pain was experienced, and continued centripetally in 5 mm steps towards the site of freeze injury until the volunteer reported a subjective perception of hyperalgesia.
Statistical analysis
Randomization was performed using a random-number table. The groups were designated as RT (subjects who received first the reference and then the test product) and TR (subjects who received first the test and then the reference product). Analysis of variance (ANOVA) for a standard 2 × 2 crossover design was used to evaluate period, sequence, and formulation effect. Statistical analysis was performed by parametric mixed-model accounting for subjects included in nested sequences as random effect. Log-natural transformed AUC0–24 and Cmax were used to evaluate the ratio and 90% confidence interval (CI) of the test drug over the reference product, according to Schuirmann’s procedure.Citation29 Furthermore, Tmax difference between the two meloxicam formulations was assessed by Mann–Whitney U test. Finally, demographic characteristics differences between the RT and TR groups were tested by one-way ANOVA or Mann–Whitney U test, according to the distribution of the variables of interest. PD subanalysis was performed by ANOVA for a standard 2 × 2 crossover design.
Tolerability assessment
Subjects were under continuous medical supervision at the study site throughout the study period. A physician, who was blind to the study treatment, was present throughout the study period to monitor the subjects and record possible adverse events (AEs). Every 2 hours, subjects were asked about and encouraged to report any unusual symptoms. Tolerability was assessed by investigators based on subject interviews, spontaneous reporting, vital signs, physical examination, and clinical laboratory tests (urinalysis, hematology, and blood chemistry) before and throughout the study period. Resting blood pressure, heart rate, and body temperature were monitored by investigators before drug administration (baseline) and every 2 hours throughout the study. AEs were considered serious if they were life-threatening or led to death, disability, inpatient hospitalization, or medical intervention to prevent permanent impairment or damage. All AEs were recorded on case-report forms by an investigator. Any serious AE suspected to be drug-related by investigators was reported to the ethics committee and to the local pharmacovigilance authority in accordance with the study protocol.
Results
Subject characteristics
The baseline demographic characteristics of the two healthy volunteer groups are reported in . No differences between the RT and TR groups in terms of age, height, weight and BMI, systolic and diastolic blood pressure, or heart rate were found, indicating the substantial homogeneity of the two study groups.
Table 1 Summary of baseline demographic characteristics in the study population of healthy male volunteers
Pharmacokinetic evaluations
Mean plasma profiles of meloxicam in the study subjects, exposed to branded (Mobic) or generic (Meloxicam Hexal) formulations, are shown in . The respective values of estimated PK parameters are reported in and .
Figure 1 Mean concentration-time profiles in plasma following the oral administration of 15 mg tablets of branded Mobic (•) or generic Meloxicam Hexal (□) formulation to healthy volunteers. Each point represents the geometric mean (95% confidence interval in vertical lines).
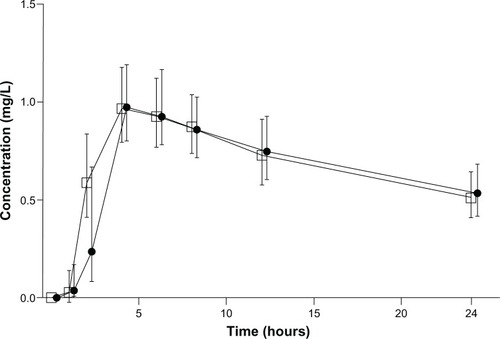
Table 2 Values of plasma pharmacokinetic parameters obtained from the study population of healthy male volunteers (n = 24) following the administration of generic (Meloxicam Hexal) and branded (Mobic) meloxicam formulations
Table 3 Values of the ratios of AUC0–5, AUC0–24, AUC0–inf, and Cmax with 90% confidence intervals
Pharmacokinetic bioequivalence assessment
All the enrolled volunteers completed the study and were included in the final study population in order to perform statistical analysis. A significant period effect was found for AUC0–24 (P = 0.03) but not Cmax (P = 0.09), with no sequence effect for either AUC0–24 or Cmax (P = 0.7113 and 0.8560, respectively), indicating a lack of formulation effect. Moreover, coefficient of variation values, estimated by ANOVA, were slightly above the recommended upper limit of 30% (34% for AUC0–24 and 33% for Cmax).
When comparing the generic formulation of meloxicam with the respective branded product, the test/reference ratios and 90% CIs were 0.99 (0.84–1.16) for AUC0–24, 0.98 (0.81–1.19) for AUC0–inf, and 1.05 (0.89–1.23) for Cmax. Moreover, no significant difference between the two meloxicam formulations was found with regard to Tmax (P = 0.472). Thus, on the basis of EMA guidelines, the 90% CI for the relevant PK parameters fell within the acceptability range of 0.80–1.25, indicating pharmacokinetic bioequivalence between the two meloxicam products. Bioequivalence was not confirmed when AUC0–5 test/reference ratio with CI was calculated: the ratio was 1.09 and CI 0.92–1.30 ().
Pharmacodynamic evaluation
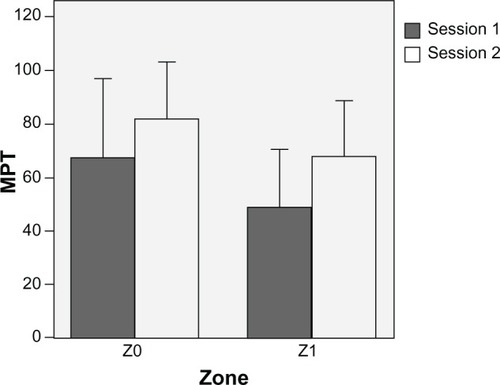
A significant difference of MPT values between control (Z0) and hyperalgesic skin areas (Z1) was observed (74.8 ± 8.7 versus 58.2 ± 7.4, P < 0.001). The MPT values for each area (Z0, Z1) at 5 hours are displayed in . The mean MPT values presented a period effect, with a reduction of both Z0 and Z1 MPT in the second period of the study. No significant differences between the two formulations of meloxicam were found: 63.7% ± 22.5% versus 52.3% ± 13.0% at baseline (P = 0.940) and 88.2% ± 34.5% versus 57.8% ± 17.5% at 5 hours (P = 0.797) for Mobic and Meloxicam Hexal, respectively.
Figure 2 Mechanical pain threshold (MPT) at 5 hours determined by a von Frey device over the two sessions on control (Z0) and hyperalgesic (Z1) skin zone. Each session was performed according to the crossover study design (period 1 and period 2). Data are expressed as means ± standard deviation (vertical lines).
Tolerability
Two not-serious AEs were recorded during the study. A skin rash with headache developed in a volunteer 30 minutes after Mobic administration, with a spontaneous recovery after 2 hours; this AE was considered as probably related to the investigational medicinal product administration. One subject reported cervical pain, which was considered as drug-unrelated.
Discussion
Concerns are being raised about the possibility that the quality of some marketed generic drugs may not fully reflect the results obtained in preregistration bioequivalence investigations.Citation30–Citation32 Substitution of branded products with generic drugs is a matter of discussion, and it is often regarded with skepticism by both health-care providers and patients. Current bioequivalence requirements are based on a measure of average bioequivalence; however, there are concerns that the use of such a measure might be inappropriate in the case of drugs with high intrasubject or intersubject variability, like analgesic medications.Citation33 Moreover, in some countries, the lack of open access to documentation of preregistration studies represents a point of weakness of the generic drug market. Indeed, prescribing physicians cannot access any information on the preregistration development of generic drugs, and they can only trust that the regulatory authority approved a specific generic formulation in full accordance with recommended procedures. If a generic drug was approved with only the minimum of regulatory requirements for its registration (ie, by an in vitro study), the postmarketing interchangeability of that generic drug with its respective branded product might not be warranted. Based on these considerations, the present study was performed: (1) to compare the PK profiles of two meloxicam formulations (branded and generic), containing different excipient ingredients, in order to verify whether the generic formulation conforms with its respective branded product, in accordance with the criteria established by EMA guidelines and Italian laws; and (2) to evaluate whether a PD end point comparison can be related to PK analysis and is suitable for improving the control of quality pursued by postmarketing studies.
In accordance with EMA guidelines, estimated PK parameters AUC0–24 and Cmax fell within the acceptability range of 0.80–1.25, indicating the interchangeability between the two meloxicam products and confirming that PK analysis is consistent with PD comparison results. Bioequivalence was not confirmed in a subanalysis of AUC0–5 due to the huge variation of standard deviation in the first phases of PK (dissolution/absorption), but PD testing at 5 hours showed that there was no difference between formulations in terms of drug effects.
It is worth noting that the AUC0–5 PK end point is not considered by current guidelines, but the present study focused attention on this end point to investigate whether it could predict the analgesic effect early after drug administration. Moreover, the PD test was considered an explorative analysis, and taking into account the huge variation obtained in this analysis, the PD results led to a more probable beta error, thus making arguable the acceptance of the null hypothesis (drug equivalence). Since large intersubject variability of AUC0–5 values and PD results hampered our analysis, this issue should be carefully considered in future studies, where the collection of plasma samples at earlier time points would allow more reliability and confidence in the results to be obtained.
The time-point collection (24 hours), which was not long enough to cover two drug half-lives, could appear as a limitation of the present study. However, the main study objective was not to investigate the complete PK profiles of meloxicam in healthy volunteers, since this information has been previously provided by other authors,Citation19–Citation22,Citation34,Citation35 but rather to compare PK and PD patterns between branded and generic meloxicam. On this basis, we performed a blood-sampling collection during the dissolution, absorption, and distribution PK phases, assuming that the elimination phase would not be significantly affected by the pharmaceutical formulation. Consistently with this assumption, when the present PK results were compared with those published earlier,Citation36 the length of time sampling appeared to be long enough to allow a good estimation of meloxicam PK.
The present results are in agreement with previous PK dataCitation19–Citation22,Citation34 obtained on branded meloxicam in healthy volunteers. When considering the study performed by Marcelín-Jiménez et alCitation35 in a Mexican female population, although the Tmax value was similar to that estimated in the present study, both Cmax and AUC0–24 values were found to be higher. However, differences in sex, body weight, genetic profile, and pharmaceutical composition of tablets are likely to explain the differences between the Italian and Mexican studies.
If two drugs are proven to be interchangeable, high-quality manufacturing standards are followed, and stringent quality controls are ensured over time, then the switch from a brand formulation to a generic one should be acceptable in most cases, without any expected noticeable variation in efficacy or tolerability.Citation37 However, postmarketing evaluations have highlighted cases of lack of equivalence between generic and branded drugs. One case is the study by Del Tacca et al,Citation2 who compared brand amoxicillin with two generic formulations available in the Italian market, and did not confirm interchangeability for one of the two generic products. In another study, the lack of postmarketing therapeutic equivalence between branded and generic cefuroxime was found to significantly influence the incidence of postoperative infections in adult patients undergoing coronary artery bypass grafting surgery.Citation6 Development of breakthrough seizures and increased seizure frequency was observed after switching to marketed generic antiepileptic drugs, possibly due to changes in serum concentrations.Citation4 Likewise, psychiatric symptom relapse in a stabilized patient, resulting in hospitalization, after a switch to a marketed generic formulation of clozapine,Citation38 has been reported. In the postmarketing setting, plasma levels of venlafaxine and its active metabolite O-desmethylvenlafaxine were found to be significantly higher in subjects given a generic product, as compared with those treated with the branded formulation.Citation11
Overall, bioequivalence studies should be considered not only as a part of the regulatory process, which allows the marketing authorization of a generic drug, but also as valuable scientific tools suitable for evaluating the “goodness” (ie, the quality) of generic drugs available in the pharmaceutical market, thus reducing the risks associated with a lack of interchangeability persistence. In particular, by means of comparative bioequivalence studies between generic products and their respective branded drugs in the postmarketing setting, we could assure for both patients and prescribing physicians not only a pharmaceutical but also a pharmacological (ie, PK and/or PD) equivalence, which would add support to the expected clinical efficacy of the marketed generic drug. Accordingly, an efficient program of postmarketing drug evaluation should take into account PK, and in some (specific) cases PD studies with single or multiple doses (when reaching a steady-state level is deemed relevant). In this respect, the present study suggests that PD evaluations may represent simple and useful tools, which might improve the quality of postmarketing comparisons. Indeed, when the PD equivalence analysis of generic and branded formulations completes the PK evaluation, the existence of actual interchangeability can be better documented.
Conclusion
The results of the present postmarketing study demonstrate that the PK profiles of the branded and generic meloxicam formulations confirm the regulatory criteria for bioequivalence. Taken together with the PD data, these results indicate that both formulations can be used interchangeably in clinical practice.
Acknowledgments
The authors wish to acknowledge Dr Catia Castiglioni for her invaluable support to the clinical management of study subjects, as well as Dr Ciro Conversano for his careful psychological evaluation of healthy volunteers before their enrollment.
Disclosure
This study was supported by unconditional funds provided by Boehringer Ingelheim. The authors report no other conflicts of interest in this work.
References
- European Medicines AgencyGuideline on the investigation of bioequivalence2010 Available from: http://www.emea.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdfAccessed July 24, 2012
- Del TaccaMPasqualettiGDi PaoloALack of pharmacokinetic bioequivalence between generic and branded amoxicillin formulations. A post-marketing clinical study on healthy volunteersBr J Clin Pharmacol200968344219660001
- CrawfordPFeelyMGubermanAKramerGAre there potential problems with generic substitution of antiepileptic drugs? A review of issuesSeizure20061516517616504545
- BergMJGrossRATomaszewskiKJZingaroWMHaskinsLSGeneric substitution in the treatment of epilepsy: case evidence of breakthrough seizuresNeurology20087152553018695164
- LeLorierJDuhMSParadisPEClinical consequences of generic substitution of lamotrigine for patients with epilepsyNeurology2008702179218618505997
- MastorakiEMichalopoulosAKriarasIIncidence of postoperative infections in patients undergoing coronary artery bypass grafting surgery receiving antimicrobial prophylaxis with original and generic cefuroximeJ Infect200856353917983660
- HalkinHShapiroJKurnikDLoebsteinRShalevVKokiaEIncreased warfarin doses and decreased international normalized ratio response after nationwide generic switchingClin Pharmacol Ther20037421522112966365
- ElkoshiZBehrDMirimskyATsvetkovIDanonAMultiple-dose studies can be a more sensitive assessment for bioequivalence than single-dose studies: the case with omeprazoleClin Drug Invest200222585592
- ShimataniTInoueMKuroiwaTXuJMienoHTazumaSAcid-suppressive effects of generic omeprazole: comparison of three brands of generic omeprazole with original omeprazoleDig Liver Dis20063855455916524789
- ReiffelJAIssues in the use of generic antiarrhythmic drugsCurr Opin Cardiol200116232911124715
- ChenuFBattenLAZernigGLadstaetterEHébertCBlierPComparison of pharmacokinetic profiles of brand-name and generic formulations of citalopram and venlafaxine: a crossover studyJ Clin Psychiatry20097095896619653973
- MeredithPAPotential concerns about generic substitution: bioequivalence versus therapeutic equivalence of different amlodipine salt formsCurr Med Res Opin2009252179218919601710
- ChassaingCSchmidtJEschalierACardotJMDubrayCHyperalgesia induced by cutaneous freeze injury for testing analgesics in healthy volunteersBr J Clin Pharmacol2006438939716542199
- MeinekeITürckDPopulation pharmacokinetic analysis of meloxicam in rheumatoid arthritis patientsBr J Clin Pharmacol200355323812534638
- SteningKErikssonOWahrenLBergGHammarMBlomqvistAPain sensations to the cold pressor test in normally menstruating women: comparison with men and relation to menstrual phase and serum sex steroid levelsAm J Physiol Regul Integr Comp Physiol2007293R1711R171617652363
- RingCVeldhuijzen van ZantenJJKavussanuMEffects of sex, phase of the menstrual cycle and gonadal hormones on pain in healthy humansBiol Psychol20098118919119393713
- KowalczykWJSullivanMAEvansSMBisagaAMVosburgSKComerSDSex differences and hormonal influences on response to mechanical pressure pain in humansJ Pain20101133034219853526
- TeepkerMPetersMVedderHSchepelmannKLautenbacherSMenstrual variation in experimental pain: correlation with gonadal hormonesNeuropsychobiology20106113114020110738
- GschwendMHErenmemişoğluAMartinWTamurUKanzikIHincalAAPharmacokinetic and bioequivalence study of meloxicam tablets in healthy male subjectsArzneimittelforschung20075726426817598697
- TangsucharitPKampanJKanjanawartSBioequivalence study of two meloxicam tablet formulations after single-dose administration in healthy Thai male volunteersInt J Clin Pharmacol Ther20094763864219825327
- HasanSMShoaibMHHassanFRehmanIUBioequivalence studies of two brands of meloxicam tablets in healthy Pakistani volunteersPak J Pharm Sci20092219920419339233
- AhmadMMurtazaGAkhtarNBioequivalence study of two brands of meloxicam tablets in healthy human Pakistani male subjectsActa Pol Pharm20116811511921485709
- PasqualettiGGoriGBlandizziCDel TaccaMHealthy volunteers and early phases of clinical experimentationEur J Clin Pharmacol20106664765320461363
- European Medicines AgencyGuideline for good clinical practice2002 Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002874.pdfAccessed July 24, 2012
- World Medical Association Declaration of Helsinki 2008Ethical principles for medical research involving human subjects2008 Available from: http://www.wma.net/en/30publications/10policies/b3/17c.pdfAccessed July 24, 2012
- BaeJWKimMJJangCGLeeSYDetermination of meloxicam in human plasma using a HPLC method with UV detection and its application to a pharmacokinetic studyJ Chromatogr B Analyt Technol Biomed Life Sci20078596973
- PolilloMTasciniCLastellaMA rapid high-performance liquid chromatography method to measure linezolid and daptomycin concentrations in human plasmaTher Drug Monit20103220020520216115
- MeyerRACampbellJNRajaSNPeripheral neural mechanisms of nociceptionWallPDMelzackRBonicaJJTextbook of PainNew YorkChurchill Livingstone19951344
- SchuirmannDJA comparison of the two one-sided tests procedure and the power approach for assessing the equivalence of average bioavailabilityJ Pharmacokinet Biopharm1987156576803450848
- ChenMLShahVPatnaikRBioavailability and bioequivalence: an FDA regulatory overviewPharm Res2001181645165011785681
- YuLXAmidonGLPolliJEBiopharmaceutics classification system: the scientific basis for biowaiver extensionsPharm Res20021992192512180542
- World Health OrganizationProposal to waive in vivo bioequivalence requirements for the WHO model list of essential medicines: immediate release, solid oral dosage forms2005 Available from: http://www.who.int/medicines/services/expertcommittees/pharmprep/QAS04_109Rev1_Waive_invivo_bioequiv.pdfAccessed July 24, 2012
- MeredithPBioequivalence and other unresolved issues in generic drug substitutionClin Ther2003252875289014693311
- TürckDRothWBuschUA review of the clinical pharmacokinetics of meloxicamBr J Rheumatol199635Suppl 113168630630
- Marcelín-JiménezGHernándezJAAngelesAPBioequivalence evaluation of two brands of meloxicam tablets (Promotion and Mobicox): pharmacokinetics in a healthy female Mexican populationBiopharm Drug Dispos20052616717115841494
- DaviesNMSkjodtNMClinical pharmacokinetics of meloxicam. A cyclo-oxygenase-2 preferential nonsteroidal anti-inflammatory drugClin Pharmacokinet19993611512610092958
- PerryRPerspectives on the bioequivalence and therapeutic equivalence of generic formulations: an overview of the landscapeClin Ther2010321796179721194603
- AlvarezCAMascarenasCTimmermanIIncreasing psychosis in a patient switched from Clozaril to generic clozapineAm J Psychiatry2006163474616585457